 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of light-responsive porous polycarbonate membranes for controlled caffeine delivery†
Lukas Baumannab, Katrin Schöllera, Damien de Courtencd, Dominik Martie, Martin Frenze, Martin Wolfc, René M. Rossia and Lukas J. Scherer*a
aEmpa, Swiss Federal Laboratories for Materials Science and Technology, Lerchenfeldstrasse 5, 9014 St.Gallen, Switzerland. E-mail: lukas.scherer@empa.ch; Fax: +41 58 7657762; Tel: +41 58 7657474
bUniversity of Basel, Klingelbergstrasse 80, 4056 Basel, Switzerland
cDivision of Neonatology, University Hospital Zurich, Frauenklinikstrasse 10, 8091 Zürich, Switzerland
dETH Zurich, Rämistrasse 101, 8092 Zürich, Switzerland
eInstitute of Applied Physics, University of Bern, Sidlerstrasse 5, 3012 Bern, Switzerland
First published on 1st October 2013
Abstract
For controlled caffeine release, light-responsive membranes were developed. It was possible to produce membranes that reduced their caffeine permeability resistance by about 97% when irradiated with UV-light compared to measurements at daylight. This was achieved by grafting polymers possessing photochromic units onto track-edged polycarbonate membranes. Covalently linked coatings on porous polycarbonate membranes were obtained by plasma activation of the membrane surface followed by plasma-induced graft polymerization. Copolymerization of spiro-compounds during the coating process as well as postmodification of preformed coatings with spiropyran resulted in photochromic membranes. For the copolymerization process, the synthesis of five photochromic methacrylic and acrylic spiropyrans and spirooxazines was successfully performed. Additionally, a spiropyran with carboxylic acid functionality was synthesized for the postmodification process. This enabled us to postmodify polymeric materials containing alcohol or amine groups to obtain photochromic materials. UV-irradiation of these light-responsive membranes resulted in a strong colouration of the membrane, in a reduction of surface tension, which resulted in a decreased caffeine permeability resistance. The membranes were characterized using XPS for the elemental composition of the coating, contact angle measurements for the surface tension, solid-state UV/VIS measurements for the determination of the kinetic and stability properties, and two-photon microscopy for the localisation of the photochromic substance in the porous membrane.
1. Introduction
Preterm neonates show an increased risk of apnea since their respiratory system is not fully developed yet.1,2 Schmidt et al. applied caffeine to preterm neonates to prevent and treat apnea.3 This caffeine treatment resulted in an increased rate of survival without disability of preterm neonates.3Caffeine is known for its ability to penetrate skin quite easily.4,5 The skin of preterm neonates represents only a minimal hindrance for caffeine directly after birth due to the undeveloped stratum corneum. This eases the transdermal caffeine delivery. The caffeine-concentration in the body after transdermal drug delivery is not only influenced by the rate of caffeine delivery, but also by the resistance of the skin towards caffeine. Since the skin properties of neonates change rather rapidly and the resistance towards caffeine increases over time, it is not suitable to develop a transdermal caffeine delivery system with a fixed delivery rate.6,7 There is also a major deviation in skin resistance comparing the skin of different patients.8 A device adapting its caffeine-delivery rate triggered by an external stimulus represents a suitable solution for the problem with different and changing skin resistances.9,10 Changing the delivery rate of the setup allows compensating the change in skin resistance.
For a transdermal drug-delivery setup, it is important to choose flexible and mechanically stable materials that allow a tight contact with the skin without limiting the agility of the person and without being damaged upon movements of the patient and which are biocompatible. A good candidate that fulfils all these requirements are thin track-edged polycarbonate (PC) membranes.11–13
Triggered and reversible changes of material properties can be achieved by integrating molecular switches into materials. Molecular switches as well as adaptive materials have been reported intensively.14–16 Temperature,17 pH,18 chemical stimuli,19 and light20–25 are known to be suitable triggers for adaptive materials. The focus of this investigation was set on light-responsive materials. Light can be applied rapidly, remotely and reversibly at the outer face of the body. Additionally, light is a clean stimulus that can easily be focused on small and defined areas. A plasma-induced photochromic surface coating with spiropyran has been reported to adjust the flow of a methanol–water-mixture through a light responsive PTFE-membrane.26 More solution was flowing through the membrane under UV-irradiation than at daylight.26 We recently showed that spirobenzopyran doped membrane surfaces regulate the permeability resistance of aqueous solutions.25
Spiropyran (SP) and spirooxazine (SO) are well known photochromic molecules. UV-irradiation of spiropyran or spirooxazine induces a heterolytic ring-opening reaction leading to a polar and coloured merocyanine (MC) state. Illuminating the MC-structure with visible light triggers the ring-closing reaction back into its initial SP-state. The surface tension of a SP containing coating depends on the actual state of the spiropyran. If spiropyran is in its nonpolar SP-state, the coated surface is rather hydrophobic. If spiropyran is switched into its MC-state, the surface becomes more hydrophilic.27 In order to improve the long-term stability of the membranes, a spirooxazine-containing photochromic coating for porous materials was developed for the first time.
A powerful and easy process to obtain covalently bound coatings on membranes is to activate the membrane surface by a plasma treatment followed by a plasma-induced graft polymerization.28 Plasma modification has some advantages compared to other surface technologies. It is a fast, dry and environmentally friendly technology, which has become an important process step in many industrial fields. It enables the tailored surface-functionalization of polymers, while maintaining their desirable bulk properties.29–31 Besides the creation of active surface species, cleaning of the surface is an additional beneficial effect, which makes plasma a promising approach for creating homogenously coated polymer-surfaces in a reproducible manner.28,32–34
Two strategies are possible to create photochromic coatings based on a plasma initiated polymerization process. The first strategy includes the creation of a surface-grafted polymeric coating with functional side chains followed by a postmodification of these functional side chains in a separate reaction step.35,36 The second strategy is the random graft-copolymerization of photochromic monomers with the main monomer in a one-step approach.26
Previous work showed that coatings with different hydrophilicity resulted in membranes with different permeability resistances.37 A modest impact of the hydrophilicity on the permeability resistance was found for coatings with contact angles below 80°. A more prominent impact was observed for coatings with a contact angle above 80°. Therefore, poly-2-hydroxyethyl methacrylate (pHEMA) (CA = 90°), poly-2-hydroxyethyl acrylate (pHEA) (CA = 95°) and polymethyl methacrylate (pMMA) (CA = 105°) are expected to be promising candidates for photochromic coatings with switchable permeability resistances. As previously reported, poly 2-aminoethyl methacrylate (pAEMA)-coatings can be easily postmodified with carboxylic acids functionalized molecules.31 Therefore, pAEMA was investigated as well despite its low contact angle for the homocoating (60°).
The goal of this study was the development and characterization of light-responsive track-edged membranes with a significant change of the caffeine permeability. Furthermore, through the closed state of the membrane should only pass small amounts of caffeine, which means that a caffeine permeability resistance of more than 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s cm−1 was desired for the closed state.
000 s cm−1 was desired for the closed state.
2. Experimental
2.1 Materials and general methods
Ethanol (EtOH, absolute, 99.0%, dried over molecular sieve), dichloromethane (DCM, 99.8%, dried over molecular sieve) and toluene (Tol, 99.8%, dried over molecular sieve) were purchased at Acros. Acetonitrile (MeCN, HPLC grade) was delivered by Fisher Chemical. Hexane (Hx, 99%) was obtained from Biosolve. Methanol (MeOH, 99%, dried over molecular sieve), 2-butanone (MEK, 99.0%), tetrahydrofuran (THF, 99.8%, dried), 2-hydroxyethyl methacrylate (HEMA, 97%), 2-hydroxyethyl acrylate (HEA, 96%) methyl methacrylate (MMA, 99%), 2-aminoethyl methacrylate (AEMA, 90%), N,N-dicyclohexylcarbodiimide (DCC, 99%), dimethyl aminopyridine (DMAP, 99%), ethyl acetate (EtOAc, puriss) and aluminum oxide (Type CG20) were obtained from Sigma Aldrich. Caffeine (reagent plus) and tert-butylmetylether (TBME, 98%) was purchased at Fluka. Poly(bisphenol A carbonate) from Sigma Aldrich was used for the PC coatings. All chemicals, unless stated otherwise, were used as delivered without further purification. Deionized water was obtained from the in-house purification system. Polycarbonate (PC) membranes cyclopore track etched (TE) (0.2 μm pore diameter) were purchased at Whatman. Argon (99.9995%) and Oxygen (99.9995%) were purchased at Alphagaz. For weighing, a Mettler Toledo AB204-S was used. Ultraviolet and visible (UV/VIS) absorption measurements of solutions were performed on a Varian 50Bio/50MPR. Scanning electron microscopy (SEM) pictures were recorded on a Hitachi S4800. All SEM-samples were coated with a gold layer of about 3 nm. Pore diameters were determined by measuring 30 pores of each sample. Only single round-shaped pores that were not fused to other pores were therefore considered. X-ray photoelectron spectroscopy (XPS) was performed on a PHI 5600 spectrometer. Investigated emission angle was 45°. XPS data were analysed using the program CasaXPS.2.2 Preparation of monomer solutions
To remove inhibitors, HEA and HEMA were dissolved in water and washed with hexane. Subsequently, the aqueous phase was saturated with sodium chloride and extracted with Et2O. The organic layers were dried over MgSO4. After removing the solvent, the monomer was distilled under reduced pressure.38 Inhibitor of MMA was removed by column chromatography over aluminium oxide. AEMA was used as delivered.2.3 Coating of PC membranes
Two membranes were positioned in the plasma chamber next to each other, with the shiny side of the membrane pointing to the gas phase. The chamber was evacuated and purged for four hours with 15 sccm argon and 2.5 sccm oxygen until a constant gas flow was obtained. After the plasma was initiated at 25 W, the power was immediately reduced to 12 W. After 4 minutes of plasma treatment, the power and the gas flow were switched off and the chamber was evacuated. Afterwards, the chamber was flooded with the prepared monomer solution and subsequently filled with argon. The flooded chamber was then stored for 12 hours at 20 °C in a conditioned room. After removal of the left-over monomer solution, the membranes were washed with ethanol and water in an ultrasonic bath for 5 minutes each to remove residual monomers. Finally, the membranes were dried in vacuo over molecular sieves for at least 2 hours before being analysed.372.4 Postmodification of coated PC membranes
2.5 Permeability measurements
All measurements were performed in a Franz diffusion-cell purchased from SES Analyse Systeme with a receptor volume of 12.0 mL and an orifice area of 1.77 cm2. Mass transfer rates of caffeine were measured under UV irradiation (366 nm, 15 W m−2) and at daylight (DL). After filling the receptor chamber with water (12.0 mL), the membrane was fixed in the diffusion cell. The donor chamber was charged with a caffeine solution (20 mM; 3.0 mL). Samples (200 μL) were collected from the receptor part of the cell after 1, 10, 20, 30, 45, and 60 minutes. The caffeine concentration of these samples were assigned by measuring its UV absorption at 293 nm.Resistance R of a membrane was calculated according to Fick's law using the formula
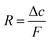 | (1) |
2.6 Contact angle measurements
All measurements were performed on a Krüss G10. A membrane was fixed with a standard tape on a metal O-ring. A drop of nanopure water (3.3 μL) was positioned on the part of the membrane surface that was not in contact with the O-ring. For measuring the impact of SP on the surface tension of the membrane, the CA was measured first at day light. Then the membrane was illuminated with UV-light (366 nm, 80 W m−2) for 1 minute and the contact angle was measured again.For measuring the repeatability of switching the surface tension, the membrane was illuminated with white light (500 W bulb) until no colouration was visible anymore before the contact angle was measured again. This cycle was repeated at least three times. The method allowed measurement with an accuracy of ±2°.
2.7 Solid state UV/VIS measurements
All measurements were performed on a Lambda 9 (Perkin Elmer) in reflection mode. UV illumination was always at 366 nm with an intensity of 80 W m−2. The untreated membrane – stored at daylight – was used for the base line measurements. After illuminating the membrane for 1 minute with UV light, the spectrum of that membrane was measured and the maximal absorbance was detected. The membrane was then illuminated with UV-light for five more minutes. Afterwards, the absorption at the assigned maximum was measured over a time span of 90 minutes to obtain information about the ring-closing kinetics of the spiro-compounds under dark conditions at room temperature. Subsequently, the membrane was illuminated for 5 hours with UV-light. After 15, 30, 45, 60, 90, 120, 180 and 300 minutes, the absorption at the assigned maximum was measured to obtain information about the speed of decomposition of the spiro-compounds (fading rate) under UV-irradiation. A linear fit was applied as an approximation. For fading rates, the measurements of the first 60 minutes were considered for the linear fitting. For ring closing rates, all measurements between minute 20 and 40 were considered for the estimation of the reaction speed.2.8 Amount of spiropyran on the membrane
A coated PC membrane was completely dissolved in DCM. The UV-absorption at 375 nm was caused by spiropyran and the amount of spiropyran that was incorporated during the modification of the membrane was determined at this wavelength. For the calibration, the corresponding spiropyran-monomer was dissolved in DCM together with an untreated PC membrane (SP2 and SP3: ε375 = 245 cm−1 M−1, SP4, SP5 and SP6: ε375 = 226 cm−1 M−1 and SO2: ε375 = 300 cm−1 M−1).2.9 XPS experiments
XPS analysis was performed using a PhI5000 VersaProbe spectrometer (ULVAC-PHI, INC.) equipped with a 180° spherical capacitor energy analyzer and a multi-channel detection system with 16 channels. Spectra were acquired at a base pressure of 5 × 10−8 Pa using a focused scanning monochromatic Al-Ka source (1486.6 eV) with a spot size of 200 μm, scanning an area of 1000 × 500 μm. The instrument was run in the FAT analyzer mode. The pass energy for survey scans was 187.85 eV and 46.95 for detailed spectra. Charge neutralisation using both a cool cathode electron flood source (1.2 eV) and very low energy Ar+-ions (10 eV) was applied throughout the analysis. Data were analyzed using the program CasaXPS (Version 2.3.15 http://www.casaxps.com). The signals were integrated following Shirley background subtraction. Sensitivity factors were calculated using published ionization cross-sections corrected for attenuation, transmission-function of the instrument and source to analyzer angle.39 As a result, the measured amounts are given as apparent normalized atomic concentration and the accuracy under the chosen condition is approximately ±10%. For the model system thin PC films were produced by spin-coating a 0.5 wt% solution of PC in dichloromethane with 3000 rpm on Si-wafers followed by plasma-induced polymerization of HEMA according to the described procedure (coating of PC membranes).2.10 Multiphoton microscopy
For multiphoton microscopy, a Nikon A1R-MP microscope was used equipped with a Chameleon Ultra II (Coherent) femtosecond pulse laser. PC membranes with pore diameters of 0.2 μm and 1.0 μm were used for this analysis. The membranes were submerged in water and imaged using a 1.4 NA, 60× oil-immersion objective (Nikon) through a cover glass at 800 nm excitation. The fluorescence signal of the spiropyran was detected with a non-descanned photomultiplier tube (with a filter block at 561 nm centre wavelength, 50 nm width). To assure that the measured fluorescence really originated from spiropyran, an original PC membrane, a plasma-treated PC membrane, and a pHEMA-coated PC membrane were measured before analyzing the SP-containing PC membranes.3. Results and discussion
3.1 Synthesis of photochromic spirocompounds
Synthetic details for all reactions can be found in the supporting information (see ESI†). The most prominently reported spiropyran including a monomeric unit is methacrylate SP2.40–42 Since copolymerization of spiropyran was not only planned with methacrylates but also with acrylates, molecule SP3 was synthesized similarly. The three step syntheses were performed with an overall yield of 23% (SP2) and 56% (SP3) (Scheme 1). Replacing acetonitrile as reported in two published syntheses41,43 by toluene increased the yield of the first step significantly. Using piperidine as base for the condensation of benzaldehyde 4 with indoline salt 3 resulted in a slightly higher yield of spiropyran SP1 than using triethylamine.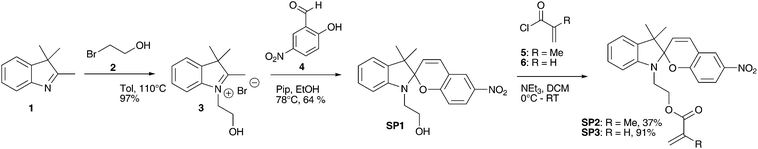 | ||
| Scheme 1 Three step synthesis of spiropyran SP7 and SP9via photochromic intermediate SP5. | ||
Spiropyran SP5 is a known photochromic monomer.44 Structure SP6 – which cannot be found in literature – was synthesized similar to molecule SP5. The three step syntheses were performed with an overall yield of 39% (SP5) and 36% (SP6) (Scheme 2). Using piperidine as base for the in situ deprotonation resulted again in slightly higher yields than using triethylamine.
 | ||
| Scheme 2 Three step synthesis of spiropyran SP5 and SP6via photochromic intermediate SP4. | ||
Molecules SP1, SP2, SP3, SP4, SP5 and SP6 were all photochromic substances at room temperature. Changes in length, structure or position of the linker from the spiropyran-unit to the (meth)acrylic unit can have a significant impact on the photochromic behaviour of spiropyrans.45
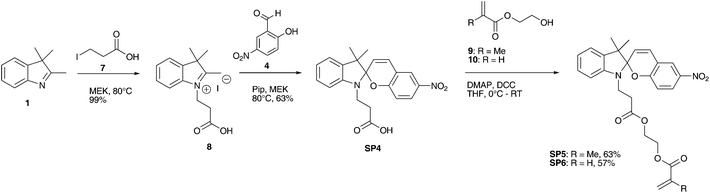
Spirooxazine SO2 was synthesized in 3 steps as reported in literature.46 This synthetic approach gave an overall yield of 36% (Scheme 3).
 | ||
| Scheme 3 Three step synthesis of methacrylic spirooxazine SO2via photochromic intermediate SO1. | ||
3.2 Postmodification
Homopolymerization was conducted with three different monomers with functional side chains.37 The aim was the introduction of either alcohols or amines on the surface of the PC membrane for the further reaction with the carboxylic acid group of SP4. It was possible to covalently link SP4 (Scheme 2) to the pHEA-, pHEMA- and pAEMA-coated membranes by creating an ester- or an amide-bond (Scheme 4). This postmodification (PM) with spirobenzopyrans transformed the homopolymer-coated membranes into photochromic membranes. UV-irradiation of the membrane resulted in a deep-blue colouration of the irradiated membrane (Fig. 1).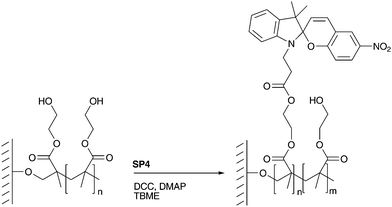 | ||
| Scheme 4 Postmodification of a HEMA-coated PC membrane via esterification. | ||
 | ||
| Fig. 1 Postmodified HEMA-coated PC membrane. Left: after UV irradiation; right: at daylight. | ||
SEM-pictures showed that the plasma-induced graft polymerization caused an increase in pore diameter from originally 0.20 ± 0.02 μm to 0.25 ± 0.03 μm.37 The postmodification process on the other hand had no detectable effect on the pore size. XPS measurements showed only very little changes comparing postmodified membranes with coated membranes (see ESI†). The N-signal corresponding to the spiropyran structure was not detected when SP was grafted onto the pHEA and pHEMA coatings respectively. However, for the pAEMA-coating, the amount of nitrogen and oxygen was lowered after reacting with SP, while the signal of C1s was significantly increased. This is a clear sign of the increased reactivity of the carboxylic group with the amine functionality compared to the free alcohol groups of the pHE(M)A coatings. Furthermore, XPS experiments revealed a higher surface density of amino functionalities after pAEMA coating than of alcohol functionalities after pHE(M)A coatings of the PC membrane. Due to the rough membrane surface and the coating thickness of bellow 10 nm, a mixture of the PC matrix and the coating was always measured, which made a quantitative analysis impossible. The layer thickness was estimated using a model system, which consisted of a thin spin-coated polycarbonate film on a silicon wafer, which was coated using the same parameters than for the membrane coating. Multi-angle XPS experiments revealed a coating thickness of 1–2 nm for the copolymerized samples (see ESI†).
Since the amount of incorporated SP4 could not be quantitatively determined using XPS experiments, UV/VIS absorption measurements at 375 nm after dissolving the coated PC membrane in DCM were performed (Table 1). The most spiropyran was bound to the pAEMA coating. pHEA-coating incorporated about the same amount of SP4 as the pHEMA-coating.
| Coating | RDL (s cm−1) | RUV (s cm−1) | CADL (°) | CAUV (°) | SP on mem. (wt%) | λmax (nm) |
|---|---|---|---|---|---|---|
| pHEMA-SP4 | 590000 ± 98000 | 15700 ± 930 | 100 | 90 | 3.1 | 552 |
| pHEA-SP4 | 101000 ± 3500 | 13800 ± 2600 | 95 | 85 | 2.9 | 545 |
| pAEMA-SP4 | 13900 ± 820 | 16200 ± 810 | 75 | 50 | 4.2 | 543 |
The different amounts of SP4 found on the pAEMA-coated membrane can be explained by the higher reactivity of amines compared to alcohols and by the presence of more functional groups on the surface of the pAEMA-coated membrane, as revealed by XPS experiments. The polymer coating and the additional SP functionalization had a significant effect on the contact angle measurements. By this means a decrease in surface tension for all photochromic coatings under UV-irradiation was measured compared to its surface tension at daylight (Table 1). pHEMA-SP4 was the least hydrophilic coating, changing its contact angle by 10° after irradiation with UV light. pHEA-SP4 showed an intermediate contact angle of 95° and a change of 10°. pAEMA-SP4 turned out to be the most hydrophilic coating with the most pronounced change in contact angle of about 25°. This CA-switching can be repeated for at least three entire cycles with recovering the initial values for all reported coatings.
For the pHE(M)A-SP4 coatings, the caffeine permeability resistance was lower under UV-irradiation than at daylight (Table 1 & Fig. 2). The largest switching potential concerning caffeine permeability resistance (97%) was found for the pHEMA-SP4 coating. Postmodification of the pHEA-coating provided a smaller but still evident change in caffeine permeability resistance. Postmodification of the AEMA-coating resulted in a photochromic membrane but the caffeine permeability resistance changed only little and a reversed switch was obtained.
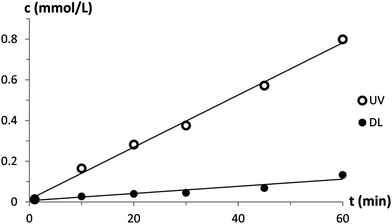 | ||
| Fig. 2 Caffeine permeability of a pHEA-coated postmodified PC membrane at daylight and under UV-irradiation. | ||
3.3 Copolymerization
A one-step approach for the production of photochromic membranes was successfully developed. Instead of creating a pHE(M)A or a pAEMA coating followed by a postmodification step, spiropyran was functionalized with an acrylate or methacrylate group and randomly copolymerized with the main coating monomer in a grafting-from process in a single step (Scheme 5). UV-irradiation after the one-step coating process induced again a deep-blue colouration of the irradiated membrane, similar to the postmodified membrane showed in Fig. 1.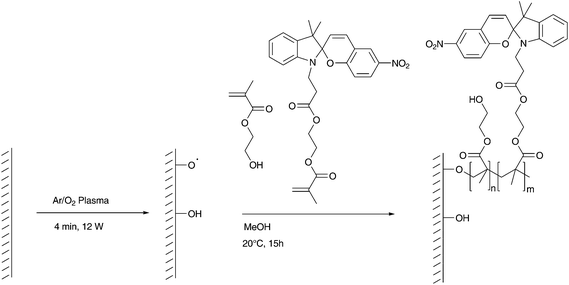 | ||
| Scheme 5 Copolymerization of HEMA and SP5 on a plasma-activated PC surface. | ||
As for the postmodified membranes, SP-modified pHEMA coatings showed the largest change in permeability resistance (Table 2). Therefore pHEMA copolymers were chosen for the following investigation. The coating process remained unchanged except for the amount of SP2. The impact of changing the amount of SP2 was found to be rather small. The largest change of permeability resistance was achieved for a concentration of 25.0 mM SP2 (Table 2). The amount of incorporated spiropyran was correlated to the switching potential of the membrane. More incorporated spiropyran resulted in a higher switching potential. Unexpectedly, the amount of incorporated spiropyran was not correlated to the concentration of spiropyran in the reaction mixture.
| Grafted monomers | SP in rxn (mM) | RDL (s cm−1) | RUV (s cm−1) | CADL (°) | CAUV (°) | SP on mem. (wt%) | λmax (nm) |
|---|---|---|---|---|---|---|---|
| PC original | 11300 ± 750 | 11600 ± 860 | 60 | 60 | — | — | |
| AEMA; SP2 | 25.0 | 15400 ± 620 | 14100 ± 540 | 60 | 55 | 1.15 | 570 |
| MMA; SP2 | 25.0 | 15000 ± 510 | 15200 ± 310 | 100 | 90 | 1.29 | 579 |
| HEMA; SP2 | 25.0 | 15200 ± 860 | 10500 ± 360 | 95 | 75 | 0.72 | 595 |
| HEMA; SP2 | 33.3 | 13800 ± 440 | 10800 ± 310 | 90 | 70 | 0.67 | 588 |
| HEMA; SP2 | 25.0 | 15200 ± 860 | 10500 ± 360 | 95 | 75 | 0.72 | 595 |
| HEMA; SP2 | 16.7 | 13300 ± 370 | 11100 ± 350 | 95 | 65 | 0.45 | 592 |
| HEMA; SP2 | 25.0 | 15200 ± 860 | 10500 ± 360 | 95 | 75 | 0.72 | 595 |
| HEMA; SP5 | 25.0 | 14200 ± 1000 | 10200 ± 700 | 70 | 55 | 1.15 | 590 |
| HEA; SP3 | 25.0 | 58000 ± 9000 | 17500 ± 1400 | 100 | 85 | 0.64 | 591 |
| HEA; SP6 | 25.0 | 15900 ± 360 | 13500 ± 2000 | 100 | 80 | 0.84 | 591 |
To study the influence of the linker length between SP unit and polymer coating on the membrane properties, the amount of spiropyran and comonomer was remained unchanged. For the copolymerization with HEA, the acrylic derivative of the corresponding spiropyran was used, whereas for approaches with HEMA the methacrylic derivative was copolymerized. It can be seen from Table 2 that using longer linkers (SP5 and SP6) resulted in an increased amount of incorporated spiropyran. A long linker allowed spiropyran to be rather far away from its reactive acrylic unit. This lowered the steric hindrance of the reactive side and facilitated the incorporation into the polymer coatings. Nevertheless, HEA in combination with the short linked SP3 resulted in the biggest change in permeability resistance followed by the HEMA coating with short-linked SP2, although less SP was incorporated. The increased linker length led to a decreased permeability resistance of the membrane.
In addition a dependence of the linker length of spiropyran on the elemental composition was found. If spiropyran with a long linker (SP5, SP6) was copolymerized, higher oxygen content and lower carbon content were detected. As for the postmodified coatings, it is assumed that the XPS signals resulted from a mixture of PC matrix, copolymer and spiropyran. Although XPS measurements showed a change in elemental distribution on the membrane surface before and after the plasma induced polymerization, FTIR- and NMR-measurements did not provide any meaningful data.
Lower surface tension was measured for all samples under UV-irradiation than under daylight, which can be correlated to the switching of spiropyran into its more hydrophilic MC-state. But the changes in permeability resistance showed no linear dependence on the contact angle changes as reported for homopolymer coatings,37 which might be due to the more complex morphology of the SP containing coatings or additional chemical interactions between SP and caffeine.
The comonomers had also an influence on the membrane properties (Table 2). Surface-induced copolymerization of methyl methacrylate (MMA) with SP2 resulted in photochromic membranes (colour change) but no change of permeability resistance was found when irradiated with UV-light. It is known from literature that low free volume caused by high rigidity of a pMMA results in reduced switching of photochromic molecules.47–49 As for the postmodified membranes, using AEMA as copolymer resulted in a membrane with only very little change in permeability resistance. Error margins showed that this change was not significant. Highest switching potential for all studied SP-copolymerized coatings was provided by copolymerization of HEA with SP3.
Not only spiropyrans but also spirooxazine SO2 was copolymerized with HEMA. The resulting photochromic membrane showed a slightly higher switching potential concerning the caffeine permeability resistance compared to its spiropyran analogue SP2 (Table 3). The incorporated amount of spiro-compounds was very similar. Copolymerization of SO2 resulted in a lower surface tension at daylight as well as under UV-irradiation indicating that SO2 is more hydrophilic than SP2.
| Grafted monomers | SP/SO in rxn (mM) | RDL (s cm−1) | RUV (s cm−1) | CADL (°) | CAUV (°) | SP on mem. (wt%) | λmax (nm) |
|---|---|---|---|---|---|---|---|
| HEMA; SP2 | 25.0 | 15200 ± 860 | 10500 ± 360 | 95 | 75 | 0.72 | 595 |
| HEMA; SO2 | 25.0 | 29300 ± 750 | 12300 ± 170 | 70 | 55 | 0.75 | 590 |
A dependence of the linker length of spiropyran on the elemental composition was found. If spiropyran with a long linker (SP5, SP6) was copolymerized, higher oxygen content and lower carbon content were detected. As for the postmodified coatings, it is assumed that the XPS signals resulted from a mixture of PC matrix, copolymer and spiropyran. Although XPS measurements showed a change in elemental distribution on the membrane surface before and after the plasma induced polymerization, FTIR- and NMR-measurements did not provide any meaningful data.
3.4 Multiphoton microscopy
Multiphoton microscopy allowed the localization of spiropyran in and on the porous membranes. A solely pHEMA-coated PC membrane did not show any fluorescence between 510 nm and 670 nm when irradiated with an 800 nm femtosecond pulse laser. Since the pHEMA coated membrane showed no auto-fluorescence, the fluorescence of the SP-coated membrane was assigned to the SP unit.50 The red fluorescence was measured on the surface of the membrane and on the inner pore walls (Fig. 3). By this means it was assured that the spirobenzopyran was located on the outside of the membrane and did not penetrate into the inside of the PC matrix. However, membranes with 1000 nm pore diameters were necessary to detect the pores. By using the membranes discussed in this study with pore diameters of 200 nm, no pores could be detected (Fig. 4). However, the depth profile of the membranes showed a clear difference between the copolymerization and the postmodification procedure (Fig. 4). While the fluorescence on the copolymerized membranes was detected through the entire membrane, spirobenzopyran was only detected in the first 2–3 μm of the postmodified membranes with the highest density on the membrane surface. TBME as a non-polar solvent could not diffuse through the entire membrane and therefore reaction occurred only on the pore walls close to the membrane surface during the postmodification process. | ||
| Fig. 3 Multiphoton microscopy image showing the SP distribution on the pore surface. In order to visualize the pores, membranes with 1000 nm pores were used. The image represents the x–y plane just below the membrane surface of a sp4 postmodified HEMA-coated membrane. | ||
 | ||
| Fig. 4 Multiphoton microscopy images showing the fluorescence intensity in a cross-section of the first 4 μm of HEMA-coated PC membrane, where SP was introduced via (a) postmodification and (b) via copolymerization; (c) mean fluorescence intensity of postmodified and copolymerized HEMA-coated PC membrane plotted vs. the depth of the membrane. The membrane surface was for both measurements at around 1 μm in the z-plane. | ||
3.5 Solid state UV/VIS measurements
To obtain information about the stability of the different photochromic coatings, the absorptions of photochromic membranes were measured after different time spans under UV-irradiation. Additionally, ring-closing kinetics under dark conditions were measured for the different coatings. A slow ring-closing kinetics allows using a pulsed instead of a permanent UV-irradiation to keep spiro-compounds in their MC-state. Using a pulsed UV-irradiation would lower the overall irradiation dose that reaches the membrane, which leads to less fading of the photochromic substance. Therefore, less UV-irradiation would extend the operating time of the membranes.As shown in Fig. 5 and 6, the postmodified membranes were more stable and showed slower ring-closing kinetics compared to the copolymerized membranes. Comparing the data of the stability measurements showed a fundamental difference from postmodified to copolymerized membranes. Whereas the postmodified samples showed a slow fading rate at the beginning and an increase over time (Fig. 5), the copolymerized samples showed a higher, constant fading rate at the beginning with a decrease towards the end of the measurement (Fig. 6).
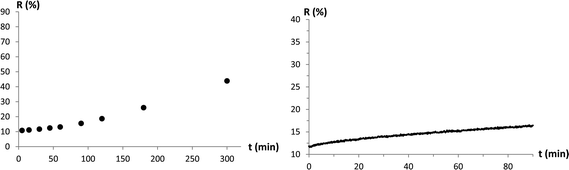 | ||
| Fig. 5 Fading-rate measurement (left) and ring-closing reaction kinetics measurement under dark conditions (right) of a postmodified, pHEA/SP4-coated PC membrane. Reflection (r in %) of the membrane at λmax was measured over time. | ||
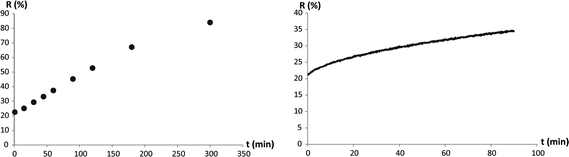 | ||
| Fig. 6 Fading-rate measurement (left) and ring-closing reaction kinetics measurement under dark conditions (right) of a copolymerized, pHEA-SP6-coated PC membrane. Reflection (r in %) of the membrane at λmax was measured over time. | ||
Since fading and ring closing reactions did not follow a known reaction mechanism,51,52 and the measured kinetic curve shapes varied for the different coatings, it was not possible to apply an appropriate model to exactly quantify the processes. To qualitatively compare the different membranes, a linear fit was applied assuming zero-order kinetics. As can be seen from the standard deviation in Table 4, the linear fit was a satisfactory approximation. For fading rates, the measurements of the first 60 minutes were considered. The slopes of all linear fits are summarized in Table 4.
| Membrane | Fading rate (ΔR%/h) | Ring closing kinetics (ΔR%/h) | λmax (nm) |
|---|---|---|---|
| PM pHEMA; SP4 | 2.3 ± 0.3 | 1.08 ± 0.06 | 552 |
| PM pHEA; SP4 | 2.5 ± 0.1 | 2.88 ± 0.06 | 545 |
| PM pAEMA; SP4 | 2.9 ± 0.4 | 0.84 ± 0.06 | 543 |
| CP pHEMA; SP2 | 15.6 ± 0.6 | 8.6 ± 0.1 | 595 |
| CP pHEMA; SP5 | 11.4 ± 0.6 | 6.2 ± 0.4 | 590 |
| CP pHEA; SP3 | 13.1 ± 0.2 | 9.6 ± 0.1 | 591 |
| CP pHEA; SP6 | 15.6 ± 0.6 | 11.5 ± 0.1 | 591 |
| CP pAEMA; SP2 | 32 ± 2 | 6.2 ± 0.1 | 570 |
| CP pMMA; SP2 | 8.9 ± 0.4 | 5.76 ± 0.06 | 579 |
| CP pHEMA; SO2 | 9.5 ± 0.5 | 13 ± 2 | 590 |
High fading rates indicated fast decomposition of the spiro-compound on the membrane. Large slopes for ring-closing-kinetics indicated that the spiro-compounds underwent a fast ring-closing reaction. There were no evident differences in fading rates between the two different linker lengths (SP2/3 and SP5/6) and between pHEMA- and pHEA-coatings when comparing the copolymerized samples. Considering the postmodified membranes, the pHEA-coated sample showed a faster ring-closing kinetics than the pHEMA-coated membrane. From all prepared samples, the pHEMA coating postmodified with SP4 showed the lowest fading rate and the lowest ring closing kinetics. Spirooxazine SO2 had – compared to Spiropyran SP2 – a slightly higher stability and a faster ring-closing-kinetics (Table 4). Surprisingly, stability measurements showed clearly that the method of production had a higher impact on the stability of the photochromic membranes than substituting spiropyrans by spirooxazines.
4. Conclusion
Spiropyrans and spirooxazine with different functionalities and linker length were synthesized. It was possible to transform pHE(M)A and pAEMA coatings in a postmodification step into photochromic coatings by linking spiropyran to functional side-chains. Switching spiropyran from one state into another state influenced the caffeine permeability rate due to changed wettability properties of the pores.Photochromic coatings were also directly applied to polycarbonate membranes in a copolymerization process. Comparing postmodified and copolymerized membranes showed that postmodified membranes had larger switching potential concerning caffeine permeability. Furthermore, postmodified membranes showed lower fading rates. The slow ring-closing kinetics of postmodified membranes would allow using a pulsed UV-irradiation. This increases the operating time of the membranes. Summing it up, postmodified pHEA and pHEMA coated membranes showed the most promising results for the use in a drug release system.
The femtosecond pulse laser could be used in future to replace the UV light to trigger the ring-opening reaction and thus to avoid harmful UV light.
Acknowledgements
This work was financially supported by Swiss National Science Foundation (NRP 62 – Smart Materials). Gratefully acknowledged is the support of P. Rupper, D. Rentsch, E. Aslan-Gürel, B. Hanselmann, B. Leuthold, and S. Al Gorani-Szigeti.Notes and references
- G. Polgar and T. R. Weng, Am. Rev. Respir. Dis., 1979, 120, 625 CAS.
- W. J. R. Daily, M. Klaus, H. Belton and P. Meyer, Pediatrics, 1969, 43, 510 CAS.
- B. Schmidt, P. J. Anderson, L. W. Doyle, D. Dewey, R. E. Grunau, E. V. Asztlos, P. G. Davis, W. Tin, D. Moddemann, A. Solimano, A. Ohlsson, K. J. Barrington and R. S. Roberts, JAMA, J. Am. Med. Assoc., 2012, 307, 275 CrossRef CAS PubMed.
- M. J. Bartek, J. A. Labudde and H. I. Maibach, J. Invest. Dermatol., 1972, 58, 114 CAS.
- I. P. Dick and R. C. Scott, J. Pharm. Pharmacol., 1992, 44, 640 CrossRef CAS.
- N. Barker, J. Hadgraft and N. Rutter, J. Invest. Dermatol., 1987, 88, 409 CAS.
- R. L. Nachman and N. B. Esterly, J. Pediatr., 1971, 79, 628 CrossRef CAS.
- M. F. Wilkosz and R. H. Bogner, U.S. Pharmacist, 2003, 28, 04 Search PubMed.
- M. Gagliardi, D. Silvestri and C. Cristallini, Drug Delivery, 2010, 17, 452 CrossRef CAS PubMed.
- R. Bengt, Int. J. Adhes. Adhes., 1999, 19, 337 CrossRef.
- D. C. Schohn, H. A. Jahn, M. Eber and G. Hauptmann, Blood Purif., 1986, 4, 102 CrossRef CAS PubMed.
- M. H. R. Magno, J. Kim, A. Srinivasan, S. McBride, D. Bolikal, A. Darr, J. O. Hollinger and J. Kohn, J. Mater. Chem., 2010, 20, 8885 RSC.
- G. Erdodi and J. P. Kennedy, Prog. Polym. Sci., 2006, 31, 1 CrossRef CAS PubMed.
- B. S. Lukyanov and M. B. Lukyanova, Chem. Heterocycl. Compd., 2005, 41, 281 CrossRef CAS.
- E. Ruel-Gariépy and J.-C. Leroux, Eur. J. Pharm. Biopharm., 2004, 58, 409 CrossRef PubMed.
- V. Giurgiutiu, J. Intell. Mater. Syst. Struct., 2000, 11, 525 Search PubMed.
- L. E. Bromberg and E. S. Ron, Adv. Drug Delivery Rev., 1998, 31, 197 CrossRef CAS.
- D. M. Lynn, M. M. Amiji and R. Langer, Angew. Chem., Int. Ed. Engl., 2001, 40, 1707 CrossRef CAS.
- Y. Lu and J. Liu, Acc. Chem. Res., 2007, 40, 315 CrossRef CAS PubMed.
- T. Shimidzu and M. Yoshikawa, J. Membr. Sci., 1983, 13, 1 CrossRef CAS.
- M. Kameda, K. Sumaru, T. Kanamori and T. Shinbo, J. Appl. Polym. Sci., 2003, 88, 2068 CrossRef CAS.
- E. Cabane, X. Zhang, K. Langowska, C. Palivan and W. Meier, Biointerphases, 2012, 7, 1 CrossRef PubMed.
- C. B. Gong, K. L. Wong and M. H. W. Lam, Chem. Mater., 2008, 20, 1353 CrossRef CAS.
- K. Kono, Y. Nishihara and T. Takagishi, J. Appl. Polym. Sci., 1995, 56, 707 CrossRef CAS.
- L. Baumann, M. Wolf, R. M. Rossi and L. J. Scherer, ACS Appl. Mater. Interfaces, 2013, 5, 5894 CAS.
- D. J. Chung, Y. Ito and Y. Imanishi, J. Appl. Polym. Sci., 1994, 51, 2027 CrossRef.
- I. Vlassiouk, C.-D. Park, S. A. Vail, D. Gust and S. Smirnov, Nano Lett., 2006, 6, 1013 CrossRef CAS PubMed.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437 CrossRef CAS PubMed.
- R. d'Agostino, in Basic Approaches to Plasma Production and Control, ed. R. D'Agostino, P. Favia, Y. Kawai, H. Ikegami, N. Sato and F. Arefi-Khonsari, Wiley-VCH Verlag GmbH & Co. KGaA, 2008 Search PubMed.
- E. M. Liston, L. Martinu and M. R. Wertheimer, J. Adhes. Sci. Technol., 1993, 7, 1091 CrossRef CAS PubMed.
- K. S. Siow, L. Britcher, S. Kumar and H. J. Griesser, Plasma Processes Polym., 2006, 3, 392 CrossRef CAS.
- D. P. Lymberopoulos and D. J. Economou, J. Vac. Sci. Technol., A, 1994, 12, 1229 CAS.
- S. K. Øiseth, A. Krozer, B. Kasemo and J. Lausmaa, Appl. Surf. Sci., 2002, 202, 92 CrossRef.
- D. Hegemann, H. Brunner and C. Oehr, Nucl. Instrum. Methods Phys. Res., Sect. B, 2003, 208, 281 CrossRef CAS.
- J. Choi, P. Schattling, F. D. Jochum, J. Pyun, K. Char and P. Theato, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4010 CrossRef CAS.
- C. T. Burns, S. Y. Choi, M. L. Dietz and M. A. Firestone, Sep. Sci. Technol., 2008, 43, 2503 CrossRef CAS.
- L. Baumann, D. Hegemann, D. de Courten, M. Wolf, R. M. Rossi, W. P. Meier and L. J. Scherer, Appl. Surf. Sci., 2013, 268, 450 CrossRef CAS PubMed.
- H. Kakwere and S. Perrier, Polym. Chem., 2011, 2, 270 RSC.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129 CrossRef CAS.
- D. S. Achilleos, T. A. Hatton and M. Vamvakaki, J. Am. Chem. Soc., 2012, 134, 5726 CrossRef CAS PubMed.
- S. Friedle and S. W. Thomas, Angew. Chem., Int. Ed. Engl., 2010, 49, 7968 CrossRef CAS PubMed.
- D. Wang, P. Jiao, J. Wang, Q. Zhang, L. Feng and Z. Yang, J. Appl. Polym. Sci., 2012, 125, 870 CrossRef CAS.
- F. M. Raymo and S. Giordani, J. Am. Chem. Soc., 2001, 123, 4651 CrossRef CAS.
- J. Chen, P. Zhang, G. Fang, P. Yi, X. Yu, X. Li, F. Zeng and S. Wu, J. Phys. Chem. B, 2011, 115, 3354 CrossRef CAS PubMed.
- A. Zelichenok, F. Buchholtz, S. Yitzchaik, J. Ratner, M. Safro and V. Krongauz, Macromolecules, 1992, 25, 3179 CrossRef CAS.
- S. Wang, M.-S. Choi and S.-H. Kim, J. Photochem. Photobiol., A, 2008, 198, 150 CrossRef CAS PubMed.
- C. D. Eisenbach, Berichte der Bunsengesellschaft für physikalische Chemie, 1980, 84, 680 CrossRef CAS.
- K. Horie, M. Tsukamoto and I. Mita, Eur. Polym. J., 1985, 21, 805 CrossRef CAS.
- R. A. Evans, T. L. Hanley, M. A. Skidmore, T. P. Davis, G. K. Such, L. H. Yee, G. E. Ball and D. A. Lewis, Nat. Mater., 2005, 4, 249 CrossRef CAS PubMed.
- M.-Q. Zhu, G.-F. Zhang, C. Li, M. P. Aldred, E. Chang, R. A. Drezek and A. D. Q. Li, J. Am. Chem. Soc., 2010, 133, 365 CrossRef PubMed.
- A. K. Chibisov and H. Görner, J. Photochem. Photobiol., A, 1997, 105, 261 CrossRef CAS.
- G. Baillet, G. Giusti and R. Guglielmetti, J. Photochem. Photobiol., A, 1993, 70, 157 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra44399j |
| This journal is © The Royal Society of Chemistry 2013 |