 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biosynthesis of natural products by microbial iterative hybrid PKS–NRPS
Katja Maria Fisch*
University of Bristol, School of Chemistry, Cantock's Close, Bristol, United Kingdom. E-mail: katja.fisch@bristol.ac.uk; Tel: +44 117 9546323
First published on 16th August 2013
Abstract
This review discusses the biosynthesis of natural products generated by iterative hybrid polyketide synthases–non ribosomal peptide synthetases (PKS–NRPS) from fungi and bacteria, the programming of the enzymes and bioengineering approaches.
 Katja Maria Fisch | Katja M. Fisch obtained her degree in Food Chemistry from WWU Münster, Germany and her PhD from the University of Bonn, Germany (under supervision of Gabriele König at the Intitute of Pharmaceutical Biology). In her first postdoctoral research she investigated possible target enzymes for bioactive natural small molecules at the University Hospital Münster, Germany. In 2006 she joined Jörn Piels group in Bonn, Germany where she successfully isolated the gene cluster for the antitumour polyketide psymberin. Since 2008 she is a Postdoctoral Research Associate at the University of Bristol, UK investigating the programming of fungal PKSs and hybrid PKS–NRPSs. |
1 Introduction
Nature creates a vast diversity of secondary metabolites by assembling acetate building blocks to polyketides using polyketide synthases (PKSs) and by assembling amino acids to peptides using non-ribosomal peptide synthetases (NRPSs). PKSs and NRPSs are megaenzymes containing domains to which specific functions can be assigned. A detailed description of the chemical reactions catalysed by the domains has been included by Evans et al.1 Exploring and exploiting these biosynthetic routes to learn about natures programme and to create new bioactive compounds are fascinating and productive research fields. This review focuses on compounds biosynthesised by microbial (bacterial and fungal) iterative hybrid PKS–NRPSs.Nature uses combined PKS and NRPS in two different ways. First, there are many known modular systems in bacteria which place PKS units (modules) and NRPS units (modules) together (in either order) in processive ‘production line’ assemblies.2 A typical example is found in BaeJ in the biosynthesis of the antibiotic bacillaene (Fig. 1A).3 BaeJ comprises of an unusual starter module followed by an NRPS module which incorporates glycine. The following PKS modules I and II have different domain architecture, i.e. KS-DH-KR-ACP-ACP vs. KS-KR-ACP. They incorporate one acetate building block each and modify the previous building block in accordance to their domain architecture (Fig. 1A).
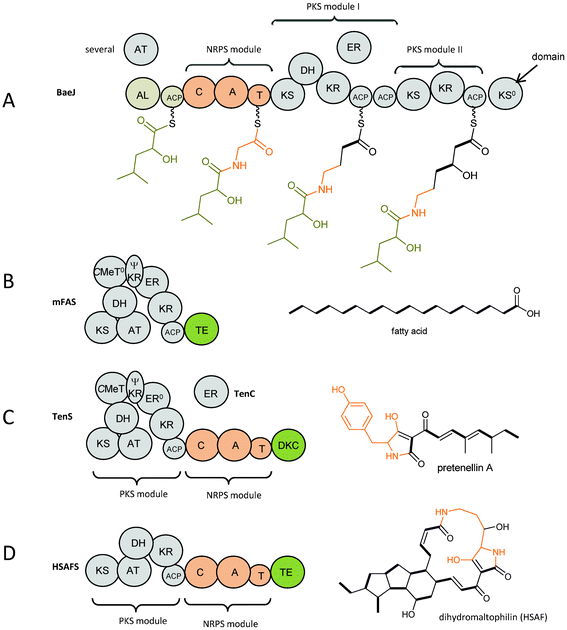 | ||
| Fig. 1 Domain architecture and biosynthesis products of (A) BaeJ as an example of a bacterial modular non-iterative hybrid PKS–NRPS with the biosynthetic intermediates attached, (B) mammalian fatty acid synthase (mFAS), (C) TenS as an example of a fungal iterative hybrid PKS–NRPS compared to (D) HSAF as an example of a bacterial iterative hybrid PKS–NRPS. Explanation of domain abbreviations is given in the text. Incorporated amino acids are shown in orange. | ||
The second way is found predominantly in fungi. Here, a single module of an iterative PKS (incorporating several acetate building blocks) is followed by a single NRPS module (fusing the polyketide chain to an amino acid) and an off-loading domain as seen in TenS (Fig. 1C).4 The domain architecture of the PKS module in fungal iterative PKS–NRPSs closely resembles that found in mammalian fatty acid synthases (mFASs, Fig. 1B and C).5 Thus the PKS module in iterative PKS–NRPSs has a general domain organisation of KS (ketosynthase), AT (acyltransferase), DH (dehydratase), CMeT (C-methyltransferase), ΨKR (a structural domain of the ketoreductase), ER (enoylreductase), KR (ketoreductase), ACP (acyl carrier protein). Therefore it belongs to the highly reducing PKS (HR-PKS) group.6 Using the same set of domains mFASs produce fully reduced non methylated fatty acids whereas the PKS modules in iterative PKS–NRPSs are programmed and produce individual and diverse polyketide chains with different reduction and methylation patterns. Although the crystal structure of mFAS shows it contains a CMeT-like domain, this domain lacks the conserved SAM binding motif possibly preventing cofactor binding and thus is not capable of carrying out any methylation.5 However, in the fungal iterative PKS–NRPSs the CMeT domain is usually active and diverse methylation patterns are observed in the produced metabolites. Another difference between mFASs and fungal iterative PKS–NRPSs lies in the ER domain. Enoyl reduction is one of the steps in polyketide biosynthesis carried out by HR-PKSs that adds diversity by occurring in a highly programmed way at different chain length of the growing chain. Usually, this step is carried out by the enoylreductase (ER) domain of a HR-PKS. From sequence alignments ER domains in all fungal iterative PKS–NRPSs are predicted to be inactive due to a variety of significant sequence differences, e.g. missing a typical GGVG motif for NADPH binding. The inactivity is indicated by a superscript zero, i.e. ER0. A trans-acting ER gene is found in the gene cluster where enoyl reduction occurs during biosynthesis, that is lovC, mokE, mlcG, eqi9/fsdC, apdC, tenC, dmbC, cheB, ccsC and eqxC as shown in Fig. 2.
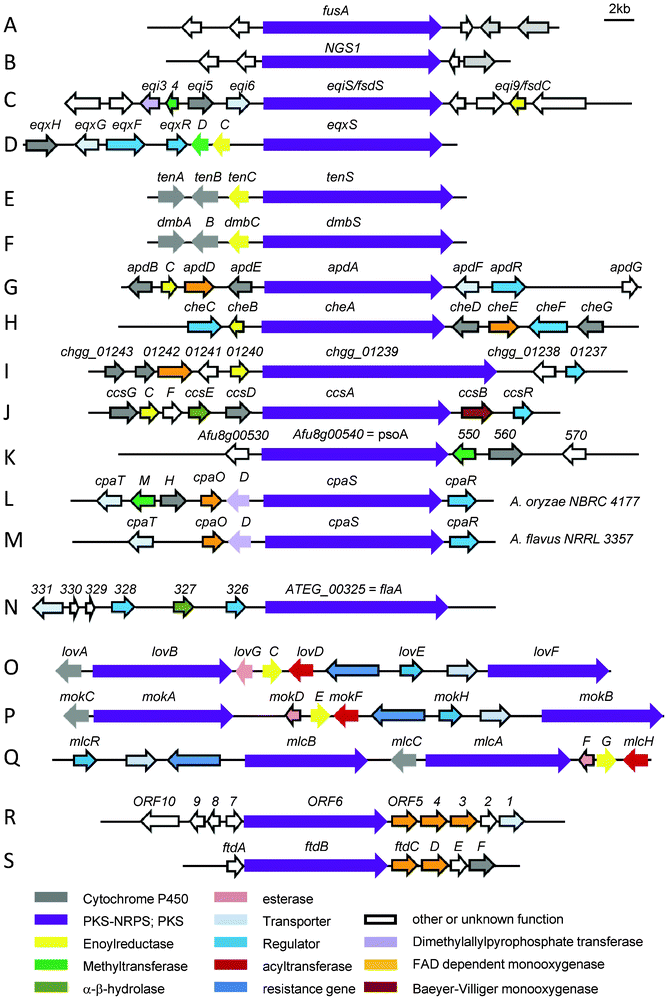 | ||
| Fig. 2 Biosynthetic gene cluster involved in the biosynthesis of (A) fusarin C in Fusarium moniliforme, (B) NG-391 in Metarhizium anisopliae, (C) fusaridione in Fusarium heterosporum, (D) equisetin in Fusarium heterosporum, (E) tenellin in Beauveria bassiana CBS 110.25, (F) desmethylbassianin Beauveria bassiana 992.05, (G) aspyridone in Aspergillus nidulans, (H) chaetoglobosins in Penicillium expansum, (I) chaetoglobosin A in Chaetomium globosum, (J) cytochalasin E and K in A. clavatus, (K) pseurotin A in A. fumigatus Af293, (L) α-CPA in A. oryzae NBRC 4177, (M) α-CPA in A. flavus NRRL 3357, (N) isoflavipucine and dihydroisoflavipucine in A. terreus, (O) lovastatin in A. terreus, (P) monacolin K in Monascus pilosus, (Q) compactin in P. citrinum, (R) dihydromaltophilin in Lysobacter enzymogenes C3, (S) frontalamide A and B in Streptomyces sp. SPB78. | ||
The presence of trans-acting ER is crucial for the biosynthesis of aspyridone, tenellin, desmethylbassianin and lovastatin and only shunt products are formed when it is absent.7–9 That confirms the action of the trans-acting ER on the enzyme bond growing polyketide chain and indicates a necessary close interaction of the PKS–NRPS and the trans-acting ER. The proteins LovB (LNKS) and LovC (trans-acting ER) coelute in size-exclusion chromatography in the absence of an in vitro substrate. This finding further supports a close interaction of both.10 However, no complex formation was detected when only the inactive ER domain from LovB was incubated with LovC.
Further evidence for the close interaction of trans-acting ER with PKS–NRPS comes from investigations with the tenellin system. Heterologous expression of TenS (PKS–NRPS) alone or silencing of TenC (trans-acting ER) leads to the production of aberrant (mis-programmed) polyketides in low titre, where the methylation fidelity has changed dramatically.9,11 However, coexpression of both TenS and TenC leads to high levels of production of correctly programmed material. This shows that the trans-acting ER is involved in programming. A deeper understanding of substrate binding and interaction of trans-acting ERs with the iterative PKS–NRPSs would enhance understanding of the programming.
Enoyl reductases in general display an unusual diversity among organisms and most of them belong to the short-chain dehydrogenase reductase (SDR) superfamily or medium-chain dehydrogenase reductase (MDR) of proteins.12 The crystal structure of the 39.5 kDa protein LovC gives insight into the protein structure of fungal trans-acting ERs interacting with iterative PKS–NRPSs and shows it to possess a (MDR) fold with a unique monomeric assembly.10 The principle difference between the monomeric LovC and the dimeric MDR proteins lies in two extended loops between the catalytic and cofactor binding domains. Sequence alignment of trans-acting ERs associated with fungal iterative PKS–NRPS shows this unique feature to be conserved among them.10
Recently the crystal structure of the ER–KR didomain from the second module of the bacterial multimodular spinosyn PKS was solved and shows the interaction of ER and KR to be different to the one in mFAS.13 Surprisingly, this didomain is monomeric as is LovC while previous studies on erythromycin domains were contradictory, i.e. EryER4 was monomeric whereas Ery(DH + ER + KR)4 was dimeric.13,14
Most known iterative PKS–NRPSs occur in fungi, but recent examples have also been observed in bacteria.15,16 The domain architecture of their PKS module is KS-AT-DH-KR-ACP and thus differs significantly from the PKS module in fungal iterative PKS–NRPSs (Fig. 1D). The absence of a CMeT and ER domain is also visible in the shorter gene size of the PKS–NRPS gene as shown in Fig. 2R and S.
Intriguingly, the bacterial iterative PKS–NRPSs are reported without an ER domain, but their products maltophilin and dihydromaltophilin contain a single bond that is proposed to arise from enoyl reduction of one of the polyketide chains (see section 11).15 No trans-acting ER is found in these biosynthesis gene clusters (Fig. 2R and S).16,17 How enoyl reduction, which the structure suggests in many PTMs, is achieved without an ER domain present in the PKS–NRPS or a trans-acting ER in close vicinity is still a mystery.
The NRPS modules of iterative PKS–NRPSs have the domain organisation of C (condensation), A (adenylation) and T (thiolation, also known as PCP peptide carrier protein). The A domains in NRPS modules are activating an amino acid and load the building block onto the adjacent T domain. They are usually highly selective for a certain amino acid, e.g. the A domain of HSAFS specifically uses L-ornithine.17 However, some flexibility has been observed, e.g. ApdA (see section 4) usually incorporates L-tyrosine, but shows about 50% efficiency also towards L-phenylalanine (L-Phe) and 4-F-Phe.8
The D-phenylalanine-activating adenylation domain (PheA) from the gramicidin S synthase from Bacillus brevis provided the first crystal structure of an NRPS adenylation domain and allowed identification of 10 core residues responsible for substrate recognition.18 X-ray structures of bacterial aryl activating A domains from bacillibactin and acinetobactin synthesizing NRPSs (DhbE and BasE) have been determined and further deepened the knowledge on bacterial A domains.19,20 In contrast, the sidorophore-producing NRPS SidN from Neotyphodium lolii is the only crystal structure of a fungal A domain solved to date. It incorporates the large unusual amino acid cis-AMHO (Nδ-cis-anhydomevalonyl-Nδ-hydroxy-L-ornithine).21 Its binding pocket is large to accommodate the large amino acid, but it is very specific to incorporation of cis-AMHO not reacting with ornithine or any of the 20 proteinogenic amino acids.
Bioinformatic tools such as the NRPS predictor, are not (yet) able to predict the amino acid incorporated by iterative PKS–NRPSs.22 However, if done with care the 10 core residues can be retrieved using these tools or a sequence alignment. The two bacterial iterative PKS–NRPS A domains incorporating ornithine show the same core residues. Blodgett et al. analyzed the 10 core residues of different FtdB homologs and found that all contain conserved motifs characteristic of ornithine binding pockets.16
Unfortunately, the 10 core residues based on bacterial sequences show very little prognostic value in fungal A domain substrate recognition.23–25 Lee et al. suggest an additional 7 residues of the binding pocket to get a better prediction of the amino acid incorporated by fungal A domains.21 Thus amino acid selection in fungal iterative PKS–NRPSs is not well understood.
Several release mechanism are known for PKS, NRPS and hybrids as reviewed recently by Du and Lou.26 An additional domain is often present to release the natural product from the enzyme, e.g. R (reductase), DKC (Dieckmann cyclase) or TE (Thioesterase). Initially the release domain of fungal iterative PKS–NRPSs was predicted to catalyse an NAD(P)H-dependent reductive release based on sequence homology to SDR (short-chain dehydrogenase/reductase) family proteins. These enzymes possess a catalytic triad of Ser-Tyr-Lys necessary for the reduction reaction. However, detailed investigations on tenellin, fusaridione and α-cyclopiazonic acid biosynthesis revealed the formation of a tetramic acid via Dieckmann cyclisation as the release mechanism as discussed in the relevant sections.9,27,28 Bacterial iterative PKS–NRPSs investigated so far harbour a TE domain that shows unusual protease-like and peptide ligase-like activities.29 Intriguingly, the TE domain is believed to releases a tetramic acid via Dieckmann cyclisation, but using an oxoester attached to itself as the substrate.17
In 2004, fusarin C synthase (FUSS) was the first iterative PKS–NRPS to be discovered in studies partly driven by the unclear origin of the nitrogen atom. A previous biosynthetic study had revealed the origins of the carbon skeleton (see below), but the origins of the nitrogen atom in the pyrrolidone ring was unclear.30 With the unexpected discovery of an NRPS module in the synthetase and implied incorporation of an amino acid, the mystery was solved.4
This was soon followed by the publication of equisetin synthase (2005),31 that has been revised very recently to be the fusaridione synthase (2013).32 Further links, in chronological order, were established between the natural product and the PKS–NRPS containing biosynthetic gene clusters for tenellin (2007),33 aspyridone A (2007),34 chaetoglobosin A from Penicillium expansum (2007),35 pseurotin (2007),36 the bacterial macrolactam dihydromaltophilin [HSAF] (2007),15 cyclopiazonic acid (2008),37 NG-391 (2010)38 that is similar to fusarin C, frontalamide A and B (2010),16 desmethylbassianin (2011)39 that is similar to tenellin, isoflavipucine and dihydroisoflavipucine (2011),40 cytochalasin E and K (2011),41 xyrrolin (2012),42 chaetoglobosin A from Chaetomium globosum (2013)43 and equisetin (2013).32 Structures are given in the relevant sections below. All of these metabolites are characterised by containing 5- or 6-membered nitrogen containing rings. Surprisingly, it was also realized, that the PKS synthesising the nonaketide moieties of lovastatin (1999) and compactin (2002) are truncated iterative PKS–NRPSs that terminate with a complete C-domain.7,44,45
Fig. 2 shows the different arrangement of clustered genes surrounding the iterative hybrid PKS–NRPS gene. Gene clusters are scaled to the same size revealing different gene sizes of the PKS–NRPSs.
Several of the biosynthetic pathways discussed here have been included in recent reviews with a different focus e.g. on the evolutionary imprint of fungal PKS–NRPS and on fungal PKS–NRPS biosynthesis by the Hertweck group, on fungal PKSs by Chooi and Tang, on polyketide biosynthesis in the genus Aspergillus by Chiang et al., on chaetoglobosins by Scherlach et al. or on secondary metabolites from entomopathogenic Hypocrealean fungi by Molnár et al.46–51 Tetramic and tetronic acids have been reviewed in 2008 by Schobert and Schlenk.52 However, no comprehensive review of the biosynthesis of natural products produced by the unique group of microbial (fungal and bacterial) iterative hybrid PKS–NRPS exists to date, despite the rapidly increasing attention they have gained.
2 Fusarin and NG-391
Fusarins are a group of mycotoxin acyl-pyrrolidones which are found in various species of Fusaria.53,54 Examples are fusarin A 1,55 C 2,55 D 355 and F 4.56 Several Z isomers of 2 have also been reported, e.g. (8Z)-fusarin C 5,55,57,58 due to the instability of 2. The closely related mycotoxin NG-391 659 which lacks the C7 methyl group of fusarin C 2 and its 8Z analogues NG-393 759 have been isolated from Metarhizium anisopliae. Lucilactaene 860 lacking the C7 methyl group of 1 from Fusarium sp. and epolactaene 961 obtained from Penicillium sp. are closely related highly bioactive compounds.62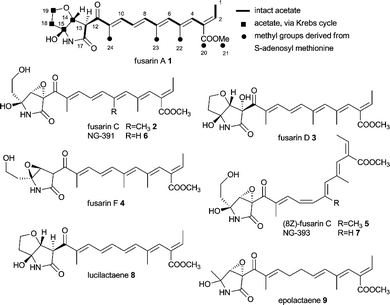
Isotopic labelling studies on fusarin A 1 proved the presence of 9 intact acetate units and that the methyl groups C20–C24 are derived from methionine.30 However, incorporation of acetate into C14–C15 and C18–C19 was significantly lower than for the other acetate units which suggested these 4 carbons derived from a Krebs cycle intermediate, e.g. oxaloacetate. The origin of the nitrogen was cryptic until the discovery of the gene cluster.
The gene cluster involved in the biosynthesis of fusarin C 2Fusarium venenatum (=F. graminearum, telemorph Gibberella zeae) was isolated using PCR primers which had been previously developed by the Bristol polyketide group for fungal CMeT domains.63 This resulted in the isolation of the first gene cluster containing a fungal PKS–NRPS hybrid, comprised of 6 putative genes (Fig. 2A). The analogous gene cluster was also identified in Fusarium moniliforme (=F. verticillioides, telemorph Gibberella fujikuroi = G. moniliformis).4
Knockouts targeting the PKS–NRPS sequences (fusA) were performed and showed no or reduced production of 2 proving the PKS–NRPS FUSS to be involved in fusarin C 2 biosynthesis.4 The function of the homologous gene in a different Gibberella zeae (anamorph F. venenatum = F. graminearum-PH1) was also later proven by a knockout strategy.64
The presence of an NRPS module suggested an amino acid as origin of the nitrogen in 2. The subsequent feeding of [1,2-13C2,15N]-L-homoserine to F. moniliforme confirmed the intact incorporation of the non proteinogenic amino acid L-homoserine into 2.65
Analysis of the fusarin synthase FUSS revealed a domain architecture of KS-AT-DH-CMeT-ΨKR-ER0-KR-ACP-C-A-T-R/DKC. The R domain was believed to catalyse a reductive release to give an aldehyde. This then would undergo an aldol reaction to form the pyrrolidone ring. An alternative route via Dieckmann condensation has been discussed at the same time. Currently no experimental proof of the exact release mechanism exists.
Tailoring steps in the biosynthesis of fusarin C 2 are believed to involve oxidation of the C20 methyl group to a carboxylate moiety and formation of a methyl ester; epoxidation of the ring and either hydroxylation alpha to the nitrogen atom or oxidation to an imine and addition of water to form the cyclic hemi-aminal as outlined in Scheme 1. These reactions would most likely be catalysed by oxidoreductases and an O-methyl transferase. At the time of the gene cluster discovery no genes with these putative activities have been found in the immediate vicinity of fusA. However, now available genome data show the presence of a methyl transferase, a Cytochrome P450 oxygenase, an aldehyde dehydrogenase and an esterase gene upstream of the published gene cluster as shown in Fig. 2A.54
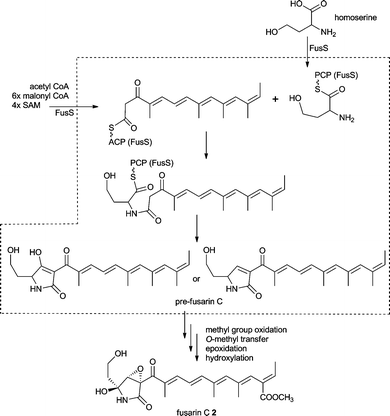 | ||
| Scheme 1 Proposed fusarin C 2 biosynthesis (structures in the dashed box are proposed compounds). | ||
In 2010 the gene cluster involved in biosynthesis of NG-391 6 and NG-393 7 was identified in the entomopathogenic fungus Metarhizium robertsii (formerly known as M. anisopliae) and proven by knockout (Fig. 2B).38 The domain organisation of the iterative hybrid PKS–NRPS is the same as in FUSS. Knockout of the PKS–NRPS gene NGS1 had no influence on virulence levels of the fungus against larvae of Spodoptera exigua or hydrogen peroxide generated oxidative stress.
3 Equisetin and fusaridione
Equisetin 10 and the N-desmethyl homolog trichosetin 11 are tetramic acids built from an octaketide and a serine moiety.66–7010 is produced by Fusarium species and 11 was found in a dual culture of Trichoderma harzianum and Catharanthus roseus Callus. The compounds inhibit HIV-1 integrase and are broadly toxic as are the phomasetin 12 and oteromycin 13.71,72 Similar compounds have been isolated from various fungi such as beauversetin 14 from Beauveria bassiana,73 ascosalipyrrolidinone A 15 and B 16 from Ascochyta salicorniae,74 Sch210972 17 from Microdiplodia sp.73 and Chaetomium globosum (Mer-0229)75 and ZG-1494α 18 from Penicillium rubrum.76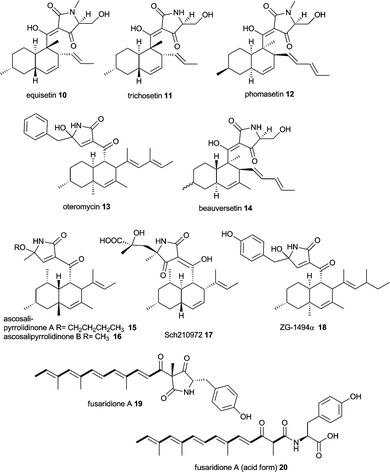
In 2005 Eric Schmidt's group published the gene cluster for equisetin 10 from Fusarium heterosporum (Fig. 2C).31 Very recently the same group showed this gene cluster to be silent under various culture conditions. Initially, degenerate primers for conserved regions in KS and AT domains of fungal iterative PKSs identified three reducing PKS gene sequences. One of them was further investigated because of a positive RT-PCR correlation to equisetin production. The correlation between the gene cluster and the natural product was thought to be confirmed by knockout mutagenesis. Increasing inconsistencies in experimental results led to whole genome sequencing of Fusarium heterosporum ATCC 74349 and revealed two iterative PKS–NRPS containing gene clusters.32 The third initially found reducing PKS lacked an NRPS portion. The original eqi genes where renamed to fsd genes in accordance with the fusaridione A 19 and 20 that was found by homologous expression of PKS–NRPS FsdS (formerly EqiS) and the new gene cluster was termed eqx.32 Both PKS–NRPSs contain the same domain organisation, i.e. KS-AT-DH-CMeT-YKR-ER0-KR-ACP-C-A-T-DKC and both clusters harbour a trans-acting ER (FsdC and EqxC, Fig. 2C and D). Both clusters also harbour genes encoding a methyltransferase (fsdD and eqxD) and a cytochrome P450 oxygenase (fsdH and eqxH). Again, the support for the newly discovered gene cluster to be the true equisetin biosynthetic gene cluster comes mainly from knockout experiments rather than heterologous expression.32
The amino acid substrate specificity of both FsdS and EqxS have been determined using pyrophosphate exchange assay in addition to the knockout experiments. All 20 proteinogenic amino acids an N-methyl-L-serine have been tested. The major amino acid activated by FsdS ACP-CATR protein was L-tyrosine, while that activated by EqxS A and ATR protein was L-serine.32 This corresponds to the amino acids found in 19/20 and 11. Furthermore, both putative regulator genes eqxF and eqxR have been overexpressed and eqxR led to efficient synthesis of 10 in 5 days in liquid culture whereas otherwise 21 days solid media was required.
The proposed biosynthesis of 10 and 11 is outlined in Scheme 2. The decalin ring system is likely to be formed analog to the well investigated biosynthesis of lovastatin (see section 10). Thus, EqxS together with EqxC is proposed to synthesise a heptaketide which then undergoes a Diels–Alder cyclisation and a further elongation to the final octaketide. After fusion to a serine moiety by the NRPS module the tetramic acid is thought to be released via a Dieckmann cyclisation. Previous detailed in vitro investigations of the FsdS (EqiS) R domain with small synthetic substrates shows that it does not perform a reduction and does not bind reducing cofactors. Instead a Dieckmann cyclisation is carried out to release a tetramic acid moiety.27 However, since these investigations were not carried out using EqxS there is no solid proof for a Dieckmann cyclisation as the release mechanism for 11. EqxD knockout transformants show high levels of production of 11 and no production of 10. Therefore, N-methylation is most likely a tailoring step in the biosynthesis of 10 following the release of 11 (Scheme 2).
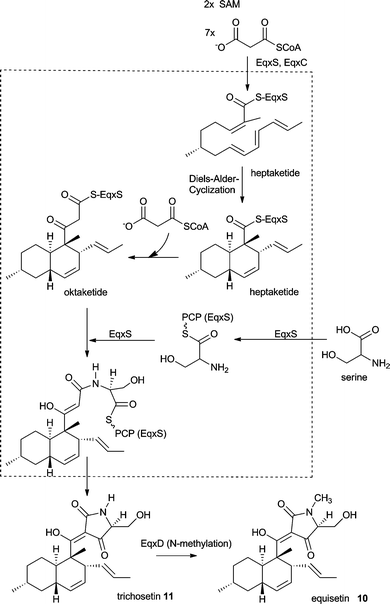 | ||
| Scheme 2 Proposed biosynthesis of equisetin 10 and trichosetin 11. | ||
4 Aspyridone, tenellin and desmethylbassianin
Aspyridone A 21 and B 22, tenellin 23 and desmethylbassianin (DMB) 24 are pyridones very similar in chemical structure with more examples in nature such as bassianin 25, militarinone A 26 and B 27 and farinosone A 28 and B 29.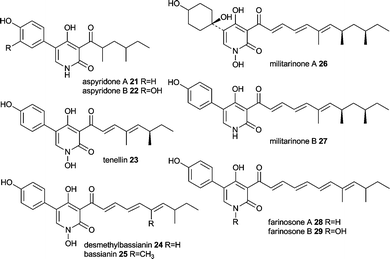
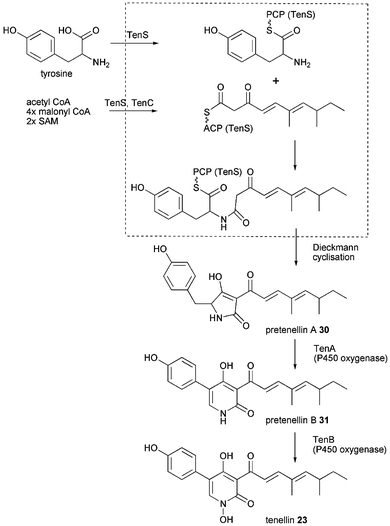
Since 23 was the first of these compounds to be linked to its genes its biosynthesis outlined in Scheme 3 stays representative for the similar biosyntheses of aspyridone A 21 and DMB 24. The 2-pyridone tenellin 23 is produced by the insect pathogen fungus Beauveria bassiana CBS 110.25 but doesn't contribute to its pathogenicity. The biosynthetic gene cluster has been found by the Cox group by systematic screening of a genomic gene library with degenerated primers based on conserved regions in the CMeT domain of highly reducing PKS and fungal PKS–NRPS hybrids.33 The 21.9 kb sequence of the positive clone revealed a gene cluster comprising of 4 putative genes, two cytochrome P450 oxygenases (TenA and TenB), a trans acting ER (TenC) and the iterative PKS–NRPS hybrid (TenS), see Fig. 2E. The domain architecture of TenS is KS-AT-DH-CMeT-ΨKR-ER0-KR-ACP-C-A-T-DKC. The involvement of the PKS–NRPS in tenellin biosynthesis has been proven by knockout experiment.33
 | ||
| Scheme 3 Proposed tenellin 23 biosynthesis. | ||
Heterologous expression in A. oryzae M-2-3 (argB−) showed the PKS–NRPS to produce pretenellin A 30 when expressed together with TenC. Whereas expression of the PKS–NRPS alone produced prototenellins A–C, tetramic acids with incorrect polyketide side chain in low titre.9 Formation of tetramic acids without involvement of any oxygenases suggests that the release domain functions as a Dieckmann cyclase rather than as reductase. The biosynthetically intriguing ring expansion from the tetramic acid precursor 30 to the 2-pyridone pretenellin B 31 is catalysed by TenA. TenB the other cytochrome P450 oxygenase catalyses the N-hydroxylation as shown in a combination of heterologous expression, RNA silencing and cell free extracts experiments, outlined in Scheme 3.77 Besides the involvement of oxidative enzymes the exact mechanism of the ring expansion remains unknown.
Aspyridone A 21 and B 22 are compounds that were discovered by genome mining of the Aspergillus nidulans genome in 2007.34A. nidulans genome revealed only one PKS–NRPS gene cluster comprising of 8 putative genes including a putative activator gene designated as apdR (Fig. 2G). C. Hertweck's group successfully overexpressed this activator in the original organism using an inducible vector and were able to prove expression of apdA mRNA in their transformants.34 Similarities in chemical structure, homolog enzymes and domain architecture of the PKS–NRPS (ApdA, KS-AT-DH-CMeT-ΨKR-ER0-KR-ACP-C-A-T-R/DKC) suggest a biosynthesis very similar to that for 23. It differs in the number of cycles leading to a tetraketide for 22vs. a pentaketide for 23. The trans-acting ER (ApdC) is most likely used two times during polyketide synthesis whereas TenC acts only in the first cycle. 21 is presumably converted into 22 by oxidation catalysed by ApdD a predicted FAD-dependent monooxygenase.
In vitro studies of intact ApdA and ApdC were carried out after reannotation of introns and revealed the C and A domains of ApdA to have some flexibility in amino acid incorporation.8 Dissected PKS and NRPS modules independently heterologously expressed are able to work together in vitro. Yeast harbouring expression plasmids for ApdA PKS, CpaS NRPS (NRPS module of cyclopiazonic acid synthase, see section 7) and ApdC is able to produce the tryptophan analog of 21.8 Intriguingly, absence of SAM or ApdC in the in vitro assays leads to the formation of pyrones with different chain lengths. This stays in contrast to in vivo experiments of heterologous expression of TenS without TenC in which tyrosine is still incorporated leading to tetramic acids with different chain length and methylation pattern.8,9
Bassianin 25 was discovered in 1968 in Beauveria tenella and B. bassiana.78 Its structural data was published in 1977.78,79 Attempts to reisolate 25 lead only to the isolation of desmethylbassianin (DMB) 24 whereas 25 was not found in 30 Beauveria species.39 The biosynthetic gene cluster of 24 has been isolated and verified by a dual KO/silencing strategy. The gene order and domain architecture of the PKS–NRPS (DMBS) are the same as in the tenellin gene cluster (Fig. 2F).39 The biosynthesis is carried out in the same way as in 21 (Scheme 3). The trans-acting ER (DMBC) acts during the first cycle as does TenC and both are interchangeable without any influence on the biosynthetic product. TenS and DMBS harbour different programmes regarding chain length and methylation pattern of the polyketide part despite their high homology (>85% identity). This enabled rational domain swaps between TenS and DMBS and revealed the CMeT domain solely responsible for the methylation pattern and the KR domain a key factor in chain length determination.80
5 Cytochalasans
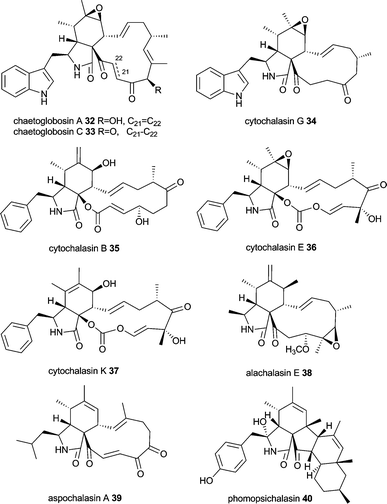
Cytochalasans have been comprehensively reviewed recently in Natural Product Reports including the biosynthesis of chaetoglobosins.49 This large group of structurally intriguing fungal metabolites is characterized by an isoindolone moiety fused to a macrocycle. They are produced by different fungal genera, e.g. Ascochyta, Aspergillus, Chaetomium, Metarhizium, Penicillium, Phoma, Rosellinia, Xylaria, Zygosporium and possess a wide range of biological activities ranging from binding to actin filaments and thus inhibiting cytokinesis to antimicrobial, anti-angiogenetic, cytotoxic and cholesterol synthesis inhibiting activity.81–84Cytochalasan biosynthesis involves in general the formation of an octa- or nonaketide chain and the incorporation of an amino acid. Intriguingly, amino acids found in cytochalasans are diverse, including tryptophan (chaetoglobosins, e.g. chaetoglobosin A 32 and C 33, cytochalasin G 34), phenylalanine (cytochalasins, e.g. cytochalasin B 35, E 36 and K 37, zygosporins), alanine (alachalasins, e.g. alachalasin E 38), leucine (aspochalasins, e.g. aspochalasin A 39) and tyrosine (scoparasins, phomopsichalasin 40, which has a tricyclic system instead of the macrolide).49,84
In 2007 the gene cluster involved in biosynthesis of chaetoglobosins has been isolated from Penicillium expansum.35 This was done by probing a gene library by dot blot hybridisation using the sequence of CMeT, KR and ACP domain of the Fusarium venenatum fusarin C synthase (fusA) as probe. The gene cluster was verified to be involved in chaetoglobosin biosynthesis by RNA silencing.35 The gene cluster comprises of 7 genes, the 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 039 kb PKS–NRPS sequence cheA, an ER sequence cheB, two sequences coding for P450 oxygenases cheD and cheG, a flavin dependent monooxygenase (FMO) cheE and two sequences encoding putative C6 transcription factors cheC and cheF (Fig. 2H).35 A gene cluster for biosynthesis of 32 has been recently identified from the genome of Chaetomium globosum (Fig. 2I) and detailed investigation of oxidative tailoring steps has been carried out.43 This gene cluster comprises of 8 genes, the PKS–NRPS sequence chgg_01239, an ER sequence chgg_01240, a sequences chgg_01241 with no function assigned, an annotated sequence chgg_01242, where close investigation revealed a P450 oxygenase chgg_01242-1 and an FMO chgg_01242-2 sequence encoded, another P450 encoding gene chgg_01243, a transposase sequence chgg_01238 and a sequences encoding a putative transcription factor chgg_01237 (Fig. 2I). The PKS_NRPS gene chgg_01239 with the common domain organisation KS-AT-DH-CMeT-ΨKR-ER0-KR-ACP-C-A-T-R/DKC is rather long, although a misassigned start codon might be the reason. Amino acid identity and similarity to CheA is only 34.3% and 52.2%.
039 kb PKS–NRPS sequence cheA, an ER sequence cheB, two sequences coding for P450 oxygenases cheD and cheG, a flavin dependent monooxygenase (FMO) cheE and two sequences encoding putative C6 transcription factors cheC and cheF (Fig. 2H).35 A gene cluster for biosynthesis of 32 has been recently identified from the genome of Chaetomium globosum (Fig. 2I) and detailed investigation of oxidative tailoring steps has been carried out.43 This gene cluster comprises of 8 genes, the PKS–NRPS sequence chgg_01239, an ER sequence chgg_01240, a sequences chgg_01241 with no function assigned, an annotated sequence chgg_01242, where close investigation revealed a P450 oxygenase chgg_01242-1 and an FMO chgg_01242-2 sequence encoded, another P450 encoding gene chgg_01243, a transposase sequence chgg_01238 and a sequences encoding a putative transcription factor chgg_01237 (Fig. 2I). The PKS_NRPS gene chgg_01239 with the common domain organisation KS-AT-DH-CMeT-ΨKR-ER0-KR-ACP-C-A-T-R/DKC is rather long, although a misassigned start codon might be the reason. Amino acid identity and similarity to CheA is only 34.3% and 52.2%.
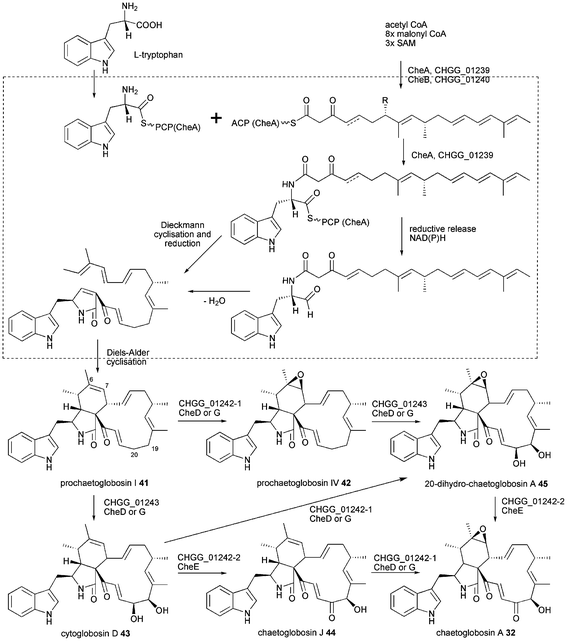
The proposed biosynthesis of chaetoglobosin A 32 is outlined in Scheme 4 based on previous P450 inhibitor studies using metyrapone and the most recent findings from chgg knockout analysis.43,85 The release mechanism and cyclisation of chaetoglobosins have not been investigated in detail leaving the discussion about reductive release vs. Dieckmann cyclisation open. However, recent results from cytochalasan biosynthesis suggest a reductive release as outlined below. A [4 + 2] Diels–Alder endo cycloaddition is predicted to occur after the release of the hybrid polyketide-amino acid molecule, because it involves the pyrrol-2-one-(5H) created during release or subsequent reduction. That differs from the lovastatin biosynthesis where the Diels–Alder reaction is proposed to occur at the hexaketide stage during polyketide synthesis.86 The first isolated intermediate is prochaetoglobosin I 41 obtained by knockout of all three oxidizing enzymes.43 Double knockout of CHGG_01242-2/CHGG_01243 led to accumulation of prochaetoglobosin IV 42 showing the P450 monooxygenase CHGG_01242-1 to perform a stereoselective epoxidation of the olefin at C6–C7. Double knockout of CHGG_01242-1/CHGG_01242 led to accumulation of cytoglobosin D 43 revealing the second P450 CHGG_01243 to carry out a stereoselective dihydroxylation of C19 and C20. Single knockout of CHGG_01242-1 showed accumulation of 43 and chaetoglobosin J 44 and knockout of CHGG_0142-2 led to isolation of 20-dihydro-chaetoglobosin A 45. Single knockout of CHGG_01243 accumulated 42 by loss of 41 compared to the triple knockout. This extensive investigation of redox enzymes proofs the three oxygenases to be sufficient for transformation of 43 into 32 and aligning the chemical reaction they catalyse.43
 | ||
| Scheme 4 Proposed biosynthesis of chaetoglobosin A 32. | ||
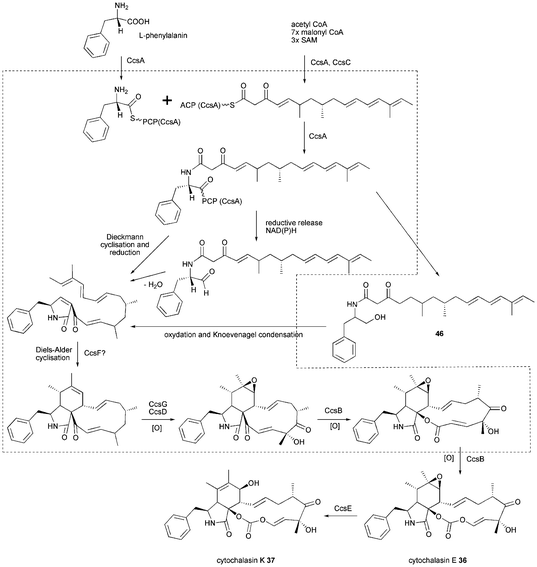
In 2011 the gene cluster involved in cytochalasin E 36 and K 37 biosynthesis has been identified by genome mining of A. clavatus using the CheA as query sequence.41 From the four PKS–NRPSs found, ACLA_004770 is orthologous to pseurotin A synthase from A. fumigatus (see section 6) and ACLA_023380 is most closely related to FsdS from F. heterosporum (see section 4). Due to the significant structural differences of pseurotin A 47 and equisetin 10 to cytochalasin E 36 and K 37 these two PKS–NRPSs were regarded as unlikely to be involved in the biosynthesis of the cytochalasins.
Earlier labelling studies on cytochalasin B 35 implied an enzymatic Baeyer–Villiger-type oxygen insertion to be involved in its biosynthesis.87,88 Accordingly, the unusual insertion of two oxygen atoms into 36 and 37 may occur via two consecutive Baeyer–Villiger oxidations. Directly downstream of the PKS–NRPS gene ACLA_078660, a gene likely encoding for a flavin dependent monooxygenase (ACLA 078650 renamed ccsB) was revealed. CcsB exhibits homology to well-characterised type I Baeyer–Villiger monooxygenases (BVMOs), especially to the recently characterised Pseudomonas sp. HI-70 cyclopentadecanone monooxygenase (CPDMO) CpdB (41% identity) and Rhodococcus ruber SC1 cyclododecanone monooxygenase (CDMO) CddA (38% identity).89 The remaining PKS–NRPS (ACLA_077660) lacks a BVMO candidate in the vicinity, but contains a gene close by that encodes most likely for an O-methyltransferase that is not required in the biosynthesis of 36 and 37. Knockout of the ACLA_078660 (ccsA) in A. clavatus abolished the production of cytochalasins 36 and 37 which proves the link between gene and biosynthesis product.41
The whole gene cluster contains 8 putative genes, the PKS–NRPS gene ccsA (domain architecture KS-AT-DH-CMeT-ΨKR-ER0-KR-ACP-C-A-T-R/DKC), the BVMO gene ccsB, a trans-acting ER gene ccsC, two cytochrome P450 oxygenase genes ccsD and ccsG, an α/β hydrolase gene ccsE, a fungal transcription factor gene ccsR and a gene ccsF where no function could be assigned (Fig. 2J). Surprisingly, CheA is less similar to CcsA than to TenS and DMBS and the tailoring enzymes in the Che and Ccs cluster do not appear to share close homology either.41
The proposed biosynthesis of 36 and 37 is outlined in Scheme 5. Very recently, ccsA and ccsC were heterologously expressed and shown to produce the acyclic alcohol 46.90 This shows the release of a linear product is possible and a reductive release is more likely than a Dieckmann cyclisation. Additionally, the Knoevenagel condensation and Diels–Alder cyclisation are predicted to occur post chain release and CcsF is a candidate for a separate Diels–Alderase. Oxidation of C17, C18 and epoxidation of double bond C6–7 is proposed to be carried out only by the two P450 oxygenases in contrast to chaetoglobosin biosynthesis. A subsequent double Baeyer–Villiger oxidation carried out by CcsB is proposed to generate the unique vinyl carbonate moiety in cytochalasin E 36 and K 37. If proven, CcsB would be the first example of a BVMO that can catalyze such a double Baeyer–Villiger oxidation. The epoxide-containing 36 is likely to be hydrolysed to 37 involving CcsE, an α/β hydrolase.
 | ||
| Scheme 5 Proposed biosynthesis of cytochalasin E 36 and K 37. | ||
6 Pseurotin
Pseurotins are characterized by the unique 1-oxa-7-aza-spiro[4,4]non-2-ene-4,6-dione skeleton and vary only slightly in their chemical structure. Pseurotin A–E (47, 49–52) were first described from Pseudeurotium ovalis.91–93 Pseurotins are also produced by different Aspergillus fumigatus, e.g. pseurotin A 47, 8-O-demethylpseurotin A 48, pseurotin D 47 and F1/F2 49/50, 11-O-methylpseurotin A 55 as well as synerazol 56.36,94–96 Additionally, Neosartorya sp. produces azaspirene 57, while Penicillium canescens and other Penicillium strains produce 47.97–99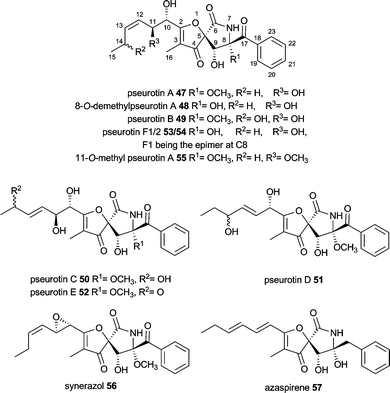
Early biosynthetic studies using labelled precursors revealed propionate as the starter unit, which is very rare in fungi, malonyl-CoA as the extender unit and phenylalanine as the incorporated amino acid.100 Since A. fumigatus strains have been shown to produce pseurotins and the A. fumigatus Af293 genome contains only one hybrid PKS–NRPS close reexamination of secondary metabolite production revealed this strain to produce pseurotin A 47.36 Knockout of the hybrid PKS–NRPS as well as its homolog overexpression confirmed the relationship between gene and natural product. The gene cluster (Fig. 2 K) comprises of genes of a PKS–NRPS hybrid Afu8g00540 (EAL85113) named psoA, two hydrolases Afu8g00530 (EAL85114) and Afu8g00570 (EAL85110), one gene with weak similarity to a methyltransferase Afu8g00550 (EAL85112) and one encoding for a P450 oxygenase Afu8g00560 (EAL85111). The involvement of tailoring enzymes outside of the gene cluster is likely, since only one P450 oxygenase gene is present in the gene cluster.36 The PKS–NRPS has the common domain architecture KS-AT-DH-CMeT-ΨKR-ER0-KR-ACP-C-A-T-R/DKC. There is no gene encoding an external ER found in close proximity, but the biosynthesis does not include any enoyl reduction step.
Simultaneous examination of culture extracts of A. clavatus NRRL1 which harbours a similar gene cluster were not able to detect pseurotins.36 No further experiments were carried out so far to provide a deeper insight into pseurotin biosynthesis leaving the biosynthesis outlined in Scheme 6 rather speculative especially regarding tailoring steps. The PKS–NRPS most likely releases a tetramic acid, although a reductive release and subsequent hydroxylation can not be excluded. The tetramic acid is proposed to be converted to 57 by EAL85111 and EAL85114 and further processed by epoxidation, oxidation and E/Z isomerisation. Hydrolysis of the epoxide by EAL85110 would lead to 48 and subsequent O-methylation by EAL85112 would give 47.
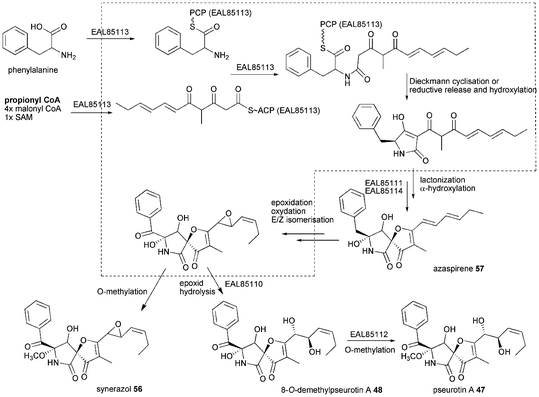 | ||
| Scheme 6 Proposed biosynthesis of pseurotin A 47. | ||
7 Cyclopiazonic acid
First isolated in 1968 from Penicillium cyclopium and later from other fungi including Aspergillus species α-cyclopiazonic acid (α-CPA) 58 is a toxic indole tetramic acid that inhibits sarcoplasmic reticulum Ca2+-ATPases resulting in cell death through apoptosis within the endoplasmic reticulum and mitochondria.101,102 Early labelling studies using P. cyclopium showed 58 to be derived from tryptophan, a C5-unit formed from mevalonic acid and two molecules of acetic acid.103 For conversion of cyclo-acetoacetyl-L-tryptophan (cAATrp) 59 to β-CPA 60 a DMAPP transferase (presumably equivalent to CpaD in A. oryzae) was predicted as well as a flavoprotein oxidocyclase (presumably equivalent to CpaO) for the conversion of 60 to 58.104–106 In 2005 the patent of Christensen showed a DMAPP transferase in A. oryzae to be involved in α-CPA 58 production.107 The enzyme CpaD has been characterised in detail and catalyses the expected reaction of 59 to 60 as shown in Scheme 7.108 The PKS–NRPS (called CpaA or CpaS) has been identified from α-CPA producing A. oryzae NBRC 4177 in 2008 by comparing the telomere-adjacent region of this strain with that of a nonproducing strain (A. oryzae RIB40) and proven by knockout which abolished production of 58.37 It has also been identified from A. flavus NRRL 3357 and heterologous expression of the PKS–NRPS produced solely 59.109 Gene clusters are shown in Fig. 2L and 2M. The size of the PKS–NRPS gene cpaS is comparable to apdS. However, DH, CMeT, ER and KR domains are non-functional due to mutations in their active sites.37,109 The release domain lacks the catalytic triad Ser-Tyr-Lys of SDR family proteins. Heterologous expression in E. coli showed it to act as Dieckmann cyclase and its loss of activity when the aspartate D3803 was mutated to alanine.28 Thus the CpaS domain organisation is KS-AT-DH0-CMeT0-ΨKR-ER0-KR0-ACP-C-A-T-DKC.28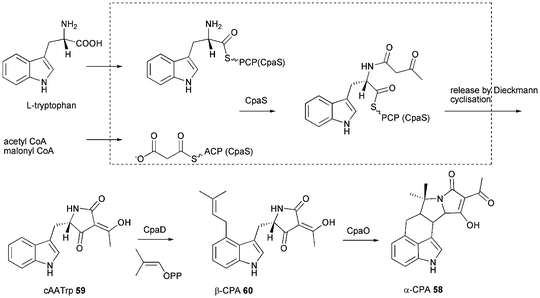 | ||
| Scheme 7 Proposed biosynthesis of α-cyclopiazonic acid (α-CPA) 58. | ||
8 Isoflavipucine and dihydroisoflavipucine
Isoflavipucine 61 (first isolated from A. flavipes) and dihydroisoflavipucine 62 (isolated for the first time during genome mining as described below, but shortly after reported in Phoma sp.) are pyridiones related to (−)-flavipucine 63 (=fruit rot toxin B) and (+)-flavipucine 64 produced by A. flavipes and other Aspergilli, fruit rot toxin A (FRT-A, Macrophoma sp.) 65, sapinopyridione 66 isolated from Sphaeropsis sapinea and rubrobramide 67 from Cladobotryum rubrobrunnescens.40,110–116 Labelling studies of flavipucine with [1-13C]-, [2-13C]- and [1,2-13C2]-acetate indicated that C1–8 are derived from acetate.117 Additionally, [1-13C]L-leucine and [5-2H3]L-leucine were incorporated into 61 by A. terreus and proved C7 to be derived from this amino acid.40 Acetate and leucine labelling of C7 are not contradictory, since leucine is biosynthesised from valine and malonyl-CoA in vitro.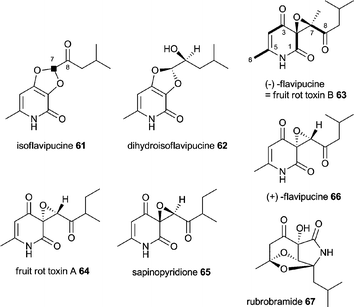
Genome mining becomes applicable as a tool since more and more fungal sequences are publicly available. After using that tool successfully to discover aspyridone A 21 and B 22 a similar approach was employed to mine compounds produced by the single PKS–NRPS (ATEG_00325) gene cluster in the pathogenic fungus A. terreus. The gene cluster shown in Fig. 2N comprises upstream of the PKS–NRPS of two regulators ATEG_00326 and ATEG_00328, an α,β-hydrolase ATEG_00327 and an MFS transporter ATEG_00331 whereas downstream only 9 genes further away (i.e. 19 kb and not shown in Fig. 2N) a ketoreductase gene is found. Induced expression of the transcription activator ATEG_00326 was not sufficient and monitoring gene expression using lacZ reporter strain under a wide range of environmental conditions was necessary to discover isoflavipucine 61 and dihydroisoflavipucine 62.40
The domain organisation of the PKS–NRPS is KS-AT-DH0-CMeT0-ΨKR-ER0-KR0-ACP-C-A-T-R and consequently a non reduced triketide is proposed to be produced from the PKS module. Labelling experiments, the genome mining study and a synthetic model showing that 63 rearranges at 139 °C in 15 min to 61, led to the proposed biosynthesis in Scheme 8.40,110
![Proposed biosynthesis of isoflavipucine 61 and dihydroisoflavipucine 62 including acetate and [1-13C]l-leucine labeling.](/image/article/2013/RA/c3ra42661k/c3ra42661k-s8.gif) | ||
| Scheme 8 Proposed biosynthesis of isoflavipucine 61 and dihydroisoflavipucine 62 including acetate and [1-13C]L-leucine labeling. | ||
Detailed investigation on the biosynthesis would be necessary to determine the release mechanism (reduction vs. Dieckmann cyclisation) and the rearrangement steps. So far speculation is possible based on the presence of the catalytic triad Ser-Tyr-Lys, an important aspartate residue adjacent to the catalytic triad and the conserved motif GxxGxxG of the R domain, all pointing towards the postulated reductive release.23,40,112 Heterologous expressed protein of the NRPS module including the R domain of ATEG_00325 produces thiopyrazines in the presence of an amino acid, DTT or SNAC, NADPH, ATP and MgCl2 which also requires a reductive release.28,40,112,118 Very recently the Yi Tang group heterologously expressed the NRPS module of ATEG00325.118 In this artificial setting the enzyme was able to form thiol-substituted pyrazines in the presence of an amino acid, DTT or SNAC, NADPH, ATP, MgCl2 and phosphate buffer from a wide range of amino acids. The NRPS modules of ApdA and CpaS did not show any thiol-substituted pyrazine formation under the same conditions. Exploring the broad substrate specificity of the A domain of ATEG00325 towards amino acids and the flexibility of the NRPS part to use different free thiols a library of 63 different thiol-substituted pyrazines was obtained in good yields.118
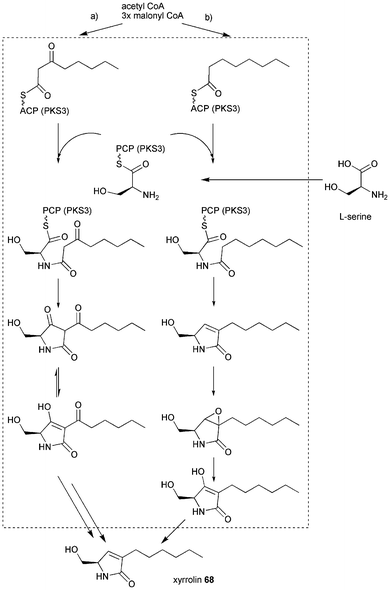
9 Xyrrolin
Xylaria sp. BCC 1067 has been reported to produce 19,20-epoxycytochalasin Q and effort was undertaken to investigate its biosynthesis.119,120 Fragments of 10 PKS, 7 NRPS and one PKS–NRPS hybrid (named Pks3) have been found in the fungus by molecular biology studies using degenerated primer. It is likely that in the molecular biology study not all gene clusters have been found and the fungus harbours more than one PKS–NRPS gene. Genome sequencing would show the whole potential of secondary metabolite biosynthesis.Knockout of Pks3 led to the absence of a single compound which was a new metabolite named xyrrolin 68 (from Xylaria pyrroline) and is not related to 19,20-epoxycytochalasin Q.42 Xyrrolin 68 shows weak cytotoxic activity against the oral cavity cancer cell line KB (IC50 36.9 mg L−1). The domain organisation of Pks3 is predicted to be KS-AT-DH-CMeT0-ΨKR-ER0-KR-ACP-C-A-T with no obvious release domain. The CMeT domain is similar to FsdS and ApdA CMeT domains and inactivity of the CMeT domain is only assumed due to the absence of a branching methyl group in 68. Inactivity of ER is assumed by comparison with known PKS–NRPSs sequences. Nothing is known about adjacent genes leaving the question open if there are trans-acting ER and tailoring genes clustered, which would be required for the proposed biosynthesis as outlined in Scheme 9.
 | ||
| Scheme 9 Proposed biosynthesis of xyrrolin 68. | ||
A tetraketide diketone (route a) or monoketone (route b) could be synthesised by the PKS part. L-serine is proposed to be selected by the A domain and the PK–NRP intermediate could be released by Dieckmann cyclisation (route a) or reductive cyclisation (route b). Dieckmann cyclisation would lead to a tetramic acid and need to undergo two unusual ketoreductions to afford the fully reduced form of 68. The reductive release as outlined in route b would still require the ketoreduction at C5. However, no putative ketoreductase sequence is found in 2 kb vicinity of the PKS–NRPS sequence.
10 Lovastatin, compactin, monacolin
Compound 69 discovered independently in 1978 as lovastatin in Aspergillus terreus and in 1979 as monacolin K in Monascus ruber is also known as meviolin and mevastatin.121 Lovastatin is related to compactin 70 (also named mevastatin and ML-236B), which has been discovered earlier (in 1976) and only lacks the C-6 methyl group.122 Compactin is produced by several fungal species but most investigations are done using Penicillium citrinum.123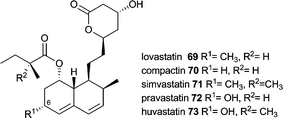
Due to its inhibitory activity towards (3S)-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase lovastatin was used as a cholesterol lowering agent and serves as the lead structure and precursor for semisynthetic statins such as simvastatin 71. Compactin possesses a similar activity and serves as a precursor for pravastatin 72. Biotechnological production and applications of statins have been reviewed recently by Barrios-González and Miranda including enhancement of production by introduction of additional copies of the compactin regulator MlcR into P. citrinum.124
The lovastatin gene cluster is amongst the best studied fungal polyketide gene clusters. Although 69 does not contain an amino acid derived moiety, the PKS responsible for biosynthesis of the nonaketide (lovastatin nonaketide synthase = LNKS = LovB) resembles the common architecture of the PKS part in a fungal iterative hybrid PKS–NRPS and contains a condensation domain at the C-terminus, i.e. domain architecture of KS-AT-DH-CMeT-ΨKR-ER0-KR-ACP-C.7 It is not yet certain if this C domain is a remaining part of an NRPS module. However, LovB that lacks the C domain is not able to produce dihydromonacolin L 74in vitro.125 In addition, the ER domain of LovB is inactive and the trans-acting ER LovC, which is present in the gene cluster (Fig. 2O), is required for the biosynthesis of 69.7 These similarities suggest biochemical characteristics of LovB and its domains as well as LovC to be similar to fungal iterative PKS–NRPSs and its trans-acting ERs and are worthy to be discussed in this context. LovC biochemical characterisations have been included into the introduction of this review, because it is the trans-acting ER studied in the most detail and most of its characteristics are believed to be general characteristics of the fungal trans-acting ERs.
Biochemical characterisation of LovB domains showed the AT domain to transfer malonyl-CoA and other acetyl groups to a range of ACPs whereas the KS domain is solely able to transfer malonyl-CoA to the same range of ACPs but to a much lower extent.126 The PKS module and the C-domain separately heterologously expressed can react together and this ability has later also been shown for the PKS and NRPS modules of aspyridone synthase ApdA.8,125 Interestingly, in vitro experiments showed that LovB is not able to release 74 but release can be achieved by interaction of LovB with different fungal TE domains.125 Very recently close investigation of LovG identified it as a multifunctional esterase involved in release of 74.127
Besides LovB and LovC, the biosynthesis of 69 (Scheme 10) involves the lovastatin diketide synthase (LDKS = LovF), which reacts non iteratively by producing the α-S-methylbutyrate side chain of 55. LovF has the domain organisation KS-AT-DH-CMeT-ΨKR-ER-KR-ACP with no obvious release domain to offload the product. Recently Yi Tang and coworkers showed protein protein interaction of LovF and the acyltransferase LovD suggesting the transfer to monacolin J 75 as an offloading mechanism.128 Earlier detailed investigations of the acyltransferase LovD revealed not only a broad substrate specificity towards the acyl substrate by using synthetic thioester but also the ability to convert 6-hydroxyl-6-desmethylmonacolin J into 72 and huvastatin 73.129
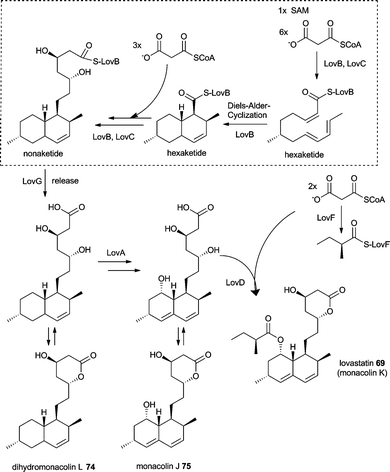 | ||
| Scheme 10 Proposed biosynthesis of lovastatin 69. | ||
The gene clusters of compactin 70 in Penicillium citrinum and monacolin K 69 in Monascus pilosus have been characterised in 2002 and 2008 and are composed of 9 genes corresponding to the ones in the lovastatin 69 gene cluster (Fig. 2P and Q).45,130 Protein similarities are between 40 and 85% and the biosynthesis of 70 is believed to resemble that of 69 (Scheme 10).130
11 Polycyclic tetramate macrolactams (PTMs)
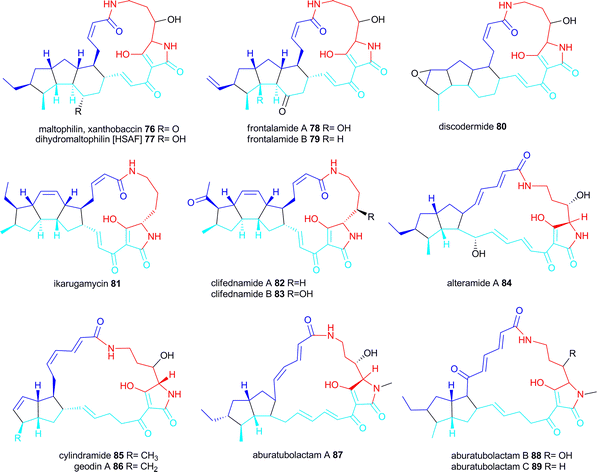
This family of biologically active small molecules comprises of a macrocyclic lactam ring (17- or 21 membered) with an embedded tetramic acid ring and a set of carbocyclic rings. Arrangements of the carbocyclic rings found so far are 5-5-6, 5-6-5 and 5-5.PTMs with a 5-5-6 ring arrangement include maltophilin 76 isolated from the γ-proteobacterium Stenotrophomonas maltophilia R3089 (later reisolated as sodium salt named xanthobaccin from a different Stenotrophomonas strain), dihydromaltophilin (=heat-stable antifungal factor HSAF) 77 first isolated from a Streptomyces sp. but also produced by the γ-proteobacterium Lysobacter enzymogenes C3 (originally called S. maltophilia C3), frontalamide A 78 and B 79 produced by Streptomyces sp. and discodermide 80 isolated from the marine sponge Discodermia dissoluta but likely to be produced by an associated symbiont.15,16,131–134
PTMs with a 5-6-5 ring arrangement are ikarugamycin 81 isolated in 1972 from a Streptomyces sp. and clifednamides A 82 and B 83 also from a Streptomyces sp.135,136
PTMs with a 5-5 ring arrangement are alteramide A 84 isolated in 1992 from Alteromonas sp. associated with the sponge Halichondria okadai, cylindramide 85 isolated from the sponges H. cylindrata, geodin 86 (as Mg-salt) isolated from the sponge Geodia sp. and the aburatubolactams A–C 87–89 isolated from a Streptomyces sp. associated with a marine mollusc.137–141
PTMs are formally created from two distinct polyketide chains and the non-proteinogenic amino acid ornithine. However, only one iterative PKS–NRPS hybrid gene has been found in dihydromaltophilin (HSAF) 77 producing L. enzymogenes (Fig. 2R).15,17 The knockout of the PKS as well as the NRPS module of HSAF synthase (HSAFS) abolished HSAF production. Knockouts of the surrounding genes ferredoxin reductase (ORF 7) and arginase (ORF 8) still produced HSAF whereas the knockout of the sterol desaturase (ORF 9) produced unidentified products instead.15 Knockouts of the three NADP/FAD-dependent oxidoreductases (ORF 3–5) also abolished HSAF production whereas knockout of the ORF 2 produced a small amount of a compound with the mass of 76 a putative precursor of HSAF.17
Genome mining of Streptomyces sp. strain SPB78 draft genome revealed homologs to ORF 6 and ORF 9 of the HSAF cluster named ftdB and ftdA (Fig. 2S) and production of frontalamide A 78 and B 79 under certain culturing conditions. Deletion of both genes abolished production of 78 and 65. Bacterial iterative hybrid PKS–NRPS genes are found to be common and conserved among phylogenetically diverse bacteria including Streptomyces.16,136
There are very limited insights into the biosynthesis of PTMs yet. Lou et al. heterologously expressed the A domain and the complete NRPS module of HSAFS in E. coli and showed that L-ornithine is the favourably activated amino acid. L-lysine is activated to a lesser extent. The NRPS module can accept two acyl chains forming two amide bonds on ornithine.17 Putative biosynthesis of 78 and 79 based on that findings is shown in Scheme 11. Nothing is known how the PKS part is programmed to produce two hexaketide chains with different reduction pattern leaving the biosynthesis scheme putative in that respect and also leaving it intriguing to see how one iterative PKS module possibly creates even penta- and hexaketide chains as expected for 80, 85 and 86. Additionally, nothing is known about programming of the cyclisation pattern and for the recently discovered clifednamides 82 and 83 even a Diels–Alder reaction is proposed to gain the 5-6-5 tricyclic arrangement.136
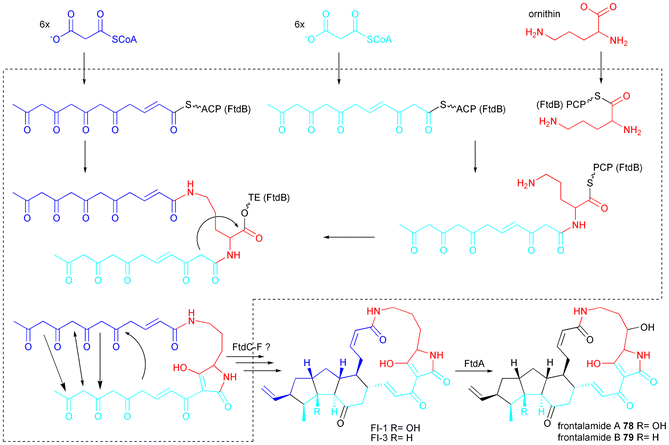 | ||
| Scheme 11 Proposed biosynthesis of frontalamide A 78 and B 79. | ||
12 Summary and further directions
Iterative PKS–NRPS generates highly diverse bioactive compounds that are used as drugs, lead structures towards drugs or are feared as mycotoxins in food and animal feed. Only a few iterative PKS–NRPSs are linked to their biosynthetic products and programming of these intriguing enzymes is poorly understood. Even more, most of the iterative PKS–NRPSs are of fungal origin where additional programming information gained from HR–PKSs and NRPSs are also limited. However, understanding of the programming is highly desirable. It would not only enable directed bioengineering towards defined compounds, but also enable prediction of compounds from genome data.The first steps have been undertaken in dissecting domains and domain swapping in iterative PKS–NRPSs and research in that direction will give insight into the mechanism, programming and interaction of domains. However, more widely accessible genome data and improvement of methods for heterologous expression as well as enzyme characterisation hold a promising future for this exciting research field. Additionally, post PKS–NRPS tailoring steps are manifold, e.g. oxidations, ring expansion or prenylation to name but a few. Functions of these tailoring enzymes are predictable from the anticipated biosynthesis and detailed investigations will gain access to new enzymes for chemical synthesis as well as new bioactive non-natural natural compounds.
Acknowledgements
The author wishes to thank Prof. Russell Cox and Prof. Tom Simpson for comments and discussions. Constructive comments from the referee are also acknowledged.References
- B. S. Evans, S. J. Robinson and N. L. Kelleher, Fungal Genet. Biol., 2011, 48, 49–61 CrossRef CAS.
- J. Piel, Nat. Prod. Rep., 2010, 27, 996–1047 RSC.
- X. H. Chen, J. Vater, J. Piel, P. Franke, R. Scholz, K. Schneider, A. Koumoutsi, G. Hitzeroth, N. Grammel, A. W. Strittmatter, G. Gottschalk, R. D. Sussmuth and R. Borriss, J. Bacteriol., 2006, 188, 4024–4036 CrossRef CAS.
- Z. Song, R. J. Cox, C. M. Lazarus and T. J. Simpson, ChemBioChem, 2004, 5, 1196–1203 CrossRef CAS.
- T. Maier, M. Leibundgut and N. Ban, Science, 2008, 321, 1315–1322 CrossRef CAS.
- R. J. Cox, Org. Biomol. Chem., 2007, 5, 2010–2026 CAS.
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas and C. Richard Hutchinson, Science, 1999, 284, 1368–1372 CrossRef CAS.
- W. Xu, X. L. Cai, M. E. Jung and Y. Tang, J. Am. Chem. Soc., 2010, 132, 13604–13607 CrossRef CAS.
- L. M. Halo, J. W. Marshall, A. A. Yakasai, Z. Song, C. P. Butts, M. P. Crump, M. Heneghan, A. M. Bailey, T. J. Simpson, C. M. Lazarus and R. J. Cox, ChemBioChem, 2008, 9, 585–594 CrossRef CAS.
- B. D. Ames, C. Nguyen, J. Bruegger, P. Smith, W. Xu, S. Ma, E. Wong, S. Wong, X. Xie, J. W.-H. Li, J. C. Vederas, Y. Tang and S.-C. Tsai, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11144–11149 CrossRef CAS.
- A. A. Yakasai, J. Davison, Z. Wasil, L. M. Halo, C. P. Butts, C. M. Lazarus, A. M. Bailey, T. J. Simpson and R. J. Cox, J. Am. Chem. Soc., 2011, 133, 10990–10998 CrossRef CAS.
- R. Massengo-Tiassé and J. Cronan, Cell. Mol. Life Sci., 2009, 66, 1507–1517 CrossRef.
- J. T. Zheng, D. C. Gay, B. Demeler, M. A. White and A. T. Keatinge-Clay, Nat. Chem. Biol., 2012, 8, 615–621 CrossRef CAS.
- A. T. Keatinge-Clay and R. M. Stroud, Structure, 2006, 14, 737–748 CrossRef CAS.
- F. G. Yu, K. Zaleta-Rivera, X. C. Zhu, J. Huffman, J. C. Millet, S. D. Harris, G. Yuen, X. C. Li and L. C. Du, Antimicrob. Agents Chemother., 2007, 51, 64–72 CrossRef CAS.
- J. A. V. Blodgett, D. C. Oh, S. G. Cao, C. R. Currie, R. Kolter and J. Clardy, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11692–11697 CrossRef CAS.
- L. Lou, G. Qian, Y. Xie, J. Hang, H. Chen, K. Zaleta-Riyera, Y. Li, Y. Shen, P. H. Dussault, F. Liu and L. Du, J. Am. Chem. Soc., 2011, 133, 643–645 CrossRef CAS.
- E. Conti, T. Stachelhaus, M. A. Marahiel and P. Brick, EMBO J., 1997, 16, 4174–4183 CrossRef CAS.
- J. J. May, N. Kessler, M. A. Marahiel and M. T. Stubbs, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12120–12125 CrossRef CAS.
- E. J. Drake, B. P. Duckworth, J. Neres, C. C. Aldrich and A. M. Gulick, Biochemistry, 2010, 49, 9292–9305 CrossRef CAS.
- T. V. Lee, L. J. Johnson, R. D. Johnson, A. Koulman, G. A. Lane, J. S. Lott and V. L. Arcus, J. Biol. Chem., 2010, 285, 2415–2427 CrossRef CAS.
- C. Rausch, T. Weber, O. Kohlbacher, W. Wohlleben and D. H. Huson, Nucleic Acids Res., 2005, 33, 5799–5808 CrossRef CAS.
- Y. Q. Xu, R. Orozco, E. M. K. Wijeratne, A. A. L. Gunatilaka, S. P. Stock and I. Molnar, Chem. Biol., 2008, 15, 898–907 CrossRef CAS.
- R. Sussmuth, J. Muller, H. von Dohren and I. Molnar, Nat. Prod. Rep., 2011, 28, 99–124 RSC.
- Y. M. Chiang, E. Szewczyk, T. Nayak, A. D. Davidson, J. F. Sanchez, H. C. Lo, W. Y. Ho, H. Simityan, E. Kuo, A. Praseuth, K. Watanabe, B. R. Oakley and C. C. C. Wang, Chem. Biol., 2008, 15, 527–532 CrossRef CAS.
- L. Du and L. Lou, Nat. Prod. Rep., 2010, 27, 255–278 RSC.
- J. W. Sims and E. W. Schmidt, J. Am. Chem. Soc., 2008, 130, 11149–11155 CrossRef CAS.
- X. Y. Liu and C. T. Walsh, Biochemistry, 2009, 48, 8746–8757 CrossRef CAS.
- L. L. Lou, H. T. Chen, R. L. Cerny, Y. Y. Li, Y. M. Shen and L. C. Du, Biochemistry, 2012, 51, 4–6 CrossRef CAS.
- P. S. Steyn and R. Vleggaar, J. Chem. Soc., Chem. Commun., 1985, 1189–1191 RSC.
- J. W. Sims, J. P. Fillmore, D. D. Warner and E. W. Schmidt, Chem. Commun., 2005, 186–188 RSC.
- T. B. Kakule, D. Sardar, Z. Lin and E. W. Schmidt, ACS Chem. Biol., 2013, 8, 1549–1557 CrossRef CAS.
- K. L. Eley, L. M. Halo, Z. Song, H. Powles, R. J. Cox, A. M. Bailey, C. M. Lazarus and T. J. Simpson, ChemBioChem, 2007, 8, 289–297 CrossRef CAS.
- S. Bergmann, J. Schumann, K. Scherlach, C. Lange, A. A. Brakhage and C. Hertweck, Nat. Chem. Biol., 2007, 3, 213–217 CrossRef CAS.
- J. Schumann and C. Hertweck, J. Am. Chem. Soc., 2007, 129, 9564–9565 CrossRef.
- S. Maiya, A. Grundmann, X. Li, S. M. Li and G. Turner, ChemBioChem, 2007, 8, 1736–1743 CrossRef CAS.
- M. Tokuoka, Y. Seshime, I. Fujii, K. Kitamoto, T. Takahashi and Y. Koyama, Fungal Genet. Biol., 2008, 45, 1608–1615 CrossRef CAS.
- B. G. G. Donzelli, S. B. Krasnoff, A. C. L. Churchill, J. D. Vandenberg and D. M. Gibson, Curr. Genet., 2010, 56, 151–162 CrossRef CAS.
- M. N. Heneghan, A. A. Yakasai, K. Williams, K. A. Kadir, Z. Wasil, W. Bakeer, K. M. Fisch, A. M. Bailey, T. J. Simpson, R. J. Cox and C. M. Lazarus, Chem. Sci., 2011, 2, 972–979 RSC.
- M. Gressler, C. Zaehle, K. Scherlach, C. Hertweck and M. Brock, Chem. Biol., 2011, 18, 198–209 CrossRef CAS.
- K. J. Qiao, Y. H. Chooi and Y. Tang, Metab. Eng., 2011, 13, 723–732 CrossRef CAS.
- S. Phonghanpot, J. Punya, A. Tachaleat, K. Laoteng, V. Bhavakul, M. Tanticharoen and S. Cheevadhanarak, ChemBioChem, 2012, 13, 895–903 CrossRef CAS.
- K. i. Ishiuchi, T. Nakazawa, F. Yagishita, T. Mino, H. Noguchi, K. Hotta and K. Watanabe, J. Am. Chem. Soc., 2013, 135, 7371–7377 CrossRef CAS.
- L. Hendrickson, C. Ray Davis, C. Roach, D. Kim Nguyen, T. Aldrich, P. C. McAda and C. D. Reeves, Chem. Biol., 1999, 6, 429–439 CrossRef CAS.
- Y. Abe, T. Suzuki, C. Ono, K. Iwamoto, M. Hosobuchi and H. Yoshikawa, Mol. Genet. Genomics, 2002, 267, 636–646 CrossRef CAS.
- D. Boettger and C. Hertweck, ChemBioChem, 2013, 14, 28–42 CrossRef CAS.
- D. Boettger, H. Bergmann, B. Kuehn, E. Shelest and C. Hertweck, ChemBioChem, 2012, 13, 2363–2373 CrossRef CAS.
- Y.-M. Chiang, B. R. Oakley, N. P. Keller and C. C. C. Wang, Appl. Microbiol. Biotechnol., 2010, 86, 1719–1736 CrossRef CAS.
- K. Scherlach, D. Boettger, N. Remme and C. Hertweck, Nat. Prod. Rep., 2010, 27, 869–886 RSC.
- I. Molnar, D. M. Gibson and S. B. Krasnoff, Nat. Prod. Rep., 2010, 27, 1241–1275 RSC.
- Y. H. Chooi and Y. Tang, J. Org. Chem., 2012, 77, 9933–9953 CrossRef CAS.
- R. Schobert and A. Schlenk, Bioorg. Med. Chem., 2008, 16, 4203–4221 CrossRef CAS.
- W. C. A. Gelderblom, P. G. Thiel, W. F. O. Marasas and K. J. Vandermerwe, J. Agric. Food Chem., 1984, 32, 1064–1067 CrossRef CAS.
- D. W. Brown, R. A. E. Butchko, M. Busman and R. H. Proctor, Fungal Genet. Biol., 2012, 49, 521–532 CrossRef CAS.
- W. C. A. Gelderblom, W. F. O. Marasas, P. S. Steyn, P. G. Thiel, K. J. Vandermerwe, P. H. van Rooyen, R. Vleggaar and P. L. Wessels, J. Chem. Soc., Chem. Commun., 1984, 122–124 RSC.
- M. E. Savard and J. D. Miller, J. Nat. Prod., 1992, 55, 64–70 CrossRef CAS.
- A. F. Barrero, J. F. Sanchez, J. E. Oltra, N. Tamayo, E. Cerdaolmedo, R. Candau and J. Avalos, Phytochemistry, 1991, 30, 2259–2263 CrossRef CAS.
- F. Eilbert, E. Thines, W. R. Arendholz, O. Sterner and H. Anke, J. Antibiot., 1997, 50, 443–445 CrossRef CAS.
- S. B. Krasnoff, C. H. Sommers, Y. S. Moon, B. G. G. Donzelli, J. D. Vandenberg, A. C. L. Churchill and D. M. Gibson, J. Agric. Food Chem., 2006, 54, 7083–7088 CrossRef CAS.
- H. Kakeya, S. Kageyama, L. Nie, R. Onose, G. Okada, T. Beppu, C. J. Norbury and H. Osada, J. Antibiot., 2001, 54, 850–854 CrossRef CAS.
- H. Kakeya, I. Takahashi, G. Okada, K. Isono and H. Osada, J. Antibiot., 1995, 48, 733–735 CrossRef CAS.
- Y. Nagumo, H. Kakeya, J. Yamaguchi, T. Uno, M. Shoji, Y. Hayashi and H. Osada, Bioorg. Med. Chem. Lett., 2004, 14, 4425–4429 CrossRef CAS.
- T. P. Nicholson, B. A. M. Rudd, M. Dawson, C. M. Lazarus, T. J. Simpson and R. J. Cox, Chem. Biol., 2001, 8, 157–178 CrossRef CAS.
- I. Gaffoor, D. W. Brown, R. Plattner, R. H. Proctor, W. Qi and F. Trail, Eukaryotic Cell, 2005, 4, 1926–1933 CrossRef CAS.
- D. O. Rees, N. Bushby, R. J. Cox, J. R. Harding, T. J. Simpson and C. L. Willis, ChemBioChem, 2007, 8, 46–50 CrossRef CAS.
- H. R. Burmeister, G. A. Bennett, R. F. Vesonder and C. W. Hesseltine, Antimicrob. Agents Chemother., 1974, 5, 634–639 CrossRef CAS.
- R. F. Vesonder, L. W. Tjarks, W. K. Rohwedder, H. R. Burmeister and J. A. Laugal, J. Antibiot., 1979, 32, 759–761 CrossRef CAS.
- E. Turos, J. E. Audia and S. J. Danishefsky, J. Am. Chem. Soc., 1989, 111, 8231–8236 CrossRef CAS.
- E. C. Marfori, S. Kajiyama, E. Fukusaki and A. Kobayashi, Z. Naturforsch. C, 2002, 57, 465–470 CAS.
- E. C. Marfori, T. Bamba, S. Kajiyama, E. Fukusaki and A. Kobayashi, Tetrahedron, 2002, 58, 6655–6658 CrossRef CAS.
- S. B. Singh, D. L. Zink, M. A. Goetz, A. W. Dombrowski, J. D. Polishook and D. J. Hazuda, Tetrahedron Lett., 1998, 39, 2243–2246 CrossRef CAS.
- D. Hazuda, C. U. Blau, P. Felock, J. Hastings, B. Pramanik, A. Wolfe, F. Bushman, C. Farnet, M. Goetz, M. Williams, K. Silverman, R. Lingham and S. Singh, Antiviral Chem. Chemother., 1999, 10, 63–70 CAS.
- K. Neumann, S. Kehraus, M. Gutschow and G. M. Koning, Nat. Prod. Commun., 2009, 4, 347–354 CAS.
- C. Osterhage, R. Kaminsky, G. M. Konig and A. D. Wright, J. Org. Chem., 2000, 65, 6412–6417 CrossRef CAS.
- S.-W. Yang, R. Mierzwa, J. Terracciano, M. Patel, V. Gullo, N. Wagner, B. Baroudy, M. Puar, T.-M. Chan, A. T. McPhail and M. Chu, J. Nat. Prod., 2006, 69, 1025–1028 CrossRef CAS.
- R. R. West, J. V. Ness, A. M. Varming, B. Rassing, S. Biggs, S. Gasper, P. A. Mckernan and J. Piggott, J. Antibiot., 1996, 49, 967–973 CrossRef CAS.
- L. M. Halo, M. N. Heneghan, A. A. Yakasai, Z. Song, K. Williams, A. M. Bailey, R. J. Cox, C. M. Lazarus and T. J. Simpson, J. Am. Chem. Soc., 2008, 130, 17988–17996 CrossRef CAS.
- S. H. E. Basyouni, D. Brewer and L. C. Vining, Can. J. Bot., 1968, 46, 441–448 CrossRef.
- C. K. Wat, A. G. Mcinnes, D. G. Smith, J. L. C. Wright and L. C. Vining, Can. J. Chem., 1977, 55, 4090–4098 CrossRef CAS.
- K. M. Fisch, W. Bakeer, A. A. Yakasai, Z. S. Song, J. Pedrick, Z. Wasil, A. M. Bailey, C. M. Lazarus, T. J. Simpson and R. J. Cox, J. Am. Chem. Soc., 2011, 133, 16635–16641 CrossRef CAS.
- J. A. Cooper, J. Cell Biol., 1987, 105, 1473–1478 CrossRef CAS.
- G. S. Pendse, Recent Advances in Cytochalasans, Chapman & Hall, London, 1987 Search PubMed.
- T. Udagawa, J. Yuan, D. Panigrahy, Y. H. Chang, J. Shah and R. J. D'Amato, J. Pharmacol. Exp. Ther., 2000, 294, 421–427 CAS.
- W. S. Horn, M. S. J. Simmonds, R. E. Schwartz and W. M. Blaney, Tetrahedron, 1995, 51, 3969–3978 CrossRef CAS.
- H. Oikawa, Y. Murakami and A. Ichihara, J. Chem. Soc., Perkin Trans. 1, 1992, 2949–2953 RSC.
- K. Auclair, A. Sutherland, J. Kennedy, D. J. Witter, J. P. Van den Heever, C. R. Hutchinson and J. C. Vederas, J. Am. Chem. Soc., 2000, 122, 11519–11520 CrossRef CAS.
- J. C. Vederas, W. Graf, L. David and C. Tamm, Helv. Chim. Acta, 1975, 58, 1886–1898 CrossRef CAS.
- J. L. Robert and C. Tamm, Helv. Chim. Acta, 1975, 58, 2501–2504 CrossRef CAS.
- H. Iwaki, S. Z. Wang, S. Grosse, H. Bergeron, A. Nagahashi, J. Lertvorachon, J. Z. Yang, Y. Konishi, Y. Hasegawa and P. C. K. Lau, Appl. Environ. Microbiol., 2006, 72, 2707–2720 CrossRef CAS.
- R. Fujii, A. Minami, K. Gomi and H. Oikawa, Tetrahedron Lett., 2013, 54, 2999–3002 CrossRef CAS.
- P. Bloch, C. Tamm, P. Bollinger, T. J. Petcher and H. P. Weber, Helv. Chim. Acta, 1976, 59, 133–137 CrossRef CAS.
- P. Bloch and C. Tamm, Helv. Chim. Acta, 1981, 64, 304–315 CrossRef CAS.
- W. Breitenstein, K. K. Chexal, P. Mohr and C. Tamm, Helv. Chim. Acta, 1981, 64, 379–388 CrossRef CAS.
- C. M. Boot, N. C. Gassner, J. E. Compton, K. Tenney, C. M. Tamble, R. S. Lokey, T. R. Holman and P. Crews, J. Nat. Prod., 2007, 70, 1672–1675 CrossRef CAS.
- O. Ando, H. Satake, M. Nakajima, A. Sato, T. Nakamura, T. Kinoshita, K. Furuya and T. Haneishi, J. Antibiot., 1991, 44, 382–389 CrossRef CAS.
- J. Wenke, H. Anke and O. Sterner, Biosci., Biotechnol., Biochem., 1993, 57, 961–964 CrossRef CAS.
- Y. Asami, H. Kakeya, R. Onose, A. Yoshida, H. Matsuzaki and H. Osada, Org. Lett., 2002, 4, 2845–2848 CrossRef CAS.
- B. V. Bertinetti, N. I. Pena and G. M. Cabrera, Chem. Biodiversity, 2009, 6, 1178–1184 CAS.
- J. C. Frisvad, J. Smedsgaard, T. O. Larsen and R. A. Samson, Stud. Mycol., 2004, 201–241 Search PubMed.
- P. Mohr and C. Tamm, Tetrahedron, 1981, 37, 201–212 CrossRef.
- C. W. Holzapfel, Tetrahedron, 1968, 24, 2101–2119 CrossRef CAS.
- P. K. Venkatesh, S. Vairamuthu, C. Balachandran, B. M. Manohar and G. D. Raj, Mycopathologia, 2005, 159, 393–400 CrossRef CAS.
- C. W. Holzapfel and D. C. Wilkens, Phytochemistry, 1971, 10, 351–358 CrossRef CAS.
- R. M. McGrath, P. S. Steyn, N. P. Ferreira and D. C. Neethling, Bioorg. Chem., 1976, 5, 11–23 CrossRef CAS.
- D. j. Steenkam, J. C. Schabort, C. w. Holzapfe and N. P. Ferreira, Biochim. Biophys. Acta, Enzymol., 1974, 358, 126–143 CrossRef.
- D. J. Steenkamp, J. C. Schabort and N. P. Ferreira, Biochim. Biophys. Acta, Enzymol., 1973, 309, 440–456 CrossRef CAS.
- United States Pat., 2005 Search PubMed.
- X. Y. Liu and C. T. Walsh, Biochemistry, 2009, 48, 11032–11044 CrossRef CAS.
- Y. Seshime, P. R. Juvvadi, M. Tokuoka, Y. Koyama, K. Kitamoto, Y. Ebizuka and I. Fujii, Bioorg. Med. Chem. Lett., 2009, 19, 3288–3292 CrossRef CAS.
- S. Loesgen, T. Bruhn, K. Meindl, I. Dix, B. Schulz, A. Zeeck and G. Bringmann, Eur. J. Org. Chem., 2011, 5156–5162 CrossRef CAS.
- C. G. Casinovi, G. Grandoli, R. Mercanti, N. Oddo, R. Olivieri and A. Tonolo, Tetrahedron Lett., 1968, 9, 3175–3178 CrossRef.
- J. A. Findlay, J. Krepinsky, A. Shum, C. G. Casinovi and L. Radics, Can. J. Chem., 1977, 55, 600–603 CrossRef CAS.
- J. A. Findlay and L. Radics, J. Chem. Soc., Perkin Trans. 1, 1972, 2071–2074 RSC.
- T. Sassa, Agric. Biol. Chem., 1983, 47, 1417–1418 CrossRef CAS.
- A. Evidente, M. Fiore, G. Bruno, L. Sparapano and A. Motta, Phytochemistry, 2006, 67, 1019–1028 CrossRef CAS.
- C. Wagner, H. Anke and O. Sterner, J. Nat. Prod., 1998, 61, 501–502 CrossRef CAS.
- G. Grandolini, C. G. Casinovi and L. Radics, J. Antibiot., 1987, 40, 1339–1340 CrossRef CAS.
- K. J. Qiao, H. Zhou, W. Xu, W. J. Zhang, N. Garg and Y. Tang, Org. Lett., 2011, 13, 1758–1761 CrossRef CAS.
- M. Isaka, A. Jaturapat, W. Kladwang, J. Punya, Y. Lertwerawat, M. Tanticharoen and Y. Thebtaranonth, Planta Med., 2000, 66, 473–475 CrossRef CAS.
- A. Amnuaykanjanasin, J. Punya, P. Paungmoung, A. Rungrod, A. Tachaleat, S. Pongpattanakitshote, S. Cheevadhanarak and M. Tanticharoen, FEMS Microbiol. Lett., 2005, 251, 125–136 CrossRef CAS.
- A. Endo, J. Antibiot., 1979, 32, 852–854 CrossRef CAS.
- A. Endo, M. Kuroda and Y. Tsujita, J. Antibiot., 1976, 29, 1346–1348 CrossRef CAS.
- R. Chakravarti and V. Sahai, Appl. Microbiol. Biotechnol., 2004, 64, 618–624 CrossRef CAS.
- J. Barrios-Gonzalez and R. U. Miranda, Appl. Microbiol. Biotechnol., 2010, 85, 869–883 CrossRef CAS.
- S. M. Ma, J. W. H. Li, J. W. Choi, H. Zhou, K. K. M. Lee, V. A. Moorthie, X. K. Xie, J. T. Kealey, N. A. Da Silva, J. C. Vederas and Y. Tang, Science, 2009, 326, 589–592 CrossRef CAS.
- S. M. Ma and Y. Tang, FEBS J., 2007, 274, 2854–2864 CrossRef CAS.
- W. Xu, Y.-H. Chooi, J. W. Choi, S. Li, J. C. Vederas, N. A. Da Silva and Y. Tang, Angew. Chem., Int. Ed., 2013, 52, 6472–6475 CrossRef CAS.
- X. Xie, M. J. Meehan, W. Xu, P. C. Dorrestein and Y. Tang, J. Am. Chem. Soc., 2009, 131, 8388–8389 CrossRef CAS.
- X. Xie, K. Watanabe, W. A. Wojcicki, C. C. Wang and Y. Tang, Chem. Biol., 2006, 13, 1161–1169 CrossRef CAS.
- Y. P. Chen, C. P. Tseng, L. L. Liaw, C. L. Wang, I. C. Chen, W. J. Wu, M. D. Wu and G. F. Yuan, J. Agric. Food Chem., 2008, 56, 5639–5646 CrossRef CAS.
- M. Jakobi, G. Winkelmann, D. Kaiser, C. Kempter, G. Jung, G. Berg and H. Bahl, J. Antibiot., 1996, 49, 1101–1104 CrossRef CAS.
- Y. Hashidoko, T. Nakayama, Y. Homma and S. Tahara, Tetrahedron Lett., 1999, 40, 2957–2960 CrossRef CAS.
- P. R. Graupner, S. Thornburgh, J. T. Mathieson, E. L. Chapin, G. M. Kemmitt, J. M. Brown and C. E. Snipes, J. Antibiot., 1997, 50, 1014–1019 CrossRef.
- S. P. Gunasekera, M. Gunasekera and P. Mccarthy, J. Org. Chem., 1991, 56, 4830–4833 CrossRef CAS.
- K. Jomon, M. Ajisaka, H. Sakai and Y. Kuroda, J. Antibiot., 1972, 25, 271–280 CrossRef CAS.
- S. G. Cao, J. A. V. Blodgett and J. Clardy, Org. Lett., 2010, 12, 4652–4654 CrossRef CAS.
- H. Shigemori, M. A. Bae, K. Yazawa, T. Sasaki and J. Kobayashi, J. Org. Chem., 1992, 57, 4317–4320 CrossRef CAS.
- S. Kanazawa, N. Fusetani and S. Matsunaga, Tetrahedron Lett., 1993, 34, 1065–1068 CrossRef CAS.
- R. J. Capon, C. Skene, E. Lacey, J. H. Gill, D. Wadsworth and T. Friedel, J. Nat. Prod., 1999, 62, 1256–1259 CrossRef CAS.
- M. A. Bae, K. Yamada, Y. Ijuin, T. Tsuji, K. Yazawa, Y. Tomono and D. Uemura, Heterocycl. Commun., 1996, 2, 315–318 CrossRef CAS.
- M. Kuramoto, H. Arimoto and D. Uemura, Mar. Drugs, 2004, 2, 39–54 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |