
Synthesis and properties of 1,3,4-oxadiazole-containing bismaleimides with asymmetric structure and the copolymerized systems thereof with 4,4′-bismaleimidodiphenylmethane
Abstract
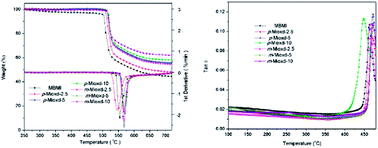
Two novel bismaleimide monomers containing 1,3,4-oxadiazole and asymmetric structure, i.e., 2-[4-(4-maleimidophenoxy)phenyl]-5-(4-maleimidophenyl)-1,3,4-oxadiazole (p-Mioxd) and 2-[3-(4-maleimidophenoxy)phenyl]-5-(4-maleimidophenyl)-1,3,4-oxadiazole (m-Mioxd), were designed and synthesized. The chemical structures of the monomers were confirmed using Fourier transform infrared spectroscopy (FTIR), 1H NMR and 13C NMR spectroscopy and elemental analysis. The thermal properties of the monomers were investigated by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). The results indicate that the incorporation of the 1,3,4-oxadiazole and asymmetric structure could improve the solubility and processability of the BMI monomers and the thermal stability of the resins. Composites composed of glass cloth and 4,4′-bismaleimidodiphenylmethane (BMDM), which were modified with 2.5, 5 and 10 wt% p-Mioxd and m-Mioxd, respectively, were also prepared. The TGA and DMA results demonstrate that the resulting composites have excellent thermal stability with high residual weight percentage at 700 °C (>45%) and Tg (>450 °C).
 Please wait while we load your content...
Please wait while we load your content...

