 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Discovery of an iridacycle catalyst with improved reactivity and enantioselectivity in the hydrogenation of dialkyl ketimines†
York Schramm, Fabiola Barrios-Landeros‡ and Andreas Pfaltz*
University of Basel, Department of Organic Chemistry, St. Johanns-Ring 19, 4056 Basel, Switzerland. E-mail: andreas.pfaltz@unibas.ch; Fax: +41 61 2671103; Tel: +41 61 2671108
First published on 9th April 2013
Abstract
Catalytically active iridacycles are formed by cyclometalation of acetophenone imines with Ir–PHOX complexes under hydrogen atmosphere. These complexes show unusually high reactivity and enantioselectivity in the hydrogenation of alkyl methyl ketimines. The structure of the cyclometalated imine has a strong effect on the conversion and enantiomeric excess.
Introduction
Chiral amines play an important role as building blocks for the synthesis of pharmaceuticals and agrochemicals. They are also of great importance as chiral auxiliaries, catalysts and resolving agents. Therefore, asymmetric hydrogenation of ketimines has received much attention as an attractive, very direct route to enantiomerically enriched amines.1 High yields, perfect atom economy and mild conditions make this approach ideal for industrial applications. This is impressively demonstrated by the multi-ton scale production of the herbicide metolachlor, based on an extremely active and productive Ir-diphosphine catalyst.2During the last two decades a wide range of chiral Ti, Rh, Ir, Pd, Ru,1 and most recently Fe1,3 complexes have been developed that catalyze the hydrogenation of various imines with high enantioselectivity. However, the scope of most catalysts is rather narrow and there are still important classes of imines that give unsatisfactory results with the available catalysts. Especially the hydrogenation of imines derived from dialkyl ketones remains a challenging problem. With the exception of the dual catalyst system reported by Xiao and co-workers,4 consisting of a chiral Ir(Cp*)–diamine complex and an elaborate chiral binaphthol-derived phosphoric acid (TRIP), most catalysts give very low enantioselectivities with these substrates. Organocatalytic asymmetric imine reduction and reductive amination has also been developed to afford chiral aliphatic amines with high enantioselectivities.5 However, these reactions generally suffer from lower yields and very long reaction times compared to transition-metal catalyzed hydrogenations. Furthermore, they require hydride donors such as dihydropyridines that generate stoichiometric waste products. So a practical readily accessible catalyst for the asymmetric hydrogenation of dialkyl ketimines remains elusive.
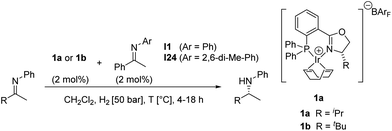
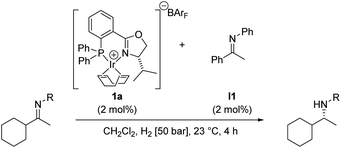
We have recently begun to reinvestigate Ir–phosphinooxazoline complexes that we originally introduced as catalysts for imine hydrogenation in 1997.6a After evaluation of a wide range of phosphinooxazoline (PHOX) derivatives and careful optimization of the reaction conditions, excellent enantioselectivities and high turnover numbers have been achieved in the hydrogenation of aryl alkyl N-arylketimines such as I1 (Scheme 1).6b
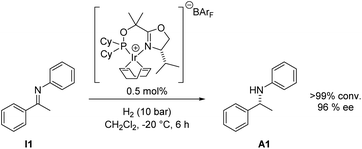 | ||
| Scheme 1 Asymmetric hydrogenation of acetophenone N-phenylimine I1. | ||
However, analogous dialkyl ketimines still gave disappointing results. We therefore decided to conduct a mechanistic study, in the hope that it would guide the development of improved catalysts with broader substrate scope.
Results and discussion
Here we report the outcome of this study that led to surprising insights into the structure of the catalytic intermediates and, ultimately, to a new catalyst system that gave promising results in the hydrogenation of dialkyl ketimines.Experimental mechanistic studies of imine hydrogenation with Ir-PHOX complexes have not been reported. However, Hopmann and Bayer7 carried out DFT calculations with catalyst 1a on nine different catalytic cycles based on proposed mechanisms for imine hydrogenations with other types of Ir complexes. The computed energies of the turnover-limiting transition states indicated a pathway involving proton transfer from an Ir–hydride complex to a free, uncoordinated imine, followed by hydride transfer to the resulting iminium ion. As all proposed mechanisms included Ir–dihydride complexes as intermediates, we decided to prepare a dihydride complex from the Ir(PHOX) precursor 1a following the procedure of Mazet et al.8 and to study its reactivity with I1 as a typical substrate (Scheme 2).
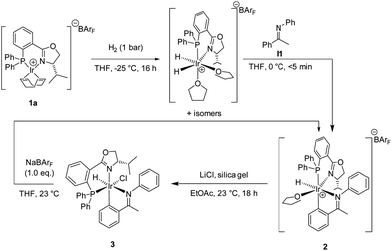 | ||
| Scheme 2 Formation of cyclometalated imine complexes. | ||
When the dihydride complex, generated by reaction of 1a with H2 in THF at −25 °C, was treated with I1 at 0 °C, rapid formation of an iridacycle 2 was observed. This complex proved to be very sensitive and rapidly decomposed upon exposure to air. Attempts to obtain suitable crystals for X-ray analysis failed and, therefore, the BArF (tetrakis[(3,5-trifluoromethyl)phenyl]borate) counterion that, in our experience, often impedes crystallization, was exchanged with chloride or hexafluorophosphate. The chloride complex 3 was readily obtained by treatment with LiCl and silica gel in ethyl acetate and purified by flash chromatography on silica gel. Ion exchange with NaBArF in THF led back to the BArF complex 2.
While crystallization attempts of 3 were unsuccessful, the hexafluorophosphate salt 5 furnished suitable crystals for X-ray analysis (Scheme 3 and Fig. 1).9 Furthermore, a preliminary crystal structure of the analogous iridacycle 7 prepared from SimplePHOX complex 6 (Scheme 4 and Fig. 2) could also be determined.10
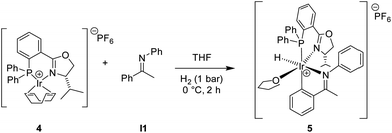 | ||
| Scheme 3 Preparation of iridacycle 5. | ||
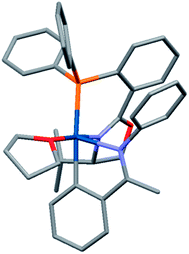 | ||
| Fig. 1 Crystal structure of 5 (PF6 counterion omitted for clarity). | ||
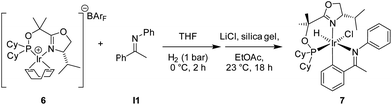 | ||
| Scheme 4 Preparation of iridacycle 7. | ||
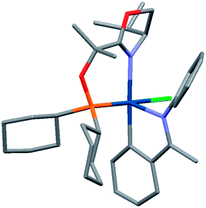 | ||
| Fig. 2 Preliminary crystal structure of 7. | ||
An analogous cyclometalation reaction of [Ir(H)2(PPh3)2(acetone)2]PF6 with benzaldehyde N-benzylimine has been reported by James and co-workers.11 The resulting iridacycle was tested in the hydrogenation of imines but showed no catalytic activity.11a
When complex 3 was tested as catalyst for the hydrogenation of I1, no reaction was observed. However, when the chloride was replaced with the non-coordinating anion BArF by addition of an equimolar amount of NaBArF, an active catalyst was generated that furnished the same enantiomer of amine A1 with identical enantioselectivity as the reduction catalyzed by the parent complex 1a (Scheme 5).
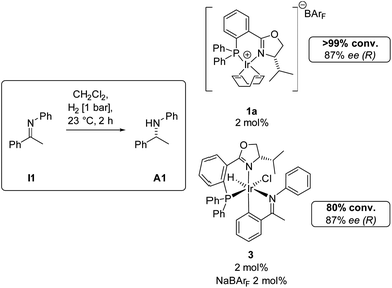 | ||
| Scheme 5 Comparison of complexes 1a and 3 as precatalysts for the hydrogenation of imine I1. | ||
What are the possible conclusions that can be drawn from these results? The iridacycle formed under hydrogenation conditions could be an inactive species outside the catalytic cycle that is in equilibrium with an active catalytic intermediate through reversible cyclometalation/reductive elimination, similar to the reaction scheme proposed by Marcazzan and James.12
Alternatively, it could be directly involved in the catalytic cycle. In this case, it could either react via reduction of the cyclometalated imine, followed by reductive elimination releasing the saturated amine, or the cyclometalated imine could serve as a stable ligand that remains bound throughout the catalytic cycle.
To distinguish between these possibilities we carried out the cross-over experiment shown in Scheme 6. If a catalytic intermediate derived from 3 would release the free imine I1, the cyclometalation product of substrate I2 along with amine A1 would be formed. However, we did not observe even traces of I1 or A1 in the course of the hydrogenation reaction by GC analysis.13,14 These results are consistent with the hypothesis that cyclometalation is irreversible and the imine remains bound to iridium throughout the catalytic reaction. The enantiomeric excess of A2 remained constant throughout the reaction.13
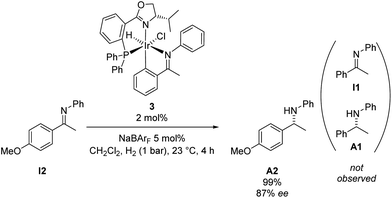 | ||
| Scheme 6 Hydrogenation of imine I2 with iridacycle 3 in combination with NaBArF. | ||
We speculated that the cyclometalated complex formed under hydrogenation conditions could be a superior catalyst compared to complex 1a and that the poor results in the hydrogenation of aliphatic ketimines could be a consequence of the inability of these substrates to form cyclometalated complexes.
Indeed, the catalyst generated in situ from the iridacycle 3 by treatment with NaBArF gave much higher conversion and enantioselectivity (Table 1, entry 1) in the hydrogenation of cyclohexyl methyl ketimine I3 than the parent Ir–PHOX complex 1a (entry 2) or iridacycle 3 alone (entry 3). Identical ee and higher conversion was obtained when the catalyst was generated by treating complex 1a with H2 and an equimolar amount of acetophenone imine I1 (entry 4). At 50 bar H2 pressure 71% ee and full conversion were observed after activation with imine I1 (entry 5), compared to 27% conversion and 35% ee with complex 1a alone (entry 6).
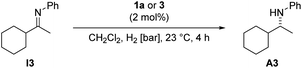
These findings clearly indicate that the new more efficient catalyst is a cationic cyclometalated complex that arises by chloride abstraction from 3 or by reaction of precatalyst 1a with H2 and imine I1 (cf. structure 2 in Scheme 2). Because cationic complexes such as 2 or 5 with very weakly coordinating anions proved to be too unstable and impractical to be used as catalysts, in situ activation of precatalyst 1a with acetophenone imine I1 or derivatives thereof was the method of choice for further experiments.
If the imine remains bound to the catalyst throughout the catalytic cycle, it is also involved in the enantiodiscriminating step. Cyclometalation of a chiral additive to an achiral complex would thus provide a chiral imine hydrogenation catalyst. We therefore prepared chiral complex (S)-11 derived from an achiral iridium–PHOX complex 8 and a chiral imine 9 (Scheme 7).
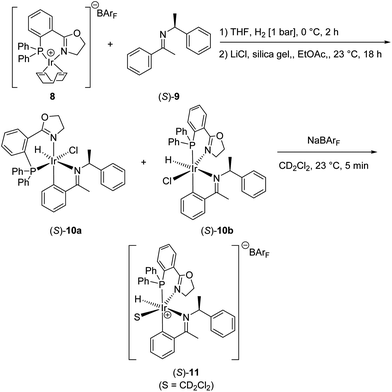 | ||
| Scheme 7 Preparation of chiral iridium complexes (S)-10 derived from achiral Ir-PHOX complex 8 and chiral imine 9. Upon addition of NaBArF, chloride abstraction results in the formation of one single hydride complex (S)-11 in solution. | ||
NMR analysis indicated the presence of two diastereomers of complex (S)-10. However, upon treatment with an equimolar amount of NaBArF in CD2Cl2, a single hydride species (S)-11 was observed.
Both enantiomers of complex 11, formed in situ from the diastereomeric mixture of (S)-10ab or (R)-10ab, were tested as catalysts for the hydrogenation of I1 (Table 2). While low conversion was observed at atmospheric hydrogen pressure, hydrogenation at 5 bar afforded A1 with 54% conversion (entry 3).
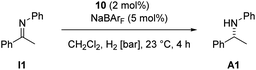
The resulting ee values showed a weak but reproducible influence of the chiral imine 9. Analogous catalysts derived from 1,3-benzoxazines were tested as well (Table 3) and gave reproducible enantioselectivities of up to 23% ee.
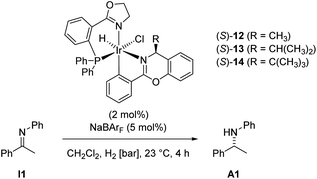
Erosion of enantioselectivity was observed when hydrogenations were conducted at 5 bar (entries 2 vs. 3 and 4 vs. 5). Furthermore, complex 14 gave higher conversion but lower enantioselectivity than complex 13 (entries 2, 4 and 6). To get a more accurate correlation of the ee with conversion, the reaction was followed by GC and HPLC analysis (Fig. 3).
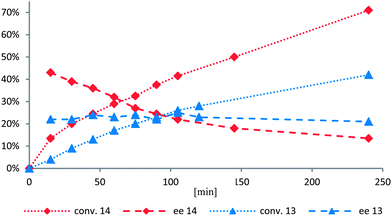 | ||
| Fig. 3 Conversion (dotted) and enantioselectivity (dashed) of A1 using iridacycle 13 (blue triangles) and 14 (red diamonds) as the catalyst. | ||
While complex 13 reacted with almost constant enantioselectivity, 14 showed a strong erosion of the enantioselectivity with increasing conversion. Furthermore, an overall higher conversion and faster reaction was observed for 14.
The following conclusions were drawn from these observations: while in 13 the benzoxazine remains bound to the iridium center throughout the reaction, complex 14 is not stable under the reaction conditions. As a consequence the benzoxazine ligand is replaced by imine I1 resulting in a complex with two achiral ligands, which produces racemic product. However, the high initial enantioselectivity of 44% ee clearly demonstrates that the cyclometalated ligand is involved in the enantiodiscriminating step of the reaction.
Attempts to improve the enantioselectivity by using catalysts prepared from a combination of chiral PHOX complexes with chiral benzoxazines gave disappointing results. However, systematic evaluation of various achiral N-aryl acetophenone imines as additives was more successful (Table 4).
| |||||
|---|---|---|---|---|---|
| Entry | Additive | R | R′ | Conv.a (%) | eeb (%) |
| a Determined by GC analysis.b Determined by HPLC analysis on a chiral stationary phase. | |||||
| 1 | I1 | H | H | >99 | 71 (R) |
| 2 | I4 | 2-Me | H | >99 | 69 (R) |
| 3 | I5 | 2-F | H | 90–95 | 71 (R) |
| 4 | I6 | 3-NO2 | H | 75–99 | 48 (R) |
| 5 | I7 | 3,5-(NO2)2 | H | 10 | 15 (R) |
| 6 | I8 | 3,5-Me2 | H | >99 | 3 (S) |
| 7 | I9 | 3,5-iPr2 | H | 34 | 42 (S) |
| 8 | I10 | 3,5-tBu2 | H | 11 | 10 (R) |
| 9 | I2 | 4-MeO | H | >99 | 67 (R) |
| 10 | I11 | 4-tBu | H | >99 | 64 (R) |
| 11 | I12 | 4-Me | H | >99 | 63 (R) |
| 12 | I13 | 4-Cl | H | >99 | 56 (R) |
| 13 | I14 | 4-F | H | >99 | 66 (R) |
| 14 | I15 | 4-CF3 | H | 95 | 50 (R) |
| 15 | I16 | 4-NO2 | H | 40 | 45 (R) |
| 16 | I17 | 4-Me | 2-Br | >99 | 68 (R) |
| 17 | I18 | H | 2-Me | >99 | 78 (R) |
| 18 | I19 | H | 2-MeO | >99 | 71 (R) |
| 19 | I20 | H | 3-MeO | >99 | 68 (R) |
| 20 | I21 | H | 4-MeO | >99 | 69 (R) |
| 21 | I22 | H | 4-CF3 | >99 | 59 (R) |
| 22 | I23 | H | 2-iPr | >99 | 81 (R) |
| 23 | I24 | H | 2,6-Me2 | >99 | 85 (R) |
The influence of substituents in the acetophenone phenyl ring showed no apparent trends that could be correlated with steric or electronic effects. Surprisingly, meta-substituents distinctly lowered the enantioselectivities (entries 4–8),15 while an ortho-methyl or ortho-fluoro substituent had essentially no effect (entries 2 and 3). Overall, introduction of substituents in the acetophenone phenyl ring did not improve the enantioselectivity. In contrast, ortho-alkyl groups in the N-aryl ring resulted in enhanced enantioselectivity (entries 17, 22 and 23). Other iridium complexes were screened as well, but none of them reached the enantioselectivities achieved with PHOX complex 1a.13
With an optimized catalyst system in hand we studied the scope for the hydrogenation of aliphatic ketimines (Table 5). Higher conversions and enantioselectivities were obtained in all cases compared to reactions using complex 1a alone. The high reactivity of this catalyst system allowed lowering the reaction temperature to −5 °C. Under these conditions imine I3 furnished the product A3 with an improved ee of 92% and full conversion (entry 4). Isopropyl methyl ketimine I25 gave 84% ee but only 33% conversion (entry 5). The ee was further improved when the more bulky complex 1b was used as precatalyst for isobutyl methyl ketimine I26 and benzyl methyl ketimine I27 (entries 8 and 10). However, for α-branched alkyl methyl ketimines such as I3, I25, I31 and I32, no reaction was observed with complex 1b. The sterically less demanding n-alkyl methyl ketimines I28, I29 and I30 gave markedly lower enantioselectivity (entries 11–13). Chemoselective hydrogenation of an imine double bond in the presence of a trisubstituted olefin can be achieved as shown in I29 (entry 12).
| ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Catalyst | Additive | T/°C | Conv.a (%) | eeb (%) |
| a Determined by GC analysis.b Determined by HPLC analysis on a chiral stationary phase after purification by flash chromatography.c Contained <3% of fully saturated product. | ||||||
| 1 |  | 1a | I1 | 23 | >99 | 71 (R) |
| 2 | 1a | I1 | −5 | >99 | 73 (R) | |
| 3 | 1a | I24 | 23 | >99 | 85 (R) | |
| 4 | 1a | I24 | −5 | >99 | 92 (R) | |
| 5 |  | 1a | I24 | −5 | 33 | 84 (R) |
| 6 | 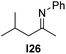 | 1a | I1 | −5 | >99 | 49 (−) |
| 7 | 1a | I24 | −5 | >99 | 70 (−) | |
| 8 | 1b | I24 | −5 | >99 | 80 (−) | |
| 9 | 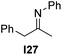 | 1a | I24 | −5 | >99 | 62 (+) |
| 10 | 1b | I24 | −5 | 94 | 72 (+) | |
| 11 | 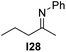 | 1b | I24 | −5 | >99 | 52 (−) |
| 12 | 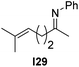 | 1b | I24 | −5 | >99c | 56 (−) |
| 13 | 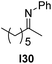 | 1b | I24 | −5 | >99 | 57 (R) |
| 14 | 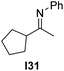 | 1a | I24 | −5 | 8 | 63 (−) |
| 15 | 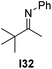 | 1a | I1 | −5 | 8 | 46 (R) |
To see whether the N-phenyl group was essential for achieving high enantioselectivity, we investigated N-alkyl imines I33–I36 as substrates (Table 6). Asymmetric hydrogenation of imines of this type has not been reported yet apart from I33. As these substrates proved to be less reactive, hydrogenations were conducted at room temperature. Using imine I1 as additive the N-benzylimine I33 furnished a moderate ee of 44% (entry 1). An even lower enantioselectivity is observed for the N-n-butylimine I34 (entry 2). On the other hand almost the same ee as for corresponding N-phenylimine I3 was observed in the hydrogenation of I35 (entry 3). The N-cyclohexyl analogue I34 reacted with even higher enantioselectivity of 77% ee (entry 4), demonstrating that purely alkyl-substituted imines are suitable substrates for this catalyst system. The more bulky complex 1b and the sterically demanding N-(2,6-dimethylphenyl)imine I24 afforded lower yields and enantioselectivities with these substrates.
| |||
|---|---|---|---|
| Entry | Substrate (R) | Conv.a (%) | eeb (%) |
| a Determined by GC analysis.b Determined by GC or HPLC analysis on a chiral stationary phase after derivatisation.c Determined after derivatisation to the 1-naphthoyl amide.d Determined after derivatisation to the acetamide. | |||
| 1 | I33 (CH2Ph) | >99 | 44 (R)c |
| 2 | I34 (nBu) | >99 | 33 (R) |
| 3 | I35 (iPr) | >99 | 73 (R)d |
| 4 | I36 (c-C6H11) | >99 | 77 (R)c |
Conclusions
We have found that the active catalyst in the hydrogenation of acetophenone-derived imines with Ir–PHOX precatalysts is an iridacycle generated under hydrogenation conditions by cyclometalation of the substrate. Cyclometalated complexes of this type, formed in situ by addition of an equimolar equivalent of acetophenone imine, show higher reactivity and better enantioselectivity in the hydrogenation of N-phenyl and N-alkyl aliphatic ketimines than the corresponding Ir-PHOX complex alone. Obviously, the reaction proceeds through a pathway that differs from the catalytic cycles proposed in the literature.7 Although at present the scope is still limited, our findings indicate many opportunities for further improvement of this catalyst system by structural variation of both the chiral P,N ligand and the cyclometalated imine.Experimental section
Screening
Imine (0.1 mmol), catalyst (2 μmol), additive (2 μmol), and a stir bar were added to an oven-dried glass vial that had been placed in an autoclave (60 mL) and purged with argon for 5 min. Anhydrous CH2Cl2 (1 mL) was added by syringe under a stream of argon and the autoclave was closed. For reactions at low temperature the autoclave was immersed in a cooling bath for 60 min before starting the reaction. The autoclave was pressurized with hydrogen gas, hydrogen was released and the autoclave pressurized again. It was then placed on a stirring plate for the time indicated. After releasing the pressure, the solvent was evaporated under a stream of nitrogen. The residue was suspended in pentane–diethyl ether (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and filtered through a short elution plug (cotton bottom, 40 × 5 mm silica gel). The crude filtrate was analysed by GC for conversion before being purified by flash chromatography (SiO2, pentane–diethyl ether (20:1), 15 × 2 cm) and analysed by HPLC on a chiral stationary phase for determination of the enantiomeric excess.
:1) and filtered through a short elution plug (cotton bottom, 40 × 5 mm silica gel). The crude filtrate was analysed by GC for conversion before being purified by flash chromatography (SiO2, pentane–diethyl ether (20:1), 15 × 2 cm) and analysed by HPLC on a chiral stationary phase for determination of the enantiomeric excess.Preparative reaction
Imine I3 (1.005 g, 5 mmol), 1a (0.1 mmol), I24 (0.1 mmol), and a stir bar were added to a 25 mL Pyrex oven-dried glass vial that had been placed in an autoclave (60 mL) and purged with argon for 5 min. Anhydrous CH2Cl2 (5 mL) was added by syringe under a stream of argon and the autoclave was closed. The autoclave was immersed in a cooling bath for 60 min at −5 °C before it was pressurized with hydrogen gas. Hydrogen was released and the autoclave pressurized again before being placed on a stirring plate for 18 h. After pressure release the reaction mixture was transferred to a 50 mL round-bottom flask and solvents removed under reduced pressure. The residue was suspended in pentane–diethyl ether (20:1) and purified by flash chromatography (SiO2, pentane–diethyl ether (10:1), 21 × 3 cm). Solvents were removed under reduced pressure and the residue was dried in vacuo to afford A3 (998 mg, 4.92 mmol, 98%).Acknowledgements
Support of this work by the Swiss National Science Foundation (SNF) and the Federal Commission for Technology and Innovation (KTI) is gratefully acknowledged. We thank Dr Markus Neuburger for the crystal structure analysis, Robin Wehlauch for synthetic contributions and Prof. Dr Klaus Dittrich from BASF for generous gifts of chemicals.Notes and references
- J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713 CrossRef CAS ; for early work on asymmetric imine hydrogenation, see: C. A. Willoughby and S. L. Buchwald, J. Am. Chem. Soc., 1992, 114, 7562 CrossRef; C. A. Willoughby and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 8952 CrossRef; C. A. Willoughby and S. L. Buchwald, J. Am. Chem. Soc., 1994, 116, 11703 CrossRef; Y. N. C. Chan and J. A. Osborn, J. Am. Chem. Soc., 1990, 112, 9400 CrossRef; F. Spindler, B. Pugin and H.-U. Blaser, Angew. Chem., Int. Ed. Engl., 1990, 29, 558 CrossRef; J. Bakos, I. Toth, B. Heil, G. Szalontai, L. Parkanyi and V. Fulup, J. Organomet. Chem., 1989, 370, 263 CrossRef ; for more recent work, see A. Trifonova, J. S. Diesen and P. G. Andersson, Chem.–Eur. J., 2006, 12, 2318–2328 CrossRef; C. Moessner and C. Bolm, Angew. Chem., Int. Ed., 2005, 44, 7564 CrossRef; T. Imamoto, N. Iwadate and K. Yoshida, Org. Lett., 2006, 8, 2289 CrossRef; S.-F. Zhu, J.-B. Xie, Y.-Z. Zhang, S. Li and Q.-L. Zhou, J. Am. Chem. Soc., 2006, 128, 12886 CrossRef; N. Mrsic, A. J. Minnaard, B. L. Feringa and J. G. de Vries, J. Am. Chem. Soc., 2009, 131, 8358 CrossRef; Z. Han, Z. Wang, X. Zhang and K. Ding, Angew. Chem., Int. Ed., 2009, 48, 5345 CrossRef.
- H.-U. Blaser, Adv. Synth. Catal., 2002, 344, 17 CrossRef CAS.
- S. Zhou, S. Fleischer, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5120 CrossRef CAS.
- C. Q. Li, C. Wang, B. Villa-Marcos and J. L. Xiao, J. Am. Chem. Soc., 2008, 130, 14450 CrossRef CAS.
- I. Storer, D. E. Carrera, Y. Ni and D. W. C. MacMillan, J. Am. Chem. Soc., 2006, 128, 84 CrossRef CAS; M. Reuping, E. Sugiono, C. Azap, T. Theissmann and M. Bolte, Org. Lett., 2005, 7, 378 Search PubMed; S. Hoffman, A. M. Seayad and B. List, Angew. Chem., Int. Ed., 2005, 44, 7424 CrossRef; K. Saito and T. Akiyama, Chem. Commun., 2012, 48, 4573 RSC.
- (a) P. Schnider, G. Koch, R. Prétôt, G. Wang, F. M. Bohnen, C. Krüger and A. Pfaltz, Chem.–Eur. J., 1997, 3, 887 CrossRef CAS; (b) A. Baeza and A. Pfaltz, Chem.–Eur. J., 2010, 16, 4003 CrossRef CAS.
- K. H. Hopmann and A. Bayer, Organometallics, 2011, 30, 2483 CrossRef CAS.
- C. Mazet, S. P. Smidt, M. Meuwly and A. Pfaltz, J. Am. Chem. Soc., 2004, 126, 14176 CrossRef CAS.
- Crystal structure: see ESI†.
- The collected data was of insufficient quality to allow accurate structure determination; see ESI†.
- (a) P. Marcazzan, B. O. Patrick and B. R. James, Russ. Chem. Bull., 2003, 52, 2715 CrossRef CAS. See also (b) J. F. van Baar, K. Vrieze and D. J. Stufkens, J. Organomet. Chem., 1975, 85, 249 CrossRef CAS; (c) M. Martín, E. Sola, S. Tejero, J. L. Andrés and L. A. Oro, Chem.–Eur. J., 2006, 12, 4043 Search PubMed.
- P. Marcazzan and B. R. James, React. Kinet. Catal. Lett., 2009, 98, 193 CrossRef CAS.
- See ESI†.
- Control experiments showed that even 0.1 mol% of I1 or A1 would have been detected by GC analysis.
- Cyclometalation of I10 was not observed in a preparative reaction, which could explain the low conversion and ee in this case.
Footnotes |
| † Electronic supplementary information (ESI) available: Protocols for procedures and experimental data. CCDC 916564. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3sc50587a |
| ‡ Current address: Yeshiva University, Department of Chemistry, New York, NY, 10033, USA. |
| This journal is © The Royal Society of Chemistry 2013 |