Biomass to biodegradable polymer (PLA)
Mamata
Singhvi
and
Digambar
Gokhale
*
NCIM Resource Centre CSIR-National Chemical Laboratory, Pune-411008. E-mail: dv.gokhale@ncl.res.in; Fax: 91-20-25902671; Tel: 91-20-25902452
First published on 6th June 2013
Abstract
Lignocellulosic biomass is renewable and cheap, and it has the potential to displace fossil fuels for the production of fuels and chemicals. Biomass derived lactic acid is an important compound that can be used as a chemical platform for the production of a variety of important chemicals on a large scale. The quality of the monomers of lactic acid and lactide, as well as the chemical changes induced during polymerization and processing, are crucial parameters for controlling the properties of the resulting polylactic acid (PLA) products. In this review, we outline the process of exploiting biomass for the production of polylactic acid, a biodegradable polymer which is well-known as a sustainable bioplastic material.
Introduction
Biomass, being renewable, is the only sustainable source of energy and organic carbon for our industrial society.1 Nature produces a vast amount of biomass by photosynthesis, 75% of which is assigned to the class of carbohydrates. Surprisingly, only 3–4% of these compounds are used by humans for food and non-food purposes.2 Biomass carbohydrates are the most abundant renewable resources available, and they are currently viewed as feedstock for the green chemistry of the future.3 The production of fuels and chemicals from biomass is beneficial concerning an environment which is associated with the reduction of the net emissions of CO2 (a greenhouse gas) into the atmosphere. In contrast to fossil fuels, biofuels are considered to be carbon neutral because any CO2 produced during fuel combustion is consumed by a subsequent biomass re-growth.4 A biomass based energy system would improve the economy of those countries determined to accept the challenges. In addition, the use of the lignocellulosic biomass does not affect the food supplies, thereby permitting a sustainable production of fuels (so-called second-generation fuels) and chemicals. Additionally, the lower cost and faster growth of the lignocellulosic biomass compared with food crops5 and its ample availability6 make this resource an attractive raw material suitable for the substitution of fossil fuels.Lactic acid (2-hydroxypropionic acid), CH3–CHOHCOOH, is the most widely occurring hydroxyl-carboxylic acid in nature. Lactic acid is frequently used in the food industry, especially for beverage production and in the pharmaceutical and chemical industry, or in medicine.7 Because lactic acid has both carboxylic and hydroxyl groups, it can also be converted into different and potentially useful chemicals such as pyruvic acid, acrylic acid, 1,2-propanediol and lactate esters.8 The recent growing interest for the manufacture of biodegradable plastic necessitates a high demand for lactic acid as the raw material for PLA production.9 Another very promising lactic acid application is the production of environmentally friendly “green” solvents (lactate esters) which can replace traditional solvents made from petrochemical feedstock.10
Poly(lactic acid) is one of the most promising biodegradable plastics.11 Much research effort is currently focused on the modifications of polylactide to make it suitable for a wider range of applications. Optically pure lactic acid is necessary to obtain high crystalline poly(lactic acid) which leads to the high strength, chemical and heat resistances properties of the polymer.12 In this review, we focus on the utilization of the lignocellulosic biomass for the production of PLA, a biodegradable polymer. Environmental, economic and safety challenges have pushed towards the partial replacement of petrochemical-based polymers with bio-based ones. The general purpose of this review is to introduce PLA, a compostable and biodegradable thermoplastic made from renewable sources (Fig. 1).
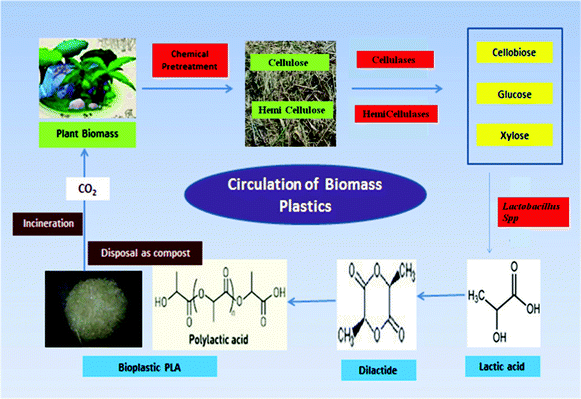 | ||
| Fig. 1 Schematic representation of the carbon cycle of bioplastics. The lignocellulosic substrate, baggase is pretreated to remove lignin and further hydrolyzed to monomeric sugars using enzymes. These sugars are further diverted to produce lactic acid using Lactobacillus sp. The produced lactic acid is used as the starting material for PLA synthesis which is a biodegradable plastic. Thus, after incineration, the released carbon can be recycled in the environment. | ||
Biomass structure
Lignocellulose, a carbohydrate source, is an interesting raw material for biotechnological processes, owing to its renewable character, widespread distribution, abundance and low price. Lignocellulosic biomass (plant biomass) is a prodigious potential resource for the production of fuels and chemicals because it is abundant, inexpensive and the production of such resources is environmentally sound. Agricultural residues are a great source of lignocellulosic biomass which is renewable, mainly unexploited and inexpensive. Such resources include: leaves, stems and stalks from corn fibre, corn stover, sugarcane bagasse, rice hulls, woody crops and forest residues. Also, there are multiple sources of lignocellulosic waste from industrial and agricultural processes, e.g. citrus peel waste, sawdust, paper pulp, industrial waste, municipal solid waste and paper mill sludge.13 The abundance of lignocellulosic biomass pinpoints to the need and potential for efficient, cost effective processes which convert it into those value added chemicals presently obtained from non-renewable resources such as fossil fuels.Biomass is a mixture of carbohydrate polymers (cellulose, hemicellulose and pectin, to varying degrees) and the non-carbohydrate polymer lignin. Cellulose consists of long microfibrils containing repeating units of cellobiose, which are dimers of glucose molecules. These hydrogen-bonded microfibrils may be quite long, up to 14,000 glucose units as observed in Arabidopsis, corresponding to a fibril length of 7 μm.14 Cellulosic materials have crystalline domains separated by less ordered amorphous regions. These amorphous regions are the potential points for chemical and enzymatic attacks. The crystalline cellulose is highly recalcitrant to chemical and enzymatic hydrolysis due to its structure in which chains of cellodextrins are precisely arranged. Cellulose is degraded by acids or enzymes known as cellulases to its monomer, glucose, that is fermented further to fuels and chemicals.
Hemicelluloses are branched polysaccharides with backbones of neutral sugars hydrogen-bonded to cellulose. Hemicellulose is a heterogeneous polymer, which varies in composition from plant to plant and also within different parts of the same plant. It is made up of mainly pentoses (D-xylose, D-arabinose), hexoses (D-mannose, D-glucose, D-galactose) and sugar acids. In hardwoods, hemicellulose contains mainly xylans, while in softwood mainly glucomannans are present. The hydrolysis of hemicelluloses requires various types of enzymes (such as xylanase, mannanase, etc.). Briefly, xylan degradation requires endo-1-4,-β-xylanase, β-xylosidase, α-glucuronidase, α-L-arabinofuranosidase, as well as acetyl xylan esterases. In glucomannan degradation, β-mannanase and β-mannosidase are required to cleave the polymer backbone. Pectins are the cement that hold the plant cell walls together and are defined by the presence of uronic acids.
Lignin is a polymer of three closely-related phenyl propane moieties.15 It is a highly cross-linked polymer built up of substituted phenols and, together with cellulose and hemicellulose, it gives strength to plants. It is present in the middle lamella and acts as cement between the plant cells. Plants are also able to store energy in products such as lipids, sugars and starch, as well as other products relatively rich in hydrogen and carbon (terpenes). Terpenes are found in essential oils that are components of resins, steroids, and rubber. This association between cellulose, hemicelluloses and lignin makes the plant cell wall resistant to mechanical and biological degradations. The processing of lignocellulosic biomass has to include the conversion of lignin to value-added products in addition to its use as fuel in order to economically transfer the biomass into useful chemicals.
Conversion of biomass to sugar
The biotechnological conversion of lignocellulosic biomass is a potentially sustainable approach to develop novel bioprocesses and products. The complex structure of lignocellulose, with its highly crystalline structure protected by lignin, confers this material with a high degree of recalcitrance, that makes its depolymerization a difficult task.16 There are four main consecutive steps involved in lactic acid production from lignocellulosic substrates: pretreatment, hydrolysis, fermentation and separation. Due to its low bioaccessibility, a number of chemical and physical methods, such as acid or base treatments and steam explosion, have been developed and used to hydrolyze lignocellulosic materials to oligosaccharide, prior to fermentation by microorganisms. Biological and biochemical pretreatment methods for the conversion of cellulosic materials into sugars appear to be attractive alternatives from both economical and environmental viewpoints.The major challenges in biomass conversion are: the relatively low rate of hydrolysis, the high cellulase costs and the little understanding of cellulase kinetics on lingo-cellulosic substrates. Two main approaches have been developed in parallel for the conversion of lignocellulosic materials to commodity chemicals- ‘‘acid based’’ and ‘‘enzyme based’’.17 Consequently, the deconstruction of lignocellulose requires an effective pretreatment step to break the lignin protection which makes it more accessible to acids or enzymes resulting in a complete hydrolysis of cellulose and hemicellulose fractions of the biomass. The morphology of the lignin network is modified in aqueous solutions at mild temperatures, which allows the hydrolysis of hemicellulose to occur under the same conditions in the presence of acids. The crystalline structure of cellulose protects its β-glycoside ether linkages from being accessed by the acid catalyst, so more severe conditions are required for a full deconstruction of this polymer.18 Several pretreatments involving physical, chemical and biological methods have been developed to depolymerize lignocellulosic materials.19 There are several advantages and disadvantages in the pretreatment processes involved in the conversion of lignocellulosic biomass (Table 1). Recently, the effects of these pretreatments on the morphology and structure of the biomass have been studied.20 In the acid-catalyzed pretreatment, the major part of the hemicellulose is degraded while in the alkali-catalyzed pretreatment part of the lignin is removed.21 This pretreated cellulose is further hydrolyzed using cellulases. Very recently, Ding et al.22 reported the use of correlative imaging in real time to assess the impact of pretreatment as well as the resulting nanometer scale changes in the cell wall structure upon digestion by the cellulase systems. From these studies, it was concluded that the complete/maximum removal of lignin from the biomass without modifying the native microfibrillar structure of carbohydrates could be the ideal pretreatment for enhancing the digestibility of the biomass.
| Pretreatment process | Advantages | Limitations and disadvantages | References |
|---|---|---|---|
| Mechanical comminution | ·Reduces both the degree of polymerization (DP) and cellulose crystallinity | ·Power consumption usually higher than inherent biomass energy | [80] |
| ·Increases the available specific surface area | |||
| Steam Explosion | ·Causes hemicellulose degradation and lignin transformation | ·destruction of a portion of the xylan fraction | [81] |
| ·Makes limited use of chemicals | ·Incomplete disruption of the lignin-carbohydrate matrix | ||
| ·Requires low energy | |||
| AFEX | ·Ammonia pretreatments have a high selectivity for reaction with lignin | ·Not efficient for biomass with high lignin | [82] |
| ·Does not produce inhibitors for downstream processes | ·The cost of ammonia basically drives the process and its application on the large scale | ||
| ·The ability to reduce, recover and recycle the ammonia used in both AFEX/ARP makes the process economically viable | ·Environmental concerns with the stench of ammonia also have a negative impact on pilot as well as industrial scale applications. | ||
| CO2 explosion | ·Increases accessible surface area | ·Does not modify lignin or hemicelluloses | [83] |
| ·Cost-effective | |||
| ·Does not cause the formation of inhibitory compounds | |||
| Ozonolysis | ·Reduces lignin content | ·Large amounts of ozone required | [84] |
| ·Does not produce toxic residues | ·Expensive | ||
| Acid hydrolysis | ·Hydrolyzes hemicellulose to xylose and other sugars | ·High cost | [85] |
| ·Alters lignin structure | ·Equipment corrosion | ||
| ·Formation of toxic substances | |||
| Alkaline hydrolysis | ·Removes hemicelluloses and lignin | ·Long residence times required | [86] |
| ·Increases the accessible surface area | ·Irrecoverable salts formed and incorporated into the biomass | ||
| Organosolv | ·Organosolv lignin is sulfur free with a high purity and low molecular weight | ·Solvents need to be drained from the reactor, evaporated, condensed and recycled | [87] |
| ·Can be used as a fuel to power pretreatment plants or further purified to obtain high quality lignin, which is used as a substitute for polymeric materials | ·High cost | ||
| ·Very effective for the pretreatment of high-lignin lignocellulose materials | ·Generation of compounds inhibitory to microorganisms | ||
| Pyrolysis | ·Produces gas and liquid products | ·High temperature | [88] |
| ·Ash production | |||
| Hot water Treatment | ·Lower temperatures minimizing the formation of degradation products | ·Down-stream processing is more energy demanding because of the large volumes of water involved. | [89] |
| ·Eliminates the need for a neutralisation | |||
| Biological | ·Degrades lignin and hemicelluloses | ·Rate of hydrolysis is very low | [90] |
| ·Low energy requirements |
The production of cellulase is a major factor in the hydrolysis of cellulose materials. Cellulase is a multi-enzyme system composed of several enzymes with numerous isozymes, which act in synergy. The basic enzymatic process for the de-polymerization of cellulose requires three types of enzymes. Endoglucanase (EG or CX) hydrolyses the internal β-1,4-glucan chain of cellulose at random, primarily within the amorphous regions and displays a low hydrolytic activity towards crystalline cellulose. Exoglucanase, i.e. exo-acting cellobiohydrolases (CBH), removes cellobiose from the non-reducing end of cello-oligosaccharide and crystalline, amorphous and acid or alkali treated cellulose. Cellobiase or β-glucosidase (BGL) hydrolyses cellobiose to yield two molecules of glucose which completes the de-polymerization of cellulose.23 Cellulases have been used for several years in food processing, feed preparation, waste-water treatment, detergent formulation, textile production and in other areas. Nevertheless, the requirement of cellulase for such uses is small compared to cellulase requirements for the bioconversion of the lignocellulosic biomass to fuel ethanol.
The insoluble recalcitrant nature of cellulose represents a challenge for cellulase systems. Cellulases are composed of an independent folding structure and functional discrete units called domains or modules.24 A general feature of most cellulases is a modular structure often including both catalytic and carbohydrate-binding modules (CBMs). The CBM affects the binding to the cellulose surface, presumably to facilitate cellulose hydrolysis by bringing the catalytic domain in close proximity to the insoluble cellulose substrate. The presence of CBMs is particularly important for the initiation and processivity of exoglucanases.25 Cellulase systems exhibit a higher collective activity than the sum of the activities of individual enzymes, a phenomenon known as synergism. Structurally fungal cellulases are simpler compared to bacterial cellulase systems, cellulosomes.26
Improvement in the cellulase production as well as the hydrolysis of cellulosic substrates can make the process more cost effective.27 Consequently it is necessary to understand the enzyme substrate interactions and both identify and quantify the contribution of various system properties to the hydrolysis process. The high cost of cellulase production is a major bottleneck in the conversion of biomass to any value added product. Hence, efforts are needed to produce cellulases at an affordable cost which can be used for hydrolyzing the biomass to monomers with high economical potential. Cellulolytic enzymes are synthesized by a number of microorganisms. Fungi and bacteria are the main natural agents of cellulose degradation. The cellulose utilizing population includes aerobic and anaerobic mesophilic bacteria, filamentous fungi, thermophilic and alkaliphilic bacteria, actinomycetes and certain protozoa. However, fungi are well known agents of decomposition of organic matter, in general, and of cellulosic substrates in particular.28
One of the most extensively studied fungi is Trichoderma reesei, which converts native as well as derived cellulose to glucose. Besides Trichoderma reesei, other fungi, like Humicola, Aspergillus and Penicillium, have the ability to secrete extracellular cellulases. Fungal cellulases are being commercially produced for biomass saccharification. The economic viability of biomass conversion depends on the pretreatment of the substrates and the cost of the enzyme. The need for lower costs triggered a search for high cellulase producing organisms using classical mutagenesis, genetic engineering and enzyme engineering techniques that included advanced biotechnological procedures, such as directed evolution and rational design studies.29 These improved enzyme preparations are expected to possess desirable properties such as higher catalytic efficiencies, increased stabilities at elevated temperatures and higher tolerance to end product inhibition. Improvements in cellulase activities by imparting desired features to enzymes by protein engineering is probably another area where cellulase research has to advance. Previously we reported cellulase production by P. Janthienllum NCIM 1171 using bagasse as the carbon source and its application in bagasse hydrolysis.30,31 The mutants of P janthinellum NCIM 1171 capable of producing enhanced levels of cellulases using submerged fermentation have been isolated,32 which also exhibited high levels of filter paper degrading activity in solid state fermentation.33 By using cellulase producing improved strains and suitable pretreatments, lignocellulosic biomass can be converted into sugars and diverted to produce value added products like ethanol, butanol succinic acid, lactic acid, etc.
Lactic acid production by microbial fermentation
Lactic acid can be produced by either microbial fermentation or chemical synthesis. Compared to chemical synthesis, the biotechnological process for lactic acid production offers several advantages, such as low substrate costs, reduced production temperature and less energy consumption.34 Although DL-lactic acid is always produced by chemical synthesis from petrochemical resources, an optically pure L(+)- or D(−)-lactic acid can be obtained by microbial fermentation when the appropriate microorganism is selected.35 The majority of the world's commercially produced lactic acid is made by the bacterial fermentation of carbohydrates, using homolactic organisms belonging to the genus Lactobacillus, which exclusively forms lactic acid. The organisms that predominantly produce the L(+)-isomer are Lactobacillus amylophilus, L. bavaricus, L. casei, L. maltaromicus and L. salivarius. Strains such as L. delbrueckii, L. jensenii or L. acidophilus produce either the D-isomer or mixtures of both. These strains show high carbon conversions from feedstocks under standard fermentation conditions, such as relatively low to neutral pH, temperatures around 40 °C and low oxygen concentrations.The optical purity of lactic acid is crucial to the physical properties of poly (lactic acid) (PLA). An optically pure L(+)- or D(−)-lactic acid can be polymerized to a highly crystalline PLA that is suitable for commercial use. Therefore, the biotechnological production of lactic acid has received a significant amount of interest recently, since it offers an alternative to environmental pollution caused by the petrochemical industry and the limited supply of petrochemical resources. Lactic acid-producing organisms, most of which are anaerobic, utilize pyruvic acid, which is the end product of the Embden–Meyerhof pathway. The conversion of pyruvic acid to lactate can be effected by either of the two enzymes, L-lactate dehydrogenase (LDH) or D-lactate dehydrogenase (LDH). The major homo-fermentative LAB used in the lactic acid production from different carbon sources are Lactobacillus delbrueckii,36L. helveticus37 and L. casei.38 Some of the homo-fermentative bacteria, like L. amylophilus, L. manihotivorans, etc., can directly consume complex carbohydrates like starch.39 Amylolytic bacteria Lactobacillus amylovorus ATCC 33622 had the efficiency of a full conversion of liquefied corn starch to lactic acid with a productivity of 2.0 g l−1 h−1.40
Poly(L-lactide) (PLLA) obtained by the polymerization of L-lactic acid has a melting temperature of 175 °C. The melting point of this polymer can be increased by blending with poly(D-lactide) (PDLA) in a solvent. Recently, it was found that the polymer blend of PLLA and PDLA produces stereo-complexes with a melting point around 230 °C. This finding has attracted more attention to the production of D-lactic acid. Currently, optically pure lactic acid is produced mainly from corn starch. However, the use of agro-waste materials for lactic acid production appears to be more attractive because they do not impact on the food chain for humans. Unfortunately, the process of converting cellulosic material into lactic acid is not yet feasible due to the high cost of cellulase enzymes involved in cellulose hydrolysis.41 In addition, cellulase inhibition by glucose and cellobiose during the hydrolysis of cellulosic material by cellulases is the main bottleneck, which remarkably slows down the rate of hydrolysis. Thus, it is advantageous to use a lactic acid producing strains that have the ability to utilize both glucose and cellobiose efficiently.42 It is known that some Lactobacillus strains utilize cellobiose as a carbon source43 but very little information is available about lactic acid production from cellobiose. We have reported the production of L-lactic acid43 and D-lactic acid44 from cellobiose with the highest productivity and yields.
The bioconversion of carbohydrate materials to lactic acid can be made much more effective by coupling the enzymatic hydrolysis of carbohydrate substrates and microbial fermentation of the derived sugars into a single step, known as the “Simultaneous Saccharification And Fermentation (SSAF)”. Different cellulosic substrates have been used for lactic acid production by the SSAF process and the comparative results are given in Table 2. SSAF eliminates the need for a complete hydrolysis of the carbon substrates prior to the fermentation. In the SSAF process, enzymatic hydrolysis, cell growth and microbial production occur simultaneously. A direct benefit of the SSAF is to decrease the inhibition caused by mono or di-saccharide accumulation, leading to an increase in the saccharification rate, consequently increasing productivity and reducing reactor volume and capital costs. We have reported the L-lactic acid45 and D-lactic acid46 production from sugarcane bagasse derived cellulose using simultaneous saccharification and fermentation process (Fig. 2). Lactic acid is produced by oxidizing NADH (nicotinamide adenine dinucleotide) generated during glycolysis with pyruvate as the electron acceptor. Zhou et al.47 examined the D(−) lactic acid production by E. coli using the D-LDH present in it. A strain of E. coli was constructed by transferring the L-LDH gene from Pediococcus, which produced L-lactic acid.48
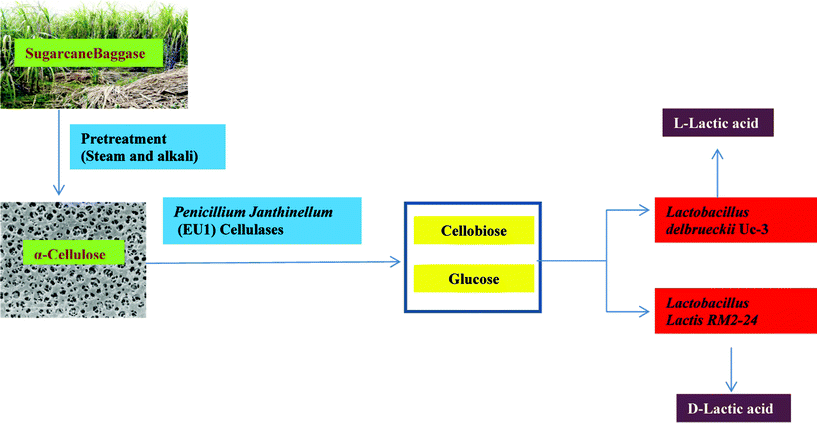 | ||
| Fig. 2 Biomass utilization through improved strains for lactic acid production using the SSAF approach. Sugarcane baggase derived cellulose is used as a substrate for lactic acid production. The cellulose is further hydrolyzed to simple sugars using cellulase produced by P. janthinellum (EU1). These sugars are diverted to produce D- and L-lactic acid by L. lactis RM2-24 and L. delbrueckii Uc3 using the SSAF process. | ||
| Organism | Substrate | Lactic acid | References | ||
|---|---|---|---|---|---|
| Concentration | Yield | Productivity | |||
| (g l−1) | (g g−1) | (g l−1 h−1) | |||
| Lactobacillus delbrueckii Uc- 3 | Molasses | 166 | 0.87 | 4.2 | [91] |
| Lactobacillus delbrueckii Uc- 3 | Cellobiose and cellotriose | 90 | 0.9 | 2.3 | [43] |
| Lactobacillus delbrueckii Uc- 3 | α-Cellulose | 67 | 0.83 | 0.93 | [45] |
| Lactobacillus lactis RM2-24 | α-Cellulose | 71 | 0.73 | 1.48 | [46] |
| Lactobacillus lactis RM2-24 | Molasses and cellobiose | 70 | 0.88 | 1.45 | [44] |
| Lactobacillus delbrueckii IFO 3202 | De-fatted rice bran | 28 | 0.28 | 0.77 | [92] |
Lactobacillus coryniformis ATCC 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 600 |
Pretreated cardboard | 23 | 0.56 | 0.49 | [93] |
| Lactobacillus delbrueckii HG 106 | Unpolished rice | 90 | 0.73 | 1.5 | [94] |
| Bacillus sp. Strain 36D1 | Solka Floc | 40 | 0. 65 | 0.22 | [95] |
| Lactobacillus delbrueckii | Sugarcane juice | 118 | 0.95 | 1.7 | [96] |
| Sporolactobacillus sp. CASD | Peanut meal, glucose | 207 | 0.93 | 3.8 | [97] |
| Lactobacillus rhamnosus strain CASL | Cassava powder | 175.4 | 0.71 | 1.8 | [98] |
Hemicelluloses are the second most abundant polysaccharide in nature due to their enormous availability, low cost and environmentally benign nature. The major fraction in hemicelluloses is pentosan and the conversion of pentose sugars is still challenging. Lactic acid bacteria are capable of fermenting glucose and other hexoses but lack the ability to ferment pentoses.49 For a complete conversion of biomass to lactic acid, lactic acid bacteria should have the capability to ferment pentoses. A genetically modified strain of E. coli containing the genes for pentose utilization and lignin degradation could be the suitable strain for hemicellulosics derived sugars fermentation.50 The lack of industrially suitable strains for the efficient conversion of xylose into lactate has been cited as a major technical obstacle for the development of the poly(lactic acid) industry. Enterococcus mundtii QU 25, a newly isolated lactic acid bacterium, efficiently metabolized xylose into L-lactate. This strain may provide an ideal wild-type microorganism for an economical L-lactate production from renewable biomass substrates.51
The use of pure sugars as the carbon source for lactic acid production which leads to an increase in cost of the product is the major bottleneck in the biotechnological production of optically pure lactic acid. This problem can be resolved through the fermentative production of lactic acid from cheaper materials such as molasses, starch, lignocellulose and wastes from agricultural and agro-industrial residues that can be used as substrates for lactic acid fermentation. However, most lignocellulose materials need to be pretreated by physicochemical and enzymatic methods because lactic acid fermenting microorganisms cannot directly use those materials due to their recalcitrance nature.52 Improvement of these microorganisms through gene modification is an essential and interesting method that has been extensively studied. The other hurdle in lactic acid production is the operating cost. For example, sterilization is necessary for the fermentative production of lactic acid. Therefore, it is difficult to avoid contamination if the medium is not sterilized. To avoid contamination, highly thermotolerant and acid tolerant strains may be useful in lactic acid production. The fermentative production of L-lactic acid by a newly isolated thermophilic strain, Bacillus sp. 2–6, has been recently reported.53 The down streaming process after fermentation also elevates the cost of lactic acid production. Owing to the inhibitory effects of low pH on cell growth and lactic acid production, CaCO3 must be added to maintain a constant pH. Fermentation of sugars to lactic acid at low pH (below 4.5) is essential to avoid the use of calcium carbonate which generates high concentrations of calcium sulfate during acid hydrolysis to liberate free lactic acid. Lactobacillus strains capable of producing lactic acid at acidic conditions have not yet been developed. The use of such acid tolerant strains will change the entire scenario of downstream processes for lactic acid purification. Recently, lactic acid (77.0 g L−1) production from 100 g L−1 cellulose equivalent of paper sludge was reported using Bacillus coagulans strains. The semi-continuous saccharification and fermentation was carried out without pH control since these strains are thermophilic and acid tolerant.54
An important step in the lactic acid production is the recovery from the fermentation broth. The conventional process for the recovery of lactic acid is still far from ideal. Indeed, it involves the precipitation of calcium lactate after the separation of micro-organisms and the conversion of the salt to lactic acid by the addition of sulfuric acid. The dilute lactic acid produced is then submitted for purification. The separation and purification stages account for up to 50% of the production cost. Moreover, reactor productivities are low and the process is unfriendly to the environment since it consumes sulfuric acid and produces a large quantity of calcium sulfate (1 ton of calcium sulfate per ton of lactic acid). Recent advances in membrane-based separation and purification technologies, particularly in micro- and ultrafiltration and electrodialysis, have led to the inception of new processes which may lead to low-cost production without the environmental problems associated with the conventional process. Biotechnology is providing new, low-cost and highly efficient fermentation processes for the production of chemicals from biomass resources.55 However, the current economic impact of lactic acid fermentation is still limited, in large part owing to difficulties in product recovery. Thus, improvements in the existing recovery technology are needed in order to allow the chemicals from fermentation to penetrate further in the organic chemical industry.
Polylactate production by the polymerisation of lactic acid
Lactic acid is a building block for the manufacture of poly lactic acid (PLA), a biodegradable polymer used as an environmentally friendly biodegradable plastic. PLA is the first commodity polymer produced from annually renewable resources. Poly(lactic acid) is a representative bio-based plastic that is used in packaging, stationery, containers, etc.56 In addition, the utilization of the polyester (polyhydroxyalkonates) has been expanded to the medical field for drug delivery, resorbable sutures and as a material for medical implants and other related applications.57 Among the biomaterials (biopolymers) used in the medical field, PLA has received significant attention. PLA and its copolymers are being used in the biomedical area in the form of implants or devices due to its biocompatibility and biodegradability.The rate of degradation of PLA depends on the degree of crystallinity. As the lactide content increases, the degradation of the graft polymer decreases.58 The PLLA is a semi-crystalline polymer with a glass transition temperature between 55 °C and 59 °C and a melting point 170 °C–180 °C. It shows good mechanical stiffness, high Young's modulus, thermal plasticity and has good processability.59 It is a relatively hydrophobic polyester, unstable in wet conditions, which can undergo chain disruptions in the human body and degrades into nontoxic byproducts, lactic acid, carbon dioxide and water, which are subsequently eliminated through the Krebs cycle and in the urine. The most widely used method for improving PLA processability is based on the melting point depression by the random incorporation of small amounts of lactide enantiomers of opposite configuration into the polymer (i.e. by adding a small amount of D-lactide to L-lactide to obtain PDLLA). Unfortunately, the melting point depression is accompanied by a significant decrease in crystallinity and crystallization rates. Recently, lactic acid consumption has increased considerably because of its role as a monomer in the production of biodegradable PLA, which is well-known as a sustainable bioplastic material. However, the global consumption of lactic acid is expected to increase rapidly in the near future.
PLAs are basically synthesized via three processes: (i) production of lactic acid (LA) by microbial fermentation, (ii) purification of LA and preparation of its cyclic dimer (lactide) and (iii) polycondensation of LA or ring-opening polymerization of lactides (Fig. 3). Fig. 3 shows the reaction mechanism for both the polycondensation of LA and the ring-opening polymerization of lactides. In direct condensation, a solvent is used and higher reaction times are required. The resulting polymer is a material with low to intermediate molecular weights. Lactide is obtained by the depolymerization of low molecular-weight PLA under reduced pressure to give L-lactide, D-lactide or meso-lactide. The different percentages of the lactide isomers formed depend on the lactic acid isomer feedstock, temperature and catalyst. Poly(lactic acid) can undergo cationic ring-opening polymerization. It has been found that trifluoromethane sulfonic acid (triflic acid) and methyl trifluoromethane sulfonic acid (methyl triflate) are the only cationic initiators to polymerize lactide. The polymerization proceeds via triflate ester end-groups instead of free carbenium ions, which yield at low temperatures an optically active polymer without racemization. The chain growth proceeds by cleavage of the alkyloxygen bond. The propagation mechanism begins with positively charged lactide ring being cleaved at alkyl-oxygen bond by an SN2 attack by the triflate anion. The triflate end-group reacts with a second molecule of lactide again in an SN2 fashion to yield a positively charged lactide that is opened. Then the triflate anion again opens the charged lactide and polymerization proceeds. Anionic lactide polymerizations proceed by the nucleophilic reaction of the anion with the carbonyl and the subsequent acyl-oxygen cleavage. This produces an alkoxide end-group, which continues to propagate.60 Ring-opening polymerization (ROP) of the lactide needs a catalyst but it results in PLA with a high molecular weight. Depending on the monomer used and the reaction conditions, it is possible to control the ratio and sequence of D- and L-lactic acid units in the final polymer. The ring-opening polymerization of lactide can be carried out in melt or solution by cationic, anionic and coordination mechanisms, depending on the initiator utilized. The most considered active initiator for the L-lactide ring-opening polymerization is stannous octoate (bis-2-ethyl hexanoate, SnOct2), which causes a low degree of racemization at high temperatures. It is catalyzed by transition metals such as tin, aluminum, lead, zinc, bismuth, ion and yttrium.61
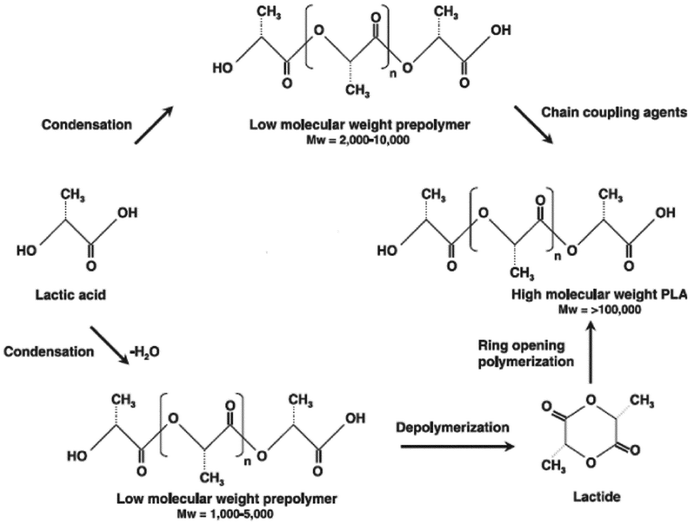 | ||
| Fig. 3 Schematic representation of PLA synthesis (Gupta et al. 2007). The process starts with the continuous condensation reaction of aqueous lactic acid to produce low molecular weight PLA prepolymers. Next, the low molecular weight oligomers are converted into a mixture of lactide stereoisomers using a catalyst to enhance the rate and selectivity of the intramolecular cyclization reaction. The molten lactide mixture is then followed by ring-opening polymerization into a high-molecular-weight lactic acid polymer. Finally, PLA high polymer is produced using an organo tin-catalyzed ring-opening lactide polymerization. | ||
PLA is chemically synthesized by the heavy metal-catalyzed ring-opening polymerization of lactide, which in turn is derived from fermentative lactate (LA). However, the trace residues of the heavy metal catalyst are unfavorable for certain applications, in particular, medical and food applications. Thus, the replacement of the heavy metal catalyst with a safe and environmentally acceptable alternative is a crucial issue. For this purpose, enzymes are attractive targets because they are natural and non-harmful catalysts that can drive the reactions under mild conditions.62 In addition, highly specific enzymatic reactions and/or whole-cell systems bearing them may be capable of synthesizing polymers with a fine structure from inexpensive raw materials. In comparison, chemical processes require extremely pure monomers, anhydrous conditions and high temperatures in order to avoid side reactions and produce high quality polymers. Therefore, a complete biosynthesis of PLA may be advantageous over the chemical process provided this challenge can be met. Thus, an ‘‘LA-polymerizing enzyme (LPE)’’, which can function as an alternative to a metal catalyst, would be desirable to establish the bioprocess. The appropriate strategy (albeit difficult) would be the discovery of a PLA-producing microorganism. There is no information available on PLA production using natural strains. However, engineered strains have been used extensively for producing LA based polyesters. Overall, the configuration of the enantiomers of the LA unit in the LA-based polyester is mainly determined by the enantio-selectivity of LDH and LPE. In that case, the Lactobacillus strain could be useful in establishing the one step process for the synthesis of LA-based polyesters. We suggest that Lactobacillus could be the host organism for the cloning and expression of the LPE gene, which directly converts lactic acid into polylactate. In this case, lactic acid is directly diverted to the synthesis of PLA, thereby reducing the requirement of a neutralizing agent to maintain the pH during fermentation.
Bacterial polyhydroxyalkanoates (PHAs) are also a major class of bio-based plastics, which are intracellularly produced by the PHA synthase-catalyzed polymerization of hydroxyacyl-CoAs. Among them, poly(3-hydroxybutyrate) [P(3HB)] is the most common PHA, and is efficiently produced from renewable carbon sources. However, in considering practical uses, there is the obstacle that P(3HB) tends to be a stiff and brittle material due to crystallinity. In addition, because of such crystallization, P(3HB) becomes opaque. These properties have limited the range of applications of these materials. Recently, a whole-cell biosynthesis system for LA-based polyester production without heavy metal catalyst has been achieved using engineered Escherichia coli.62,63 In this biological system, LA synthesized in the cell is directly converted into the polymer without any extraction and purification processes. The discovery of LPE,63 an engineered polyhydroxyalkanoate (PHA) synthase,64 was a key to develop the first microbial system. To date, the E. coli platform has been used to produce various LA-based polymers incorporating 3-hydroxybutyrate (3HB), 3-hydroxyvalerate (3HV), and 3-hydroxyhexanoate (3HHx).65,66 Most recently, the successful incorporation of new 2-hydroxy acids, such as 2- hydroxybutyrate and glycolate, using LPE has been reported.67,68 PLA homopolymer and its copolymer, poly(3-hydroxybutyrate-co-lactate) have been produced by the direct fermentation by metabolically engineered E. coli.69 The engineered E. coli strain was constructed by introducing heterologous metabolic pathways involving engineered propionate CoA-transferase and polyhydroxyalkanoate (PHA) synthase. This resulted in the efficient generation of lactyl-CoA which is incorporated into the polymer. However, the PLA and poly(3-hydroxybutyrate-co-lactate) were synthesized with low frequency in engineered E. coli. This strategy of combined metabolic engineering and enzyme engineering could be useful for developing other engineered organisms capable of producing different unnatural polymers by direct fermentation from the biomass.
Song et al.70 designed metabolic pathways in C. glutamicum to generate monomer substrates, lactyl-CoA (LA-CoA) and 3-hydroxybutyryl-CoA (3HBCoA), for the copolymerization catalyzed by LPE. LA-CoA was synthesized by D-LDH and propionyl-CoA transferase, while 3HB-CoA was supplied by β-ketothiolase (PhaA) and NADPH (nicotinamide adenine dinucleotide phosphate)-dependent acetoacetyl-CoA reductase (PhaB). The functional expression of these enzymes led to a production of P(LA-co-3HB) with high LA fractions (96.8 mol%). The newly engineered C. glutamicum potentially serves as a food-grade and biomedically applicable platform for the production of a poly(lactic acid)-like polyester.
Tajima et al.71 cloned a pha locus from a thermotolerant bacterium, Pseudomonas sp. SG4502, which is capable of accumulating polyhydroxyalkanoate (PHA) even at 55 °C, as a source of thermostable enzymes and identified two genes encoding PHA synthases (PhaC1SG and PhaC2SG). Two mutations, Ser324Thr and Gln480Lys, corresponding to those of a LPE from mesophilic Pseudomonas sp. 61-3, were introduced into PhaC1SG to evaluate the potential of the resulting protein as a “thermostable LPE”. The mutated PhaC1SG [PhaC1SG(STQK)] showed a high thermal stability in synthesizing P(LA-co-3HB) in an in vitro reaction system under a range of high temperatures which could be useful in synthesizing LA based copolymers.
Biodegradation of polymer
Biodegradable polymers have received much more attention in the last decades due their potential applications in the fields related to environmental protection and the maintenance of physical health. Two classes of biodegradable polymers can be distinguished: synthetic or natural polymers. There are polymers produced from feedstock derived either from petroleum resources (non renewable resources) or from biological resources (renewable resources). To improve the properties of biodegradable polymers, a lot of methods have been developed, such as random and block copolymerization or grafting. For example, THE anchoring of minute quantities of saccharide moieties onto polyolefins improved their rates of biodegradation.72,73 These methods improve both the biodegradation rate and the mechanical properties of the final products. Physical blending is another route to prepare biodegradable materials with different morphologies and physical characteristics.Biodegradation takes place through the action of enzymes and/or chemical deterioration associated with living organisms. This event occurs in two steps. The first one is the fragmentation of the polymers into lower molecular mass species by means of either abiotic reactions, i.e. oxidation, photodegradation or hydrolysis, or biotic reactions, i.e. degradations by microorganisms. This is followed by THE bioassimilation of the polymer fragments by microorganisms and their mineralization. Biodegradability depends not only on the origin of the polymer but also on its chemical structure and the environmental degrading conditions (Fig. 4). The degradation of PLA has been studied for several years, but understanding oF the mechanisms involved is still incomplete. Several reports concluded that PLA degradation occurs strictly through hydrolysis with no enzymatic involvement.74,75 Other reports suggested that enzymes have a significant role in PLA degradation.76 In the last years, a new type of material called nanocomposites, have been developed.77 Fukushima et al.78 reported PLA degradation and its nanocomposites in composts under controlled conditions. Recently, Arena et al.79 reported that the degradation of PLA and its composites by Bacillus licheniformis is due to the extracellular esterase activity.
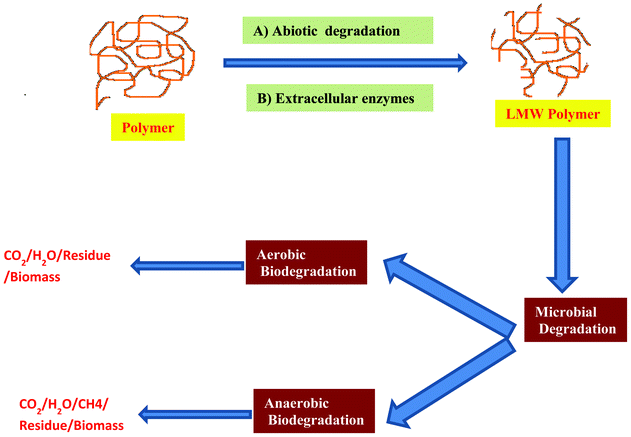 | ||
| Fig. 4 Schematic representation of the chemistry of biodegradation of the polymer. Biodegradation takes place in two stages: the first stage is the depolymerization of the macromolecules into shorter chains. Extra-cellular enzymes (endo or exo-enzymes) and abiotic reactions are responsible for the polymeric chain cleavage. The second step corresponds to the mineralization. Once sufficiently small sized oligomeric fragments are formed, they are transported into cells where they are bioassimilated by the microorganisms and then mineralized. Biodegradation takes place in aerobic and anaerobic condition depending on the presence or absence of oxygen. | ||
Conclusion and future perspectives
The complete conversion of hexoses and pentoses derived from lignocellulosic materials into fuels and chemicals would make the process commercially viable. As lactic acid contains two reactive functional groups, a carboxylic group and a hydroxyl group, it can undergo a variety of chemical reactions to yield potentially useful chemicals. Lactic acid is a commonly occurring organic acid, which is valuable due to its wide use in food and food-related industries. Additionally, it can be polymerized to yield biodegradable and biocompatible polylactate polymers. The PLA could be a better substitute for many petrochemical-based polymers for almost all pharmaceutical and direct food contact packaging materials in the near future. The biotechnological route for the synthesis of bio-based lactic acid derivatives may replace the chemically derived methods in the near future. Owing to environmental concerns and the limited availability of petrochemical feedstock, a completely green process would be the preferred method for the production of lactic acid derivatives. Future technologies for the synthesis of PLA are expected to be developed which will use Lactobacillus strains with ability to divert lactic acid directly to PLA by expressing the LPE gene in it. The construction of such strains is possible through metabolic engineering, system biology, genetic engineering, etc. The development of robust strains capable of synthesizing PLA from sugars derived from the biomass in a one step process could be the ultimate solution for a commercially viable PLA technology.References
- J. Chheda, G. Huber and J. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164 CrossRef CAS
.
- H. Roper, Starch/Staerke, 2002, 54, 89 CrossRef CAS
- F. Lichtenthaler and S. Peter, C. R. Chim., 2004, 7, 65 CrossRef CAS
-
Natl. Sci. Found (NSF), G. W. Huber, Workshop, 2008
-
D. Klass, In Encyclopedia of Energy, ed. C. J. Cleveland, London, Elsevier, 2004, vol. 1, p. 193 Search PubMed
- G. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044 CrossRef CAS
-
T. Vick Roy, In: M. M. Young (ed.) Comprehensive Biotechnology., Pergamon Press, New York, 1985, vol. 3, p. 761 Search PubMed
- Y. Fan, C. Zhou and X. Zhu, Catal. Rev. Sci. Eng., 2009, 51, 293 CAS
- A. Södergård and M. Stolt, Prog. Polym. Sci., 2002, 27, 1123 CrossRef
- J. Yang, J. Neng Tan and Y. Gu, Green Chem., 2012, 14, 3304 RSC
- K. Sakai, M. Taniguchi, S. Miura, H. Ohara, T. Matsumoto and Y. Shirai, J. Ind. Ecol., 2004, 7, 63 CrossRef
- J. Lunt, Polym. Degrad. Stab., 1998, 9, 145 CrossRef
- M. Maki, K. Leung and W. Qin, Int. J. Biol. Sci., 2009, 5, 500 CrossRef CAS
- D. Sommerville, R. Bradley and D. Mailley, Trees, 2004, 18, 608 CrossRef
-
P. Mishra and P. Singh, In: S. Neidlemen, A. I. Laskin (ed.), Adv. App. Microbiol., 1993, vol. 39, p. 351 Search PubMed
- N. Mosier, C. Wyman, B. Dale, R. Elander, Y. Lee, M. Holtzapple and M. Ladisch, Bioresour. Technol., 2005, 96, 673 CrossRef CAS
- B. Hahn-Hagerdal, M. Galbe, M. Gorwa-Grauslund, G. Liden and G. Zacchi, Trends Biotechnol., 2006, 24, 549 CrossRef CAS
- J. Lange, Biofuels, Bioprod. Biorefin., 2007, 1, 39 CrossRef CAS
- P. Kumar, D. Barrete, M. Delwiche and P. Stroeve, Ind. Eng. Chem. Res., 2009, 48, 3713 CrossRef CAS
- R. Kumar, B. Gaurav Mago, C. Venkatesh Balan, E. Charles and D. Wyman, Bioresour. Technol., 2009, 100, 3948 CrossRef CAS
- K. Kovacs, S. Macrelli, G. Szakacs and G. Zacchi, Biotechnol. for Biofuels., 2009, 14, 1 Search PubMed
- S. Ding, Y. Lui, Y. Zeng, M. Himmel, J. Baker and E. Bayer, Science, 2012, 338, 1055 CrossRef CAS
-
M. Himmel, J. Baker and R. Overend, In: Enzymatic Conversion of Biomass for Fuel Production., 1994, ACS symposium series p. 566 Search PubMed
- B. Henrissat, T. Terri and R. Warren, FEBS Lett., 1998, 425, 352 CrossRef CAS
- T. Teeri, Biochem. Soc. Trans., 1998, 26, 173 CAS
- Y. Zhang, M. Himmel and J. Mielenz, Biotechnol. Adv., 2006, 24, 452 CrossRef CAS
- M. Himmel, S. Ding, D. Johnson, W. Adney, M. Nimlos, J. Brady and T. Foust, Science, 2007, 2207, 804 CrossRef
- L. Lynd, P. Weimer, W. Van Zyl and I. Pretorious, Microbiol. Mol. Biol. Rev., 2002, 66, 506 CrossRef CAS
- M. G. Adsul, M. Singhvi, S. Gaikaiwari and D. Gokhale, Bioresour. Technol., 2011, 102, 4304 CrossRef CAS
- M. G. Adsul, J. Ghule, R. Singh, H. Shaikh, K. Bastawadw, D. Gokhale and A. Varma, Carbohydr. Polym., 2004, 57, 67 CrossRef CAS
- M. G. Adsul, J. Ghule, R. Singh, H. Shaikh, K. Bastawadw, D. Gokhale and A. Varma, Carbohydr. Polym., 2005, 62, 6 CrossRef CAS
- M. G. Adsul, K. Bastawde, A. Varma and D. Gokhale, Bioresour. Technol., 2007, 98, 1467 CrossRef CAS
- M. G. Adsul, A. Terwadkar, A. Varma and D. Gokhale, Bioresources., 2009, 4, 1670 CAS
- R. Datta and M. Henry, J. Chem. Technol. Biotechnol., 2006, 81, 1119 CrossRef CAS
- K. Hofvendahl and B. Hahn-Hägerdal, Enzyme Microb. Technol., 2000, 26, 87 CrossRef CAS
- S. R. Kadam, S. Patil, K. Bastawde, J. Khire and D. Gokhale, Process Biochem., 2006, 41, 120 CrossRef CAS
- M. Tango and A. Ghaly, Appl. Microbiol. Biotechnol., 2002, 58, 712 CrossRef CAS
- P. Rojan, K. Noomphhothiri, A. Nair and A. Pandey, Biotechnol. Lett., 2005, 27, 1685 CrossRef CAS
- B. J. Naveena, M. Altaf, K. Bhadriah and G. Reddy, Bioresour. Technol., 2005, 96, 485 CrossRef CAS
- D. Zhang and M. Cheryan, Biotechnol. Lett., 1991, 10, 733 CrossRef
- R. Yanez, A. Moldes, J. Alonso and J. Parajo, Biotechnol. Lett., 2003, 25, 1161 CrossRef CAS
- A. Moldes, J. Alonso and J. Parajo, Bioprocess Engineering, 2000, 22, 175 CrossRef CAS
- M. G. Adsul, J. Khire, K. Bastawde and D. Gokhale, Appl. Environ. Microbiol., 2007, 73, 5055 CrossRef CAS
- D. S. Joshi, M. Singhvi, J. Khire and D. Gokhale, Biotechnol. Lett., 2010, 32, 517 CrossRef CAS
- M. G. Adsul, A. Varma and D. Gokhale, Green Chem., 2007, 9, 58 RSC
- M. S. Singhvi, D. Joshi, M. Adsul, A. Varma and D. Gokhale, Green Chem., 2010, 12, 1106 RSC
- S. D. Zhou, T. Causey, A. Hasona, K. Shanmugam and L. Ingram, Appl. Environ. Microbiol., 2003, 69, 399 CrossRef CAS
- S. D. Zhou, K. Shanmugam and L. Ingram, Appl. Environ. Microbiol., 2003, 69, 2237 CrossRef CAS
- M. Patel, M. Ou, R. Harbrucker, H. Aldrich, M. Buszko, L. Ingram and K. Shanmugam, Appl. Environ. Microbiol., 2006, 72, 3228 CrossRef CAS
- T. B. Grabar, S. Zhou, K. Shanmugam, L. Yomano and L. Ingram, Biotechnol. Lett., 2006, 28, 1527 CrossRef CAS
- M. Rehman, Y. Tashiro, T. Zendo, K. Hanada, K. Shibata and K. Sonomoto, Appl. Environ. Microbiol., 2011, 77(5), 1892 CrossRef
- K. Okano, T. Tanaka, C. Ogino, H. Fukuda and A. Kondo, Appl. Microbiol. Biotechnol., 2010, 85, 413 CrossRef CAS
- J. Qin, B. Zhao, X. Wang, L. Wang, B. Yu and Y. Ma, PLoS One., 2009, 4, 43 Search PubMed
- N. Budhavaram and Z. Fan, Bioresour. Technol., 2006, 100, 5966 CrossRef
- S. Varadarajan and D. Miller, Biotechnol. Prog., 1999, 15, 845 CrossRef CAS
- M. Nampoothiri, R. Nair and P. John, Bioresour. Technol., 2010, 101, 8493 CrossRef
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835 CrossRef CAS
- G. Luckachan and C. Pillai, Carbohydr. Polym., 2006, 24, 254 CrossRef
-
R. Auras, S. Selke and H. Tsuji, Poly (lactic acid): Synthesis, structure, properties, processing and applications, 2010, Wiley & Sons, Inc Search PubMed
- D. Garlotta, J. Polym. Environ., 2002, 9, 63 CrossRef
- A. Agrawal and R. Bhalla, J. Macromol. Sci., Part C, 2003, 43, 479 CrossRef
- K. Matsumoto and S. Taguchi, Appl. Microbiol. Biotechnol., 2009, 85, 921 CrossRef
- S. Taguchi, M. Yamada, K. Matsumoto, K. Tajima, Y. Satoh and M. Munekata, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17323 CrossRef CAS
- S. Taguchi and Y. Doi, Macromol. Biosci., 2004, 4, 145 CrossRef CAS
- F. Shozui, K. Matsumoto, T. Nakai, M. Yamada and S. Taguchi, Appl. Microbiol. Biotechnol., 2010, 85, 949 CrossRef CAS
- M. Yamada, K. Matsumoto, T. Nakai and S. Taguchi, Biomacromolecules, 2009, 10, 677 CrossRef CAS
- X. Han, Y. Satoh, T. Satoh, K. Matsumoto, T. Kakuchi, S. Taguchi, T. Dairi, M. Munekata and K. Tajima, Appl. Microbiol. Biotechnol., 2011, 92, 509 CrossRef CAS
- K. Matsumoto, A. Ishiyama, K. Sakai, T. Shiba and S. Taguchi, Biotechnol., 2011, 156, 214 CAS
- Y. Jung, T. Kim, S. Park and S. Lee, Biotechnol. Bioeng., 2010, 105, 161 CrossRef CAS
- Y. Song, K. Matsumoto, M. Yamada, A. Gohda, J. Christopher, J. Brigham, J. Anthony, J. Sinskey and S. Taguchi, Appl. Microbiol. Biotechnol., 2012, 93, 1917 CrossRef CAS
- K. Tajima, X. Han, Y. Satoh, A. Ishii, Y. Araki, M. Munekata and S. Taguchi, Appl. Microbiol. Biotechnol., 2012, 94, 365 CrossRef CAS
- P. Galgali, A. Varma, U. Puntambelar and D. Gokhale, Chem. Commun., 2002, 23, 2884 RSC
- P. Galgali, U. Puntambelar, D. Gokhale and A. Varma, Carbohydr. Polym., 2004, 55(4), 393 CrossRef CAS
- S. Li, H. Garreau and M. Vert, J. Mater. Sci.: Mater. Med., 1990, 1, 123 CrossRef CAS
- M. Hakkarainen, A. Albertsson and S. Karlsson, Polym. Degrad. Stab., 1996, 52, 283 CrossRef CAS
- A. Hoshino and Y. Isono, Biodegradation, 2002, 13, 141 CrossRef CAS
- S. Sinha Ray and M. Bousmina, Prog. Mater. Sci., 2005, 50, 962 CrossRef
- K. Fukushima, C. Abbate, D. Tabuani, M. Gennari and G. Camino, Polym. Degrad. Stab., 2009, 94, 1646 CrossRef CAS
- M. Arena, C. Abbate, K. Fukushima and M. Gennari, Environ. Sci. Pollut. Res., 2011, 18, 865 CrossRef CAS
- J. Zhua, G. Wang, X. Pan and R. Gleisner, Chem. Eng. Sci., 2009, 64, 474 CrossRef
- R. Chandra, R. Bura, W. Mabee, A. Berlin, X. Pan and J. Saddler, Adv Biochem Eng Biotechnol., 2007, 108, 67 CrossRef CAS
- M. Galbe and G. Zacchi, Adv. Biochem. Eng./Biotechnol., 2007, 108, 41 CrossRef CAS
- T. Hendricks and G. Zeeman, Bioresour. Technol., 2009, 100, 10 CrossRef
- J. Quesada, M. Rubio and D. Gomez, J. Wood Chem. Technol., 1999, 19, 115 CrossRef CAS
- N. Mosier, C. Wyman, B. Dale, R. Elander, Y. Lee and M. Holtzapple, Bioresour. Technol., 2005, 96, 673 CrossRef CAS
- C. Wyman, B. Dale, R. Elander, M. Holtzapple, M. Ladisch and Y. Lee, Bioresour. Technol., 2005, 96, 1959 CrossRef CAS
- Y. Zhang, J. Ind. Microbiol. Biotechnol., 2008, 35, 367 CrossRef CAS
- R. Zwart and H. Boerrigter, Energy Fuels, 2006, 20(5), 2192 CrossRef CAS
- N. Mosier, R. Hendrickson, M. Brewer, N. Ho, M. Sedlak and R. Dreshel, Appl. Biochem. Biotechnol., 2005, 125, 77–97 CrossRef CAS
- L. Magnusson, R. Islam, R. Sparling, D. Levin and N. Cicek, Int. J. Hydrogen Energy, 2008, 33, 5398 CrossRef CAS
- A. Dumbrepatil, M. Adsul, S. Chaudhari, J. Khire and D. Gokhale, Appl. Environ. Microbiol., 2008, 74, 333 CrossRef CAS
- T. Tanaka, M. Hoshina, S. Tanabe, K. Sakai, S. Ohtsubo and M. Taniguchi, Bioresour. Technol., 2006, 97, 211 CrossRef CAS
- R. Yanez, J. L. Alonso and J. C. Parajo, J. Chem. Technol. Biotechnol., 2005, 80, 76 CrossRef CAS
- Z. Lu, M. Lu, F. He and L. Yu, Bioresour. Technol., 2009, 100, 2026 CrossRef CAS
- M. A. Patel, M. S. Ou, L. O. Ingram and K. T. Shanmugam, Biotechnol. Prog., 2005, 21, 1453 CrossRef CAS
- B. Calabia and Y. Tokiwa, Biotechnol. Lett., 2007, 29, 1329 CrossRef CAS
- L. Wang, B. Zhao, F. Li, K. Xu, C. Ma and F. Tao, Appl. Microbiol. Biotechnol., 2011, 89, 1009 CrossRef CAS
- L. Wang, B. Zhao, B. Liu, C. Yang, B. Yu and Q. Li, Bioresour. Technol., 2010, 101, 7895 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2013 |