 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bringing D-limonene to the scene of bio-based thermoset coatings via free-radical thiol–ene chemistry: macromonomer synthesis, UV-curing and thermo-mechanical characterization†
Mauro
Claudino
a,
Jeanne-Marie
Mathevet
a,
Mats
Jonsson
b and
Mats
Johansson
*a
aDepartment of Fibre and Polymer Technology, School of Chemical Science and Engineering, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden. E-mail: matskg@kth.se; Tel: +46-8-790 92 87
bDepartment of Chemistry, School of Chemical Science and Engineering, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden
First published on 2nd December 2013
Abstract
The increasing pursuit for bio-based plastic materials led us to investigate the potential use of the monoterpene limonene in thermoset synthesis using the free-radical mediated thiol–ene reaction. The high efficiency of this reaction to prepare multifunctional ene-terminated resins, as intermediary macromolecular precursors, for thermosets synthesis was demonstrated under thermal and photoinitiated conditions. Although an excess of terpene favors formation of well-defined macromonomers in organic solution, the characteristic low-vapor pressure of limonene hinders its simple removal (or recycling) via evaporation after synthesis. Alteration to an initial thiol–ene stoichiometry of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 enables production of high molecular weight resins in the form of ‘hyperbranched oligomeric-like’ structures having moderate polydispersity. UV-curing of these polyfunctional resins combined with equal mole compositions of multifunctional alkyl ester 3-mercapto propionates yields highly sticky, amorphous and flexible elastomers with different thermo-mechanical properties. These can be further modulated by varying the amount of unreacted thiol occluded within the networks working as a plasticizer. Introduction of a renewable cycloaliphatic structure into the materials offers a convenient way to enhance the glass-transition temperature and stiffness of traditional thiol–ene networks. The materials synthesized may be considered potentially useful as sealants and adhesives in a wide variety of applications including organic coatings. The versatility of UV-irradiation over thermal initiation makes this method particularly suitable for green industrial synthesis processes via thiol–ene chemistry using limonene and multifunctional thiols. The thiol–ene system evaluated herein serves as a model example for the sustainable incorporation of natural diolefinic monomers into semi-synthetic thiol–ene networks exhibiting a range of thermo-mechanical properties.
:0.5 enables production of high molecular weight resins in the form of ‘hyperbranched oligomeric-like’ structures having moderate polydispersity. UV-curing of these polyfunctional resins combined with equal mole compositions of multifunctional alkyl ester 3-mercapto propionates yields highly sticky, amorphous and flexible elastomers with different thermo-mechanical properties. These can be further modulated by varying the amount of unreacted thiol occluded within the networks working as a plasticizer. Introduction of a renewable cycloaliphatic structure into the materials offers a convenient way to enhance the glass-transition temperature and stiffness of traditional thiol–ene networks. The materials synthesized may be considered potentially useful as sealants and adhesives in a wide variety of applications including organic coatings. The versatility of UV-irradiation over thermal initiation makes this method particularly suitable for green industrial synthesis processes via thiol–ene chemistry using limonene and multifunctional thiols. The thiol–ene system evaluated herein serves as a model example for the sustainable incorporation of natural diolefinic monomers into semi-synthetic thiol–ene networks exhibiting a range of thermo-mechanical properties.
1. Introduction
The development of plastic materials derived from renewable resources for replacement of petroleum-based ones continues gaining growing interest both in industry and in the scientific research community. Also, by using renewable feedstocks all the biosynthetic capabilities of Nature are fully exploited in a straightforward fashion. Terpenes represent a plentiful and inexpensive class of non-polar substrates with enormous potential for the synthesis of bio-based polymeric materials.1–6 They occur naturally as unsaturated cyclic hydrocarbons made up of C5 isoprene units based on 2-methyl-1,3-butadiene, having the general formula C10H16, and exist mostly as ingredients of essential oils obtained from widespread sources of plants, trees, and fruits where the most common is pine-wood turpentine.1,2 One of such ‘green’ constituents is D-limonene, an optically active monoterpenic fragrance also present abundantly in the peels of lemons, oranges and other citrus fruits.3,7 Worldwide annual production of D-limonene from the citrus fruit processing industry has been estimated to be around (30–70) × 103 tons.8–11 Despite being available naturally in a large scale almost in a stereochemically pure form (over 95% from orange peel oil),12D-limonene also incorporates in its structure a chiral center and two distinct unconjugated electron-rich C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double-bonds with different degrees of substitution readily available for radical polymerization: an internal (endocyclic) double-bond (i.e., 1-methyl-cyclohexene moiety) and a terminal (exocyclic) aliphatic bond (i.e., vinylidene or isopropenyl moiety) (Scheme 1).
C double-bonds with different degrees of substitution readily available for radical polymerization: an internal (endocyclic) double-bond (i.e., 1-methyl-cyclohexene moiety) and a terminal (exocyclic) aliphatic bond (i.e., vinylidene or isopropenyl moiety) (Scheme 1).
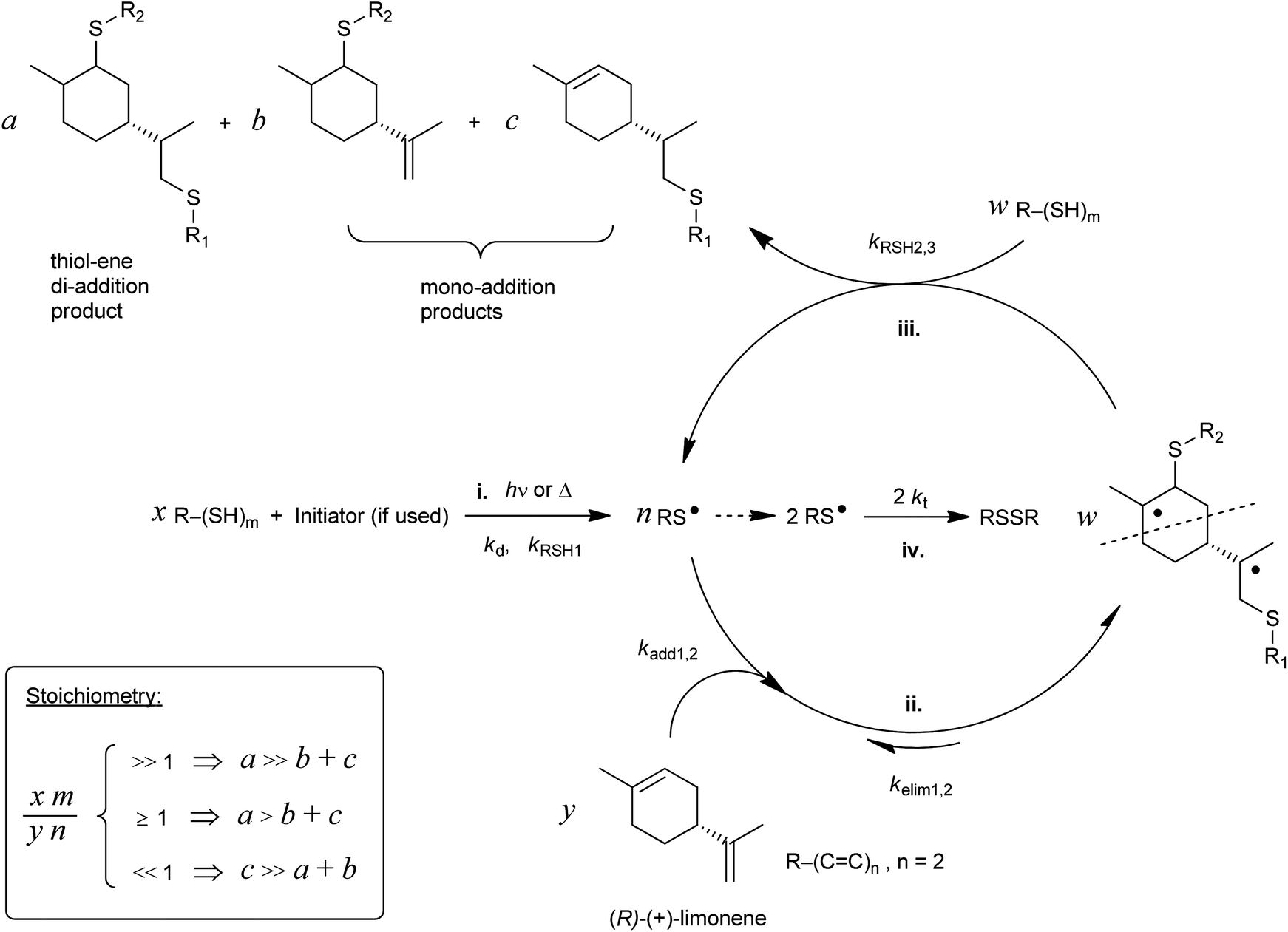 | ||
| Scheme 1 Stepwise thiol–ene reaction scheme involving the two unconjugated double-bonds of limonene with: initiation (i), reversible propagation (insertion–elimination) (ii), chain-transfer (hydrogen-abstraction) (iii), and thiyl self-termination (homocoupling) (iv) steps. Ideally, w = a + b + c. The inset box denotes the kinetic effect of initial thiol–ene stoichiometry on the outcome of the coupling reaction.73 | ||
Significant efforts have been conducted throughout the years to polymerize limonene either by itself or with other monomers using a variety of chemistries. For instance, this terpene was originally homopolymerized into poly(D-limonene) by Roberts and Day (1950)13 and Marvel and coworkers (1965)14 using a Friedel–Crafts catalyst and Ziegler–Natta catalyst, respectively. However, low average molecular weights in the order of 1200 g mol−1 were obtained.13 Later, Doiuchi et al. (1981) proposed an inter/intra-molecular cyclopolymerization of limonene with maleic anhydride yielding optically active 1:2 alternating copolymers upon thermal decomposition of the azo compound 2,2′-azobis(2-methylpropionitrile) (AIBN).15 The suggested structure was subsequently questioned by Maslinska-Solich et al. (1994) proposing instead a 1:1 alternating poly(limonene-co-maleic anhydride) sequence with pendant 4-methyl-3-cyclohexenyl residues.16 Copolymerization of limonene with acrylonitrile17 as well as N-vinyl pyrrolidone18 and synthetic vinyl monomers such as methyl methacrylate,19 styrene,20 butyl methacrylate21 and vinyl acetate22 has also been reported by Sharma and Srivastava using thermally initiated free-radical chemistry, among others.1 Direct copolymerization of limonene with ethylene in the presence of a titanium-based complex as the catalyst has also been achieved by Shiono and co-workers (2012); although the reaction proceeded much slower, with only ∼4.0 mol% of monoterpene incorporation, low molecular weight and broader distribution of molecular weight than ethylene homopolymerization and the alternating copolymerization of ethylene with isobutylene.23 Recently (2012), limonene was homo- and co-polymerized with indene into amorphous rigid polymers via cationic polymerization with a Friedel–Craft catalyst. Among the several olefins tested, including styrene, indene, and 5-ethylidene-2-norbornene, limonene resulted to be the most active chain-transfer agent, although an increase in the initial feed concentration resulted in low molecular weight oligomers with reduced polydispersities and exhibiting low Tg's.24 Following a different line, Nakatani et al. (2009) used limonene to modify poly(1-butene) via a free-radical graft polymerization route with the purpose of incorporating functional groups along the polymer backbone.25 Xu and co-workers (2004) synthesized a structural hybrid epoxy resin in a three-step procedure containing both naphthyl and cycloaliphatic moieties from 1-naphthol and limonene. The epoxy polymer, composed of a mixture of stereoisomers and geometric isomers, was cured thermally with AIBN affording a much higher Tg and enhanced physical properties when compared to that of diglycidyl ether of bisphenol A type epoxy resin.26 Mathers et al. (2009) have described a general method for the synthesis of functional hyperbranched polymers exhibiting complex structures by ring-opening metathesis polymerization (ROMP) of dicyclopentadiene (DCPD) with limonene, limonene oxide (LO) or β-pinene, using the Grubb's second generation catalyst.27 In two former studies the authors clearly demonstrated the efficient utilization of limonene acting both as a renewable solvent and a chain-transfer agent in ROMP of selected alkene macromonomers28 as well as in the metallocene/methylaluminoxane (MAO) catalyzed polymerization of α-olefins.29 More recently (2011), the same group reported on the covalent attachment of limonene and other monoterpenes during ROMP of DCPD with the purpose of adjusting the thermo-mechanical properties of the resulting networks by means of controlling the degree of crosslinking.30 Kamigaito and co-workers (2010) successfully copolymerized D-limonene with maleimide via controlled/living radical polymerization yielding chiral and high-Tg linear copolymers with exceptional end-to-end sequence regulation.31 Following a similar approach, the same authors also reported on the free-radically induced copolymerization of limonene and β-pinene (or their functional derivatives possessing hydroxyl and other related moieties) with functional maleimide (MI) derivatives in fluorinated alcohol [PhC(CF3)2OH] affording a linear ABB-type monomer sequence, where A stands for terpene and B denotes the maleimide monomer (or their functionalized derivatives). The copolymers were found to be particularly well arranged on a 1:2- or 2:1-alternating series, as a consequence of the characteristic bulky structures of such terpenes in combination with effects of hydrogen-bonding interactions between the fluorinated solvent and MI units.32,33 Periodically carbamate-functionalized and grafted copolymers resulting from hydroxyl- and chlorine-functionalized pre-copolymers were subsequently synthesized via a polymer reaction with isocyanate and metal-catalyzed living radical copolymerization, respectively.33 The final structures of these bio-based copolymers are claimed to convey novel functions and properties attributed to the highly ordered sequence, presence of chirality and backbone rigidity.32
Chemical modifications of limonene and derived compounds to make them suitable for more standard polymerization chemistries can be found throughout the literature. In the classic example reported by Marvel and Olson (1957), D-limonene was transformed into di-pentene dimercaptan (DD) by free-radical thiol–ene photo-addition with thiolacetic acid followed by hydrolysis of the resulting dithiol ester.34 The terpene-based dithiol was subsequently polymerized with original limonene into poly-alkylene sulfides via thiol–ene chemistry affording low melting, highly viscous sticky materials. Recently (2012), DD was introduced into thiourethane thermosets derived from sucrose soya ester (SSE) to increase the bio-renewable content in these materials.35 One of the most common modifications of limonene is the conversion into its monoxide (LO) or dioxide (LDO) analogs.36 This methodology offers the possibility for subsequent cationic and/or free-radical polymerizations of the bio-derived diepoxides upon exposure to visible and/or sunlight radiation by means of advanced photoinitiating systems.37–45 In a previous study, Aikins and Williams (1981) reported on the radiation-induced cationic polymerization of neat LO at room-temperature by ring-opening of the epoxide moiety yielding a low molecular weight 1,2-trans polyether (7.6 < DP < 18.7) accompanied by very high monomer conversions into polymer (at least 80%).46 Duchateau and co-workers (2013) synthesized partially bio-based alternating polyesters by catalytic ring-opening copolymerization of LO and phthalic anhydride via a series of metal t-Bu–salophen complexes combined with different cocatalysts and various mono-/di-/tri-functional chain-transfer agents (CTA)s. Chromium-chloride based catalysts provided the best results without any major drop in catalytic activity. Inclusion of (CTA)s enabled molecular weight reduction of the copolymers while retaining low polydispersities.47 In another study, partially bio-based polymers bearing reactive epoxy groups in the side-chains were synthesized by Morinaga and co-workers (2013) via thermally induced radical copolymerization of LO with methyl acrylate.48 A pioneering study by Bähr et al. (2012) also employed LDO as a difunctional precursor of cyclic limonene dicarbonate via halide catalysis with CO2. This newly derivatized carbonated monomer was further reacted with di- and poly-functional amines to prepare linear, non-toxic (isocyanate-free), oligo-hydroxyurethanes and thermosetting polyurethanes, respectively.49 Diepoxides of limonene have also been obtained by catalytic oxidation using immobilized Mn(Salen)Cl complexes, although the 1,2-epoxide is formed primarily due to a higher regioselectivity.50–52 Coates et al. (2004) reported on the synthesis of alternating polycarbonate copolymers issued from the cis and trans diastereomers of D-limonene monoxide (LO) and CO2 using β-diiminate zinc acetate complexes as catalysts; although, the reaction proceeded selectively on the trans monomer affording a regioregular polycarbonate structure.53 This work represents an excellent example of combination of two very different sources of renewable substances in the creation of linear polymers. Nucleophilic thiylation of limonene-8,9-oxide into different thioterpenols has also been described after synthesis of the initial monoxide diastereomers from racemic limonene.54 Another very interesting modification refers to the synthesis of sulfur containing terpenoids such as 1- or 2-mercapto-p-menth-8-ene from thioepoxidation of cis/trans-(+)-limonene 1,2-epoxides via hydride reduction of the corresponding epi-sulfides.55 Analogously, Janes et al. (1993) prepared thioterpineol (p-menth-1-en-8-thiol), commonly known as grapefruit mercaptan, by reacting either limonene or α-pinene with gaseous hydrogen sulfide in the presence of aluminium trihalide.56 Although most of the thiol-modified terpenes described were prepared primarily with the purpose of creating new fragrances of potential olfactory interest commercially, they could be viewed also as important substrates for future free-radical thiol–ene addition reactions since one of the two olefinic bonds in D-limonene is retained intact. More recently, Meier and co-workers (2012) demonstrated the efficient thiol–ene coupling of cysteine hydrochloride to R-(+)- and S-(−)-limonene in the preparation of new diamine functionalized renewable monomers which served as platform precursors for polyamide and polyurethane synthesis.10 In an early report, the authors developed optimal experimental conditions for the selective functionalization of the terminal vinylidene group with methyl thioglycolate as a model compound further enabling the synthesis of a diversity of monomer precursors and ensuing polymers. Direct polymerization of limonene with dithiols was also reported in this study.57 In another paper the authors also successfully described the attachment of alcohol and ester functionalized thiols to the two enantiomers of limonene via thiol–ene coupling affording monofunctional, homo-difunctional and hetero-difunctional terpene-modified monomers. The difunctional addition products were subsequently homo- and co-polymerized with short-chain diols using cyclo guanidine 1,5,7-triazabicyclododecene (TBD) as the polycondensation organocatalyst because of its high transesterification activity. The same strategy was employed to synthesize high molecular weight polyesters (from 9 up to 25 kDa) based on limonene and fatty-acid methyl ester derivatives.9 These three recent reports illustrate the extreme versatility of the thiol–ene reaction in the design of new linear polymers issued from limonene in combination with distinct classes of renewable monomers (natural or modified) and chemistries.
Thiol–ene chemistry, although extensively studied in the past, has witnessed a spiraling development over the last 10–15 years and has recently been recognized as a ‘click’ method due to the high yields and reduced by-products attained.58–62 This reaction (videScheme 1 exemplified with limonene) presents well-established key advantages in thermoset synthesis, including a reduced sensitivity to oxygen inhibition, self-initiation without the need for a photoinitiator (enabling the cure of thick coatings), relatively rapid reaction rates leading to highly crosslinked products, improved curing control, and a free radical step-growth mechanism resulting in polymeric network materials with homogenous properties across all spacial dimensions. Moreover, because of the stepwise nature of radical growth, viscosity builds-up slower throughout the liquid phase resulting in much higher conversions at gel-point than with conventional acrylic formulations.63 This results in decreased property change with time. Quite importantly, the thiol–ene reaction is also considered an eco-friendly synthesis tool since it can proceed in neat (solventless), under environmentally clean and mild conditions (if photoinduced) and that rejects potentially toxic transition-metal catalysts so commonly in use in other ‘click’ reactions.58,64 Another additional advantage of thiol–ene photopolymers is that they are optically clear and exhibit improved physical properties such as flexibility and good adhesion to various substrates, which makes this chemistry ideal for organic coating applications (e.g., clear protective coatings).
Despite the plethora of olefinic substrates (natural and synthetic) available for the thiol–ene reaction, the rate of addition has shown to be strongly dependent on the type of thiol and structure of the ene (e.g., internal, isolated, conjugated, non-conjugated, and substituted), with thiyl radicals inserting generally faster to electron-rich α-olefins (terminal and monosubstituted) and norbornenes than to electron-deficient ones (e.g., (meth)acrylates) and sterically hindered multi-substituted olefins.58,59 This difference in reactivity drastically affects the effectiveness of thiol–ene additions and overall kinetics; and, denomination with the term (‘click’) should thus be used with care when dealing with less reactive double bonds. For example, our recent studies using this reaction have clearly demonstrated the efficient utilization of internal main-chain alkenes from natural resources in the synthesis of thermosetting elastomers despite the sluggish reactivity of the 1,2-disubstituted ene moiety.65,66 This was attributed essentially to a reversible cis/trans-isomerization process associated with poor hydrogen abstractibility of the intermediate alkyl radical adduct during chain-transfer to the thiol.65 Following the same principle, we would like to examine if the internal 1,1,2-trisubstituted unsaturation of limonene is also feasible in thermoset synthesis, at the image of 1,2-disubstituted olefins, except that this double-bond does not isomerize. To the best of our knowledge, and from a thermosetting film-formation perspective, very little has been published regarding limonene as a direct building-block co-monomer to obtain permanent networks.30,67 Only a few cases presented in the literature make use of either its dioxide analog or DD, as already mentioned. Furthermore, documented work of its combined utilization with free-radical thiol–ene chemistry is practically inexistent within the context of thermosetting polymers. Our major interest in this simple unmodified monoterpene stems from the idea that the two naturally occurring alkene groups can easily couple to sulfur compounds via a free-radical thiol–ene mechanism and this way be used, for the first time, in the formation of multi-chiral crosslinked poly(thioether) networks which could be of relevance to the field of polymer coatings.9,10 The second reason is related to the rigid structure and bulkiness of the alicyclic ring which may help introduce stiffness to the final network structure in a way similar to well-known stiff aromatics. Thiol–ene networks usually lack this feature when compared to their acrylate counterparts as a consequence of the formation of a more flexible S–C thioether bond (1.8 Å) versus the C–C bond (1.2–1.5 Å).68–71 At last, limonene is a naturally pleasant fragrance and hence could help neutralize the nasty repellent scent so distinctive of thiol-based formulations.
Recently, we have investigated in great detail the kinetics of free-radical thiol–ene photo-additions involving D-limonene and two mono-/tri-functional thiols sharing a propionate ester moiety.72,73 We found that thiol–ene coupling at the exo-olefinic bond proceeds about 6.5 times faster than at the endocyclic one under organic solution conditions. This observed value for the selectivity obtained from empirical kinetics was confirmed by the relative reactivity based on the detailed two-route linear cyclic mechanism at steady-state, further revealing that contribution of the chain-transfer routes to the double-bond selectivity is negligible, this being explained mostly by the two reversible propagation steps (addition–elimination) directly involving each unsaturation. The difference in double-bond reactivity was ascribed predominantly to a higher relative energy of the tertiary β-thioether carbon-radical intermediate resulting from thiyl-radical insertion across the endocyclic double-bond; and, partially to steric hindrance effects controlling thiyl radical addition onto the two double bonds. These two effects support evidence for the relatively low conversion rates observed for the two multi-substituted enes when compared to more reactive olefins attributed to higher extents of reversibility in each propagation step in addition to a chain-transfer rate-controlling step that slows down the overall reaction.72 Manipulations in the initial co-reactants stoichiometry, as represented in Scheme 1, indicated that thiol–ene coupling at the external isobutylene functionality could be addressed regioselectively by increasing the relative concentration of monoterpene over the thiol.73 Now, we wish to expand further this concept, taking advantage of the difference in ene reactivity, toward the synthesis of multifunctional limonene-terminated precursors based on two polyfunctional mercaptan ester propionates, which through a second-step will serve as intermediary alkene macromonomers in thermoset synthesis using the original multifunctional thiols as network crosslinkers (Scheme 2). Finally, the thermal and viscoelastic properties of the semi-synthetic photocured materials are reported and discussed thoroughly.
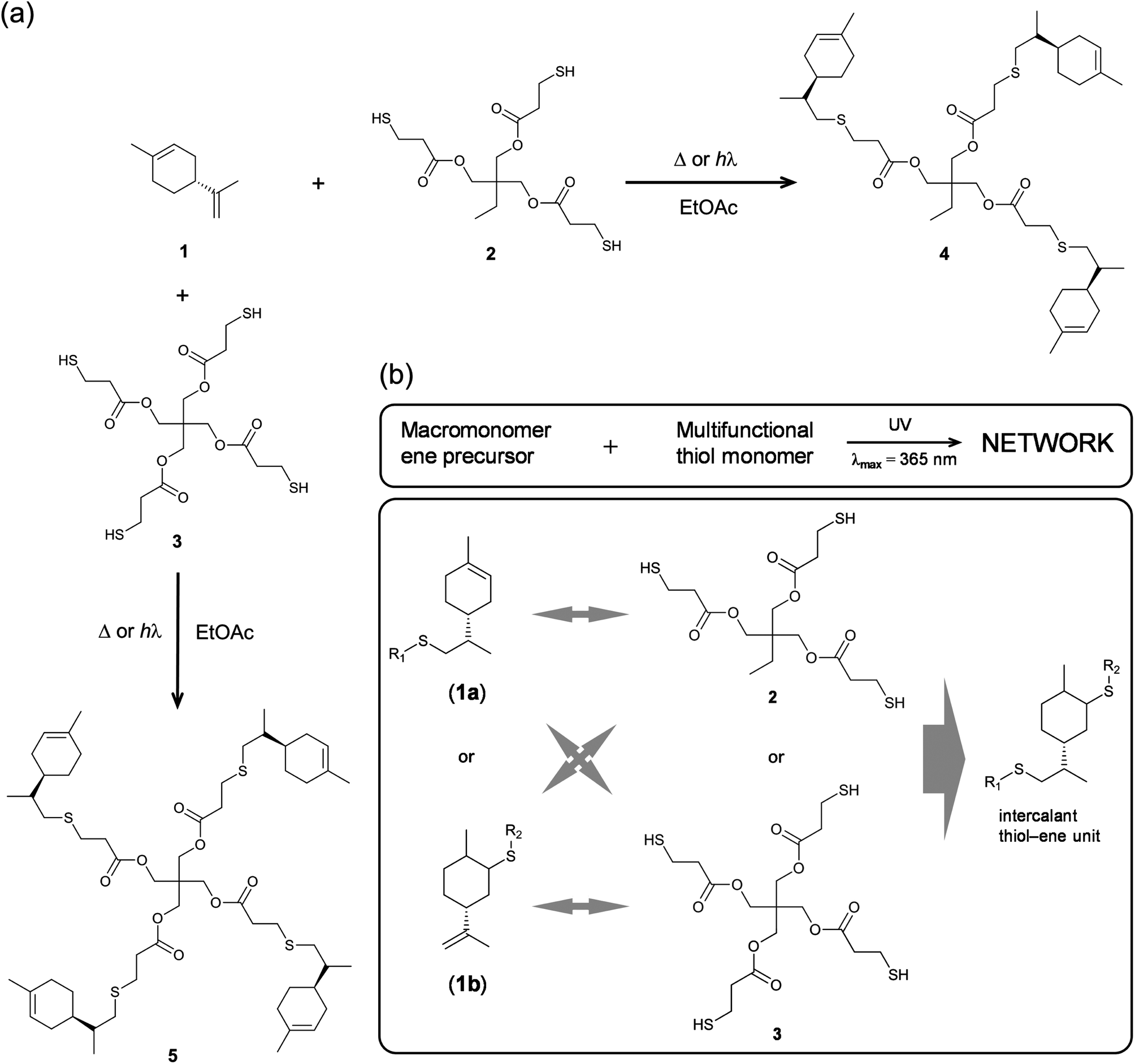 | ||
| Scheme 2 Illustrative organic synthesis routes of the limonene-terminated precursors 4 and 5 induced either thermally (AIBN, 70 °C) or photochemically (Irgacure 651/DMPA, λmax = 365 nm, r.t.) (a) and network formation diagram based on equimolar amounts of multifunctional groups (b). Bidirectional grey arrows denote photoinduced thiol–ene coupling between multifunctional thiols (2 and 3) and the two possible arrangements of pendant limonene residues (1a and 1b) attached in the two intermediary macromonomers/resins (4 and 5). | ||
2. Results and discussion
The notable features inherent to free-radical thiol–ene addition prompted us to investigate the synthesis of bio-based thermosets using R-(+)-limonene 1, as a renewable diolefinic substrate, together with TMPMP 2 as the primary synthetic thiol crosslinker. Limonene represents an exceptionally versatile monomer to introduce new chemical functional groups which can be directed towards specific polymerization techniques. In spite of the obvious attributes of functionalization, this is an approach that generally entails multistep synthesis reactions, involving numerous precursors and different chemistries, some of them hardly following the principles of green chemistry, alongside with laborious intermediate purifications, also depending on the route of synthesis and physical–chemical features of the desired polymers. Instead, the reaction system proposed in this work offers the possibility to synthesize partially bio-based (‘semi-synthetic’) crosslinked materials in a simple and environmentally benign fashion via a two-step reaction procedure using only limonene and TMPMP 2 as multifunctional comonomers in the presence of minute amounts of a radical initiator to speed-up the reaction. We also attempt to increase the crosslink density within the final material(s) by introducing a tetra-functional thiol monomer, PETMP 3, and compare the final properties with those obtained from the standard thiol–ene formulation involving the tri-functional thiol. Finally, the physicomechanical properties of the film coatings obtained from crossed over thiol–ene resins with the multifunctional thiols, mixed in different stoichiometric levels, are evaluated.2.1. Synthesis and characterization of multifunctional macromonomer resins
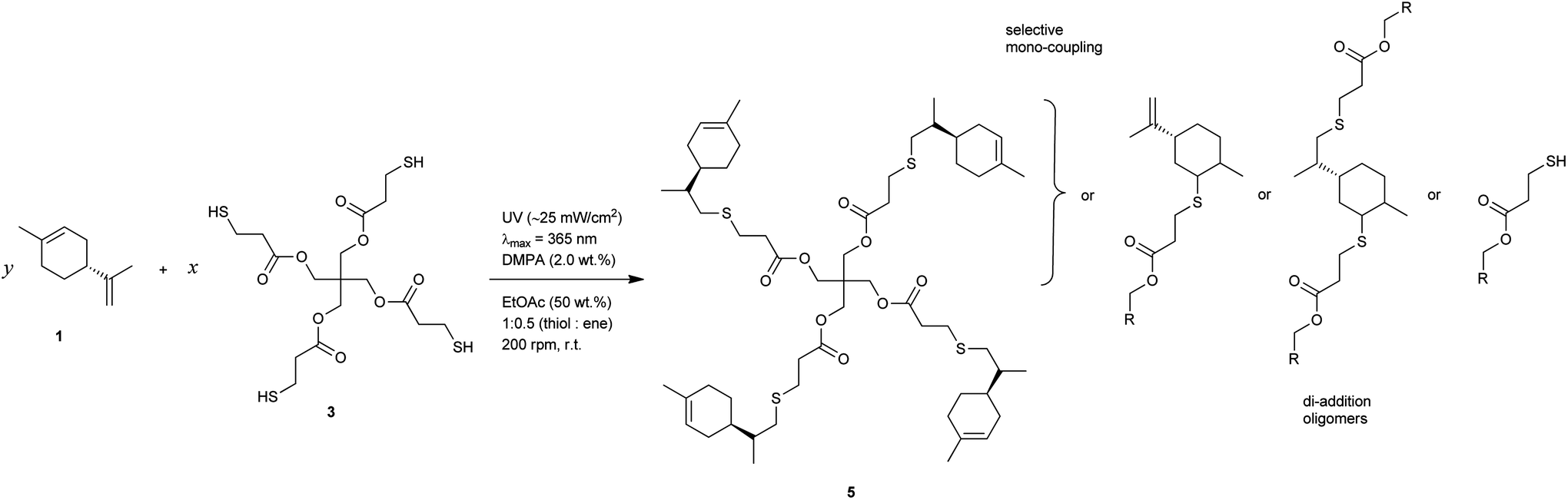
Multifunctional macromonomers 4 and 5 depicted in Scheme 2 were synthesized via free-radical thiol–ene coupling between R-(+)-limonene 1 and one of the multifunctional thiol propionate esters (TMPMP, 2 or PETMP, 3) in the presence of a small amount of initiator. Ethyl acetate (EtOAc, 50 wt%) was added to the mixtures to ensure complete reactant miscibility since the thiol–ene monomers are originally incompatible at room-temperature which prevents any direct synthesis of macromolecular thiol–ene materials in the bulk based on these compounds. Preliminary syntheses of 4 and 5 were performed under thermal conditions in an inert atmosphere by means of addition of AIBN as an initiator, for a period of 24 hours, using two different starting thiol–ene stoichiometries between co-reactants. The results shown in Fig. 1–3 obtained from DMF-SEC and FT-Raman measurements illustrate well the kinetic effect of initial thiol–ene stoichiometry on the overall outcome of the reaction. Favored formation of mono-addition products selectively at the exo-vinylidene bond of limonene occurs when the monoterpene is charged in relative excess with respect to the thiol; whereas at a thiol–ene stoichiometry of 1:0.5 formation of di-addition products, mainly in the form of high molecular weight ‘hyperbranched-like’ oligomers, takes place alongside with formation of macromonomers 4 and 5 (main bands appearing at 28–30 ml). The same pattern in the formation of thiol–ene coupled products has been verified under photochemical conditions as reported in a previous kinetic study.73 Although the first composition stoichiometry is kinetically attractive as a direct means to synthesize compounds 4 and 5 in high yields, this route was not preferred experimentally because of the low relative vapor pressure of limonene, 3.39 × 10−2 (EtOAc/b.p., 77.1 °C),11,74 which hampers removal of the unreacted terpene fraction via evaporation. Moreover, from a macromolecular materials synthesis standpoint, it is not absolutely necessary to have the two macromonomers obtained in pure form since the aliphatic ring structure of limonene mediating the two sulfur groups works as a crosslinker unit between propionate ester arms once the two thioether linkages are effectively formed creating a thiol–ene network. The photoinduced synthesis of multifunctional thiol–ene resins 1 and 2 (designated by TE-C1 and TE-C2, respectively), is illustrated in Scheme 3 using an initial thiol–ene stoichiometry of 1:0.5. The course of the reactions was monitored via1H-NMR and FT-Raman spectroscopy by following the disappearance of individual double bonds and thiol functional groups, respectively. After solvent evaporation, the two resins were obtained as clear, faintly yellow, highly viscous liquids exhibiting a fresh citreous odor note.
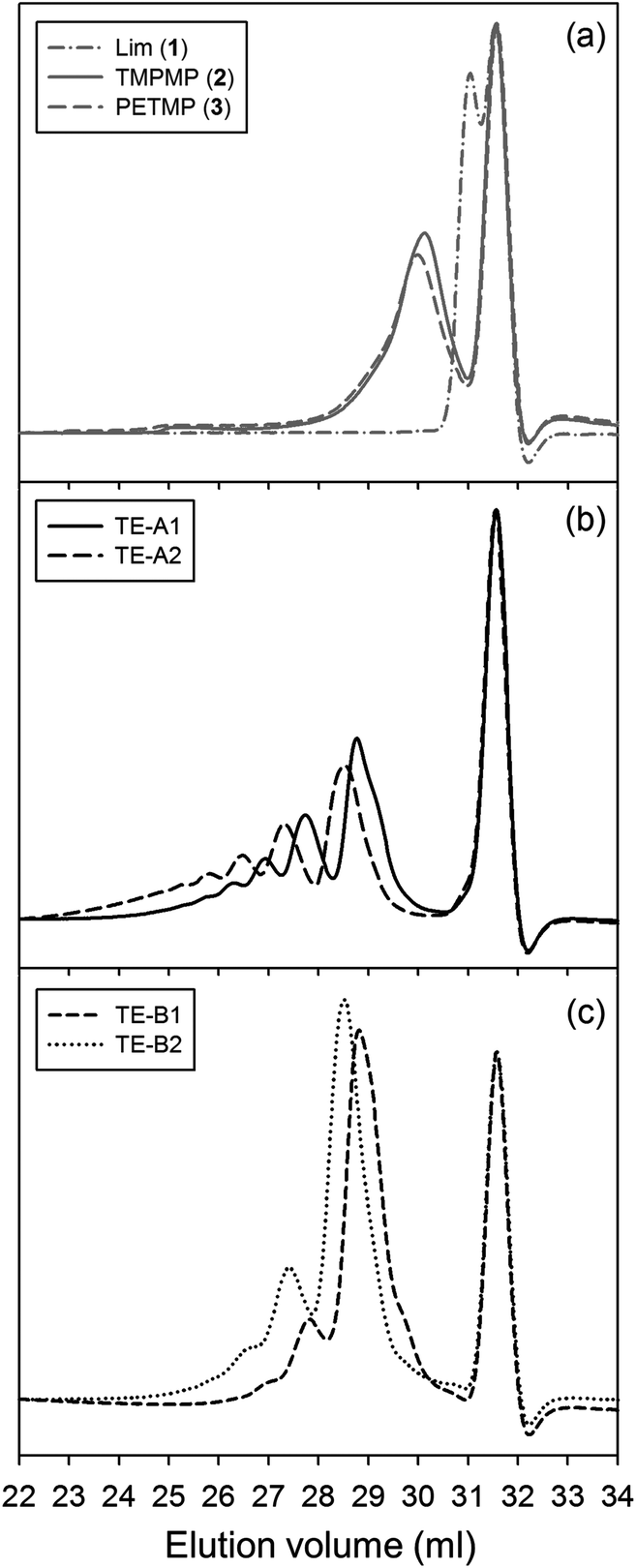 | ||
| Fig. 1 Normalized DMF-SEC traces of the synthesised crude resins containing 4 and 5 prepared via thermally initiated thiol–ene chemistry using two different mole compositions with respect to thiol–ene functional groups: (a) pure thiol–ene reactants (grey), (b) 1:0.5 stoichiometry, and (c) 1:10 stoichiometry. Main bands appearing in the range of 28–30 ml correspond majorly to products 4 and 5. Successive peaks in the direction of lower retention volumes represent dimers, trimers, tetramers and other high molecular weight oligomers (in the same order) of the respective multifunctional macromonomers. The flow-rate marker (toluene) was set at 31.5 ml. | ||
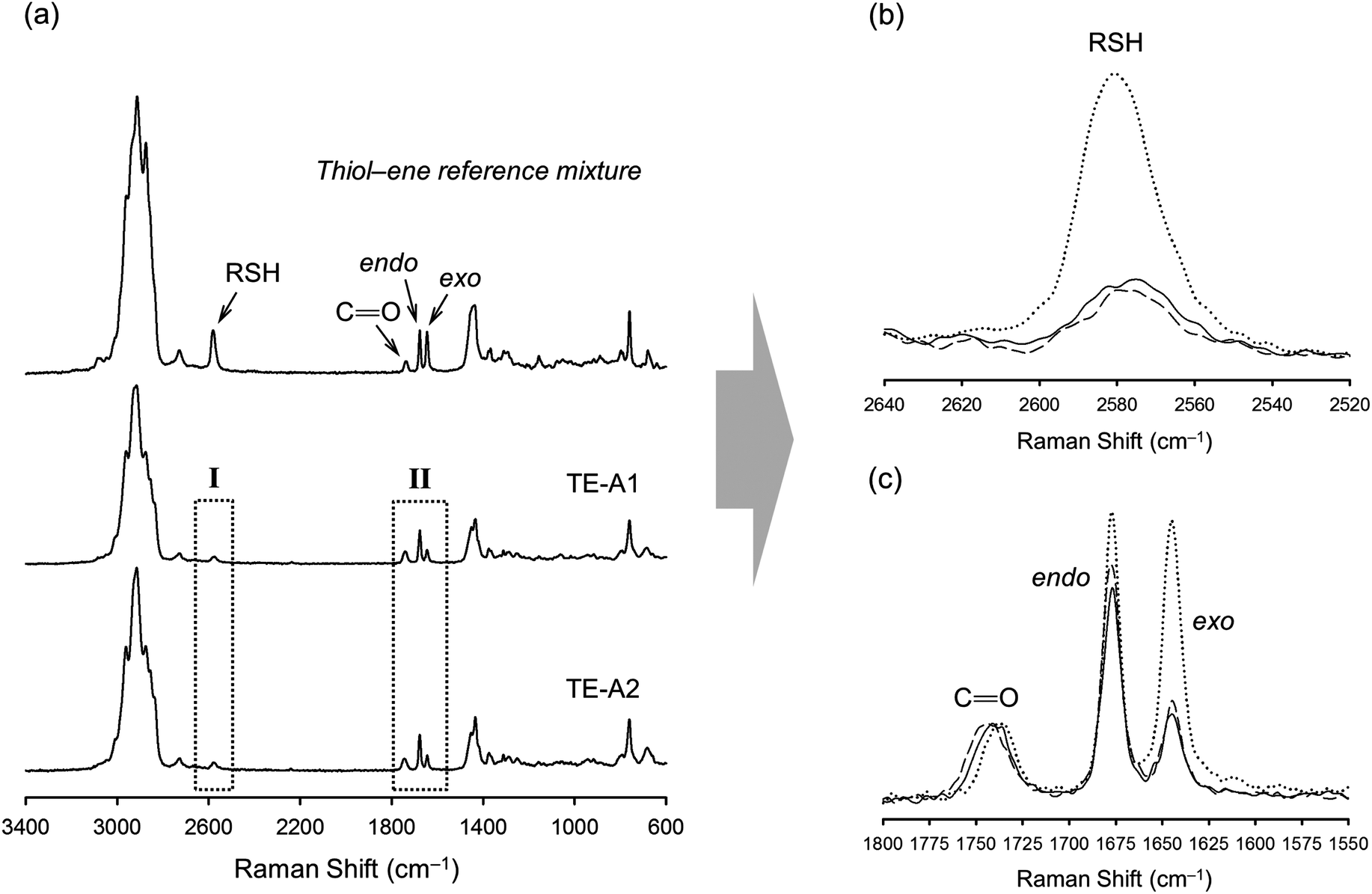 | ||
| Fig. 2 Normalized spectral FT-Raman profiles of finished syntheses products resulting from the thermal thiol–ene reaction on a 1:0.5 thiol–ene group stoichiometry. (a) Overall spectra in comparison to a reference mixture prepared with monothiol (iso-tridecyl 3-mercapto propionate) on a 1:1 thiol–ene mole ratio with respect to functional groups; (b) zoom-in of region I showing the consumption of thiol, and (c) enhancement of region II showing the disappearance of unsaturations with favored coupling at the external double-bond position. The dotted line refers to the reference mixture, the solid line is from the TE-A1 product, and the dashed line the spectrum obtained from the TE-A2 product. The carbonyl band (CO, 1735 cm−1) was used as an internal reference in the spectral normalization process. | ||
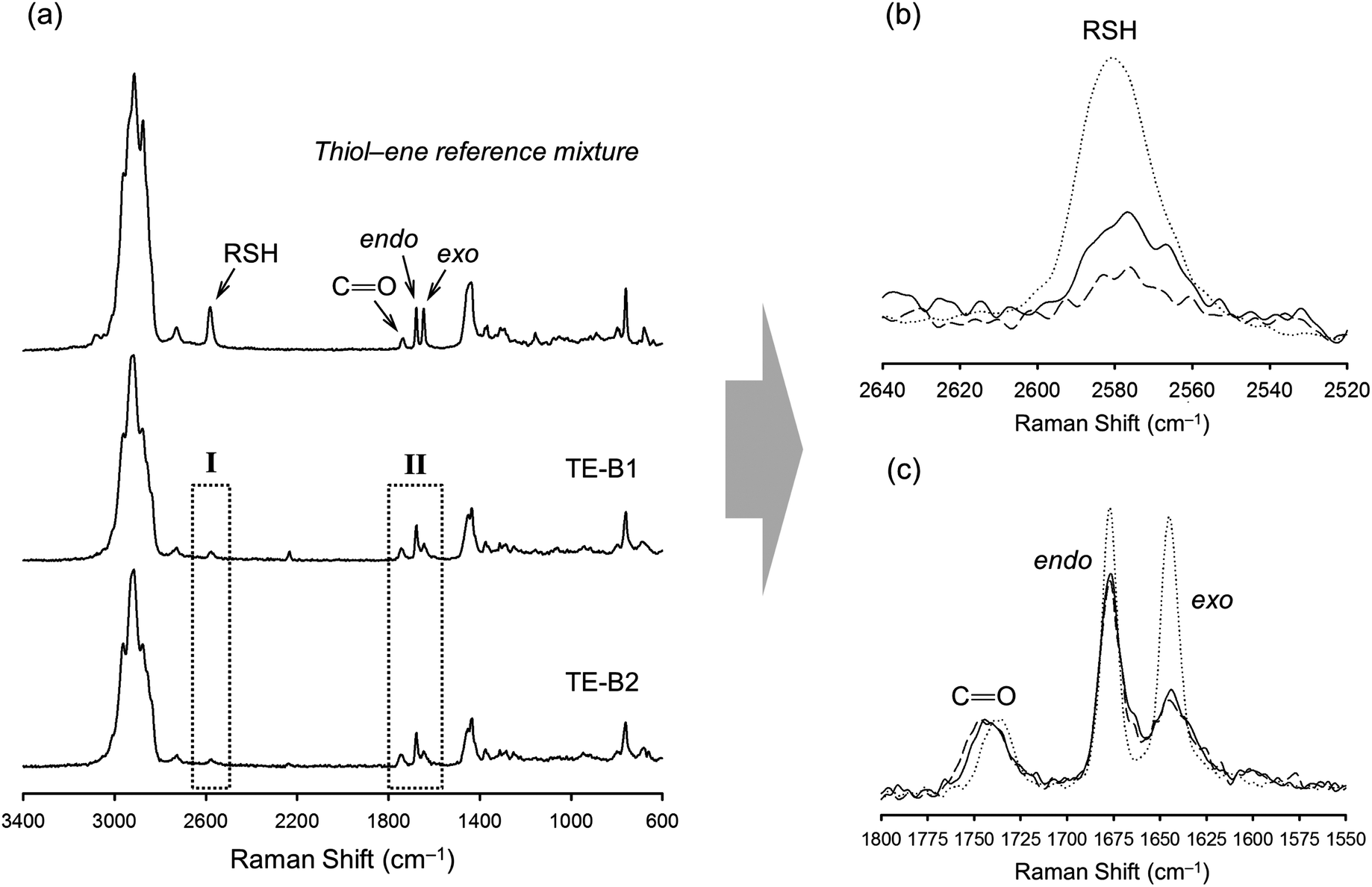 | ||
| Fig. 3 Normalized spectral FT-Raman profiles of finished syntheses products resulting from the thermal thiol–ene reaction with 10 mole excess of limonene. (a) Overall spectra in comparison to a reference mixture prepared with monothiol(iso-tridecyl 3-mercapto propionate) on a 1:1 thiol–ene mole ratio with respect to functional groups; (b) zoom-in of region I showing the consumption of thiol, and (c) enlargement of region II showing the disappearance of unsaturations with preferential coupling at the external double-bond position. The dotted line refers to the reference mixture, the solid line is from the TE-B1 product, and the dashed line the spectrum obtained from the TE-B2 product. The carbonyl moiety (CO, 1735 cm−1) was used as an internal reference band in the spectral normalization process. | ||
 | ||
| Scheme 3 Synthesis of resin 2 (TE-C2) issued from limonene 1 (n = 2) and tetrathiol 3 (m = 4). Also valid for the synthesis of resin 1 (TE-C1) issued from compounds 1 and 2 (m = 3). Non-ideal synthesis of resins comprises a mixture of all possible combinations (complete or partial). Initial thiol–ene stoichiometry: xm/yn = ½ (or 1:0.5). | ||
Photo-initiated transformation of individual thiol–ene functional groups into resins 1 and 2 after 6 hours of reaction achieved conversions of 81.8 ± 1.6% for the exo-unsaturation, 18.2 ± 1.4% for the endocyclic ene and, 100% (overall) for the thiol group as determined by 1H-NMR (enes) and FT-Raman (thiol) (Fig. 4 and 5, respectively). A significant decrease of the doublet signal (b) at 4.7 ppm assigned to the terminal protons of the exo-vinylidene bond was accompanied by a small reduction of signal (a) at 5.4 ppm attributed to the single proton of the 1,1,2-trisubstituted 1-methyl-cyclohexene functional group. The decrease of these two signals was also followed by a significant loss of triplet signal (f) at 1.75 ppm characteristic of the methyl protons adjacent to the terminal ene and practically no decrease in the triplet signal (g) located at 1.65 ppm; thus, indicating favored coupling at the exocyclic double-bond position. Moreover, proton signals (e) and (d) located between the ester and thiol groups (∼1.5 ppm) and centered at 2.66 and 2.78 ppm respectively were slightly shifted followed by the formation of two new neighboring signals: a faint singlet (solid star) at ∼2.92 ppm and a strong quadruplet (white diamond) at 2.25–2.5 ppm assigned to the CH proton at position 2 and CH2 at position 10, respectively. Two novel signals at 0.98–1.20 ppm (white star) and 0.88–0.98 ppm (two doublets, solid diamond) ensuing from the coupling reactions were also detected corresponding to the methyl groups located at positions 7 and 9, respectively. The numbering terminology of each nucleus used for the assignment of 1H-NMR signals was adopted from a previous reference.9 Characterization by FT-Raman spectroscopy represented qualitatively in Fig. 5 indicates a significant reduction of the exocyclic ene band over that of the endocyclic ene with practically no vestigial amounts of thiol detected within the system after synthesis. This evidence supports the 1H-NMR results and is in conformity with previous kinetic studies (experimental and simulated).73 Indeed, the double-bond conversion ratio obtained experimentally yielded a value of ∼4.5 in favor of the exocyclic ene which is extraordinarily well explained by its homologous ratio obtained from simulation, ∼5.2, after a running time of 6 hours (see ESI† for more details). DMF-SEC analyses of the two resins derived from limonene shown in Fig. 6 confirm the effective presence of multifunctional high molecular weight oligomers as a result of the combined effect of primary and/or secondary coupling reactions acting on limonene alone and pending ene structures, respectively. Dispersity index (Đ)75 was 2.1 for resin 1, 2.7 for resin 2 and, 1.1 for the two individual macromonomers 4 and 5 which are characterized by the first bands appearing immediately after the flow rate marker. Dispersities of the first three individual bands corresponding to the oligomers took unit values although moving in the direction of low elution volumes an increasing loss of resolution (separation capacity) is verified. The relative proportion of individual compounds 4 (MWtheo/MWSEC = 0.76) and 5 (MWtheo/MWSEC = 0.81) including partially coupled macromonomers in each of the corresponding resins was 31.4 ± 4.4%; i.e., ∼25% of 4 or 5 and ∼6.3% of partial (incomplete) thiol–ene additions in both peaks if an averaged molecular weight ratio, MWtheo/MWSEC, of 0.8 is considered. This indicates that approximately 68.6% of the overall synthesis mixtures were constituted by primary (mono-addition) and secondary (di-addition) coupling products (complete and partial oligomers). In 25% of individual macromonomers 4 or 5, about 21% results from coupling at the exo-unsaturation and only ∼4.6% from coupling at the endo-unsaturation. In the same manner, of the 68.6% corresponding to ‘hyperbranched-like’ oligomers, about 56.5% of these result from coupling at the exo-unsaturation whereas only ∼12.6% corresponds to coupling at the endo-unsaturation. Since the simulation ratio of mono-to-di-addition products after 6 hours was [P]f/[D]f ≈ 5.6, with [P]f = [P1]f + [P2]f, and the double-bond conversion ratios obtained from 1H-NMR measurements and simulation agree relatively well with each other, than is anticipated that both resins will contain similar distribution of mono- and di-addition products. The fact that resin 2 yielded a slightly higher dispersity index over resin 1 associated with a much broader molecular weight distribution curve is very likely to be attributed to the influence of an extra mercapto propionate ester arm which increases the probability for high order coupling combinations in the first synthesis step. This effect is obviously translated into development of much higher viscosity for resin 2 when compared to resin 1 observed during and after synthesis. All these results demonstrate the effectiveness of the free-radical thiol–ene reaction in the preparation of multifunctional polydisperse macromonomer resins based on limonene suitable for the synthesis of polymeric thermoset materials.
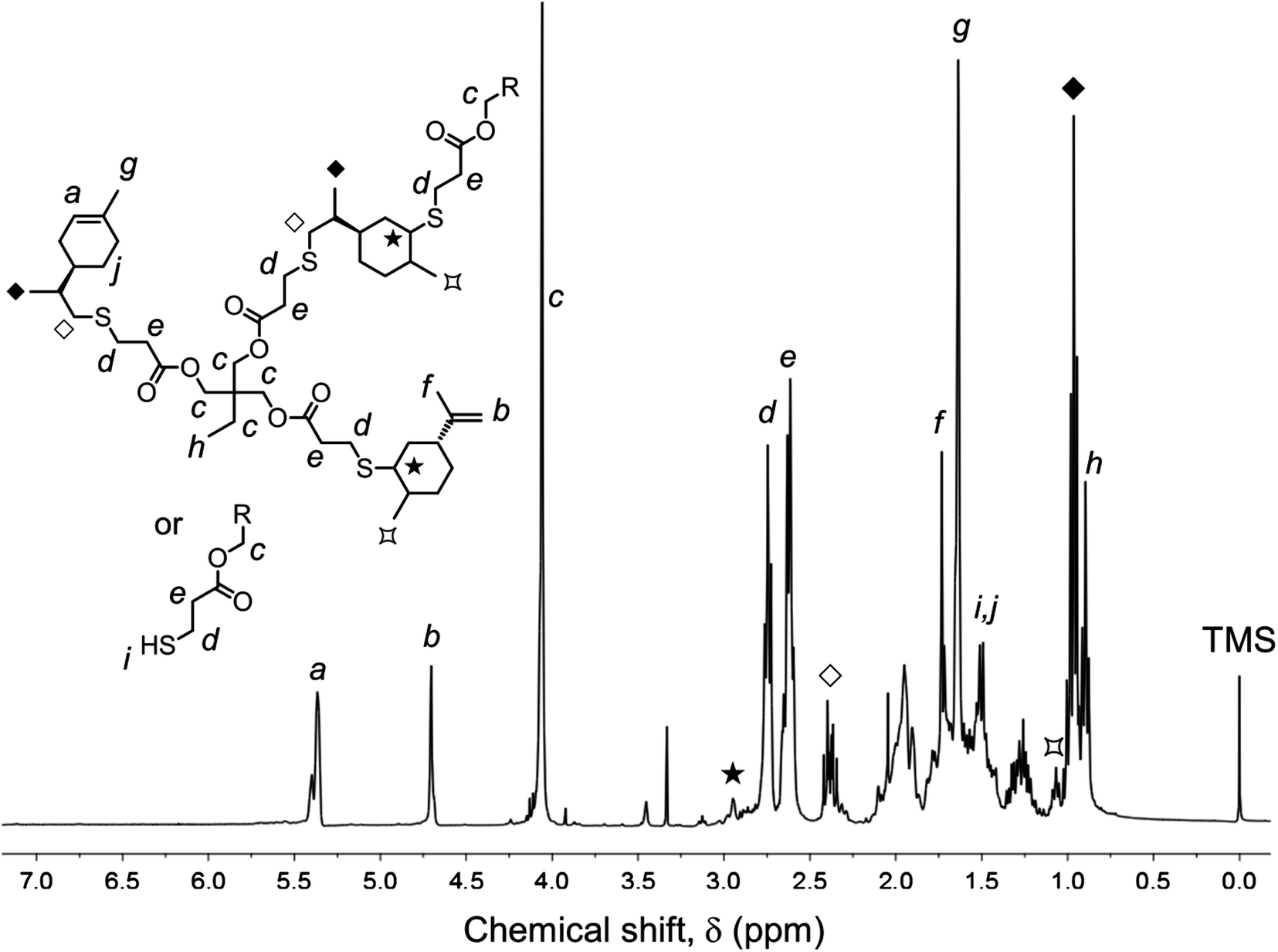 | ||
| Fig. 4 Representative 1H-NMR (400 MHz, CDCl3) spectrum of resin 1 (TE-C1) with principal signal assignments resulting from photoinduced thiol–ene coupling between compounds 1 and 2 reacted at an initial stoichiometry of 1:0.5. The synthesis reaction was conducted for a period of 6 hours at room temperature in EtOAc solution. Integration value of the ethyl ester proton signal (c) was used as a reference peak for the calculation of double-bond conversion. | ||
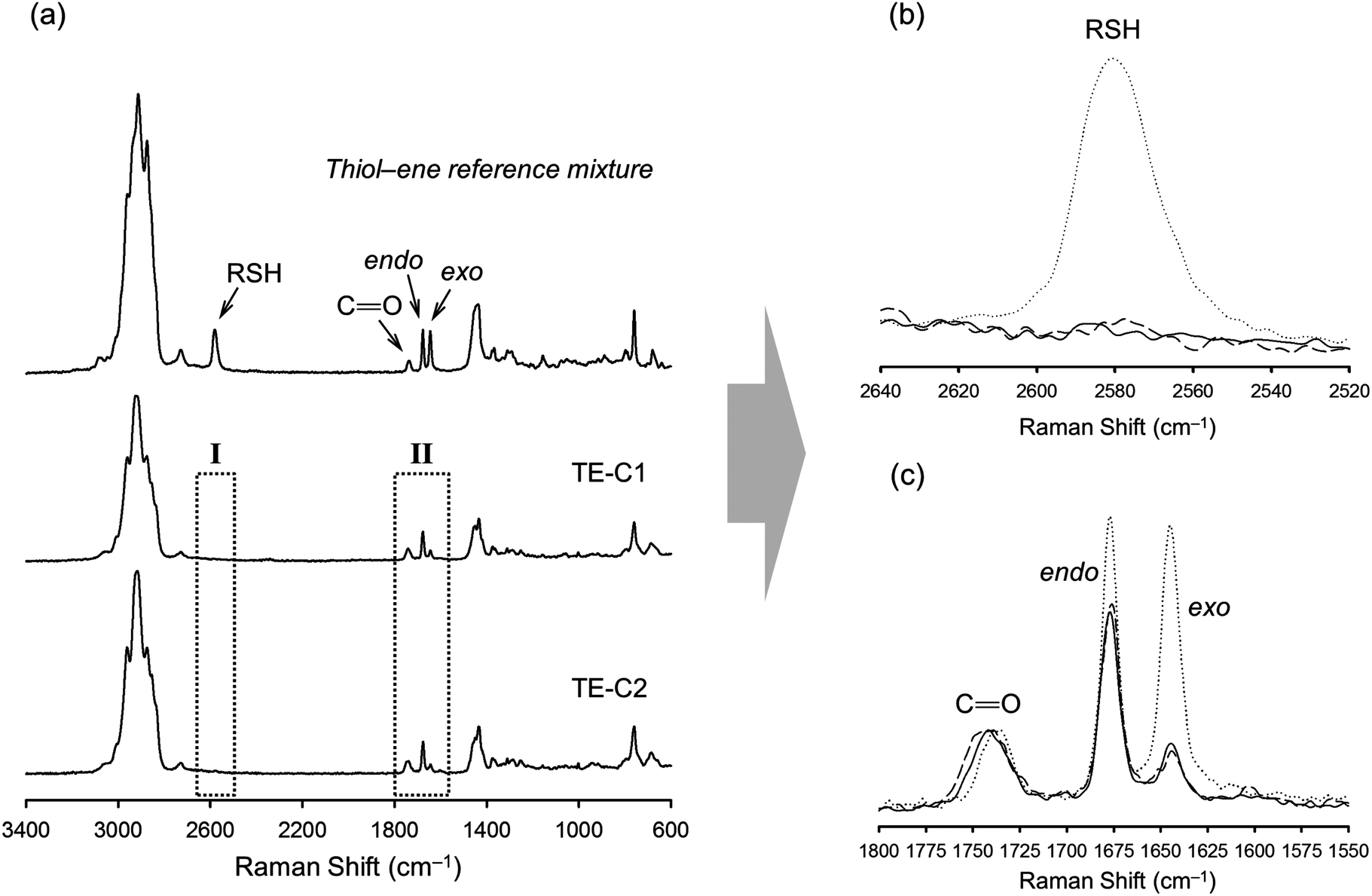 | ||
| Fig. 5 Normalized spectral FT-Raman profiles of finished synthesis products resulting from the photoinduced thiol–ene reaction on a 1:0.5 thiol–ene functional group stoichiometry. (a) Overall spectra in comparison to a reference mixture prepared with monothiol (iso-tridecyl 3-mercapto propionate) on a 1:1 thiol–ene mole ratio with respect to functional groups; (b) zoom-in of region I showing the full consumption of thiol, and (c) amplification of region II showing the disappearance of unsaturations with preferential coupling at the external double-bond position. The dotted line refers to the initial reference mixture, the solid line is from the TE-C1 product (resin 1), and the dashed line the spectrum obtained from the TE-C2 product (resin 2). The carbonyl group (CO, 1735 cm−1) was used as an internal reference band in the spectral normalization process. | ||
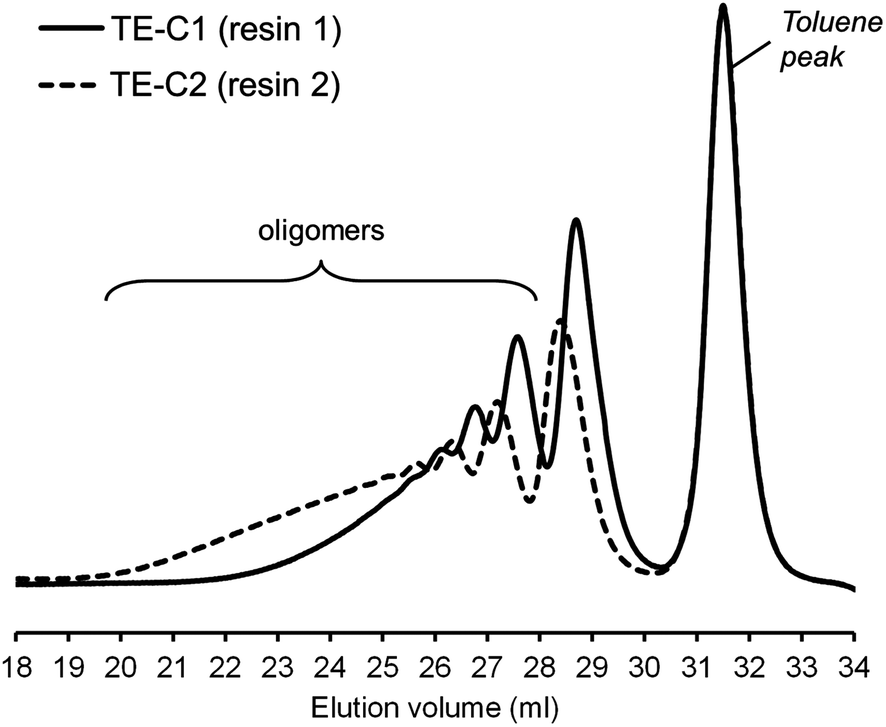 | ||
| Fig. 6 Superimposition of normalized DMF-SEC traces of the two multifunctional resins synthesized via photoinduced thiol–ene coupling. | ||
2.2. Thiol–ene crosslinking
Limonene represents an exceptional diene monomer to prepare bio-based thiol–ene networks. Not only we have observed that the two alkene structures preserve enough reactivity necessary for the thiol–ene coupling process72 but the existence of a slightly rigid cycloaliphatic ring may also turn advantageous at conferring extra mechanical strength to the final thermoset materials.One particular prerequisite involving typically known thiol–ene thermosets is the need for full stoichiometric control in order to ensure optimal thermo-mechanical properties with very few or no free reactive groups remaining within the material. In this case, equimolar mixtures of multifunctional thiol and ene components taking the forms xR1–(SH)m and yR2–(‘ene’)n require that xm = yn, where x and y represent the number of molecules of each monomer and m and n denote the number of thiol and ene functionalities per molecule, respectively. Alternatively, off-stoichiometric thiol–ene networks (OSTEs) can now be formulated by manipulating these parameters, ensuring that xm ≠ yn, as recently demonstrated in microfluidic device applications.76–78 This strategy allows for the fine-tuning of the viscoelastic properties and glass-transition temperatures of the final networks as well as other potential uses such as post-functionalization either at the film surface and/or in the bulk of the material. Here this novel concept is revisited by introducing in the reaction system small gradual amounts of thiol relative to the ene (xm/yn = 1.10, 1.25, and 1.45) to inspect its relative impact on the physicomechanical properties of the ensuing limonene-based thiol–ene thermosets.
A series of crosslinked thiol–ene networks were synthesized through photopolymerization of the two multifunctional macromonomer resins with the two polyfunctional thiol monomers. Different cross-over combinations of the monomers, with and without an excess of thiol functional groups at different levels, were prepared leading to a total of 16 different compositions. The nomenclature adopted is represented in Scheme 4 and Table 1. Network formation was accomplished by UV-irradiating liquid thiol–ene samples spread onto glass slides. All photocured thiol–ene films were obtained as clear (amorphous) homogeneous and flexible materials without any discernible thiol odor. However, films with low functionality (M1 + T3) resulted to be much stickier to the glass surface than those with higher functionality (M2 + T4) which were also tack-free. This difference in adhesion capacity may be ascribed to different crosslink densities achieved for the two materials. In general, the higher the crosslink density, the greater the stress build-up during cure,79 even if this occurs via a stepwise growth mechanism (after gelation), resulting in higher contraction energies developed at the film–substrate interface and peeling is facilitated.
 | ||
| Scheme 4 Multifunctional thiol–ene combinations and nomenclature adopted in the preparation of thermoset films based on limonene. | ||
| Formulation/sample | Stoichiometry | T g (°C) | T g (°C) | E′Rubb (MPa) | Height (MPa) of tan(δ) peak | tan(δ) fwhm (°C) |
|---|---|---|---|---|---|---|
| a Determined by DSC. b Determined by DMTA. | ||||||
| M1 + T3 | 1:1 |
−6.0 | 35.0 | 2.7 | 1.27 | 17.05 |
| 1:1.45 |
−8.0 | 24.6 | 0.4 | 2.10 | 17.04 | |
| M1 + T4 | 1:1 |
−2.0 | 43.1 | 5.3 | 0.73 | 33.83 |
| 1:1.45 |
−5.0 | 27.1 | 3.9 | 1.39 | 19.12 | |
| M2 + T3 | 1:1 |
5.0 | 30.3 | 5.9 | 0.98 | 19.71 |
| 1:1.45 |
−2.0 | 32.0 | 3.3 | 1.65 | 14.44 | |
| M2 + T4 | 1:1 |
12.4 | 37.8 | 8.9 | 0.73 | 28.43 |
| 1:1.45 |
11.5 | 45.8 | 8.3 | 1.19 | 16.78 | |
2.3. Thiol–ene network characterization
To gain a first assessment of the curing degree and density of crosslinks within the networks achieved after photopolymerization, sol-content determinations were performed by immersing the films in an appropriate organic solvent. Generally, the higher the crosslink density of the cured film the lower the fraction of the unbound material occluded within the network; and, therefore, the lower the sol-content. Leaching tests results are shown in Fig. 7. Thiol–ene films with incremental mole amounts of thiol functional groups resulted in higher fractions of soluble material trapped inside because once the network is chemically locked at the gel point the remaining unreacted portion becomes occluded into the network. Upon swelling the soluble fraction is released and a reduction in mass is detected gravimetrically. This was particularly evident from networks resulting from the two intermediate cross-combinations of high and low functionality components, M1 + 4T and M2 + T3, respectively. Conversely, the low (M1 + T3) and high (M2 + T4) functionality formulations afforded materials with relatively high and very low (or none) sol-content, respectively. An increase in functionality of the reacting system leads to a higher number of chemical crosslinks per unit volume of material and, therefore, the chances to extract any fraction of unreacted material occluded are lower. Indeed, it was observed that networks having the highest functionality will reach full crosslinking upon UV-cure regardless of the amount of thiol used and practically no leaching of the unbound fraction was detected. In contrast, materials having the lowest average number functionality showed much higher tendency to leach out upon swelling in a very good solvent, such as THF, even if full thiol–ene cure is achieved with respect to the limiting enes. It is remarkable the differences in extraction capacity observed between systems with and without the presence of one extra propionate ester arm (on average)! Statistically, the probability for di-coupling reactions between macromonomers and multifunctional thiols having the same average number functionality in reactive groups is higher for the combination M1 + T3 than for the combination M2 + T4 which dramatically reduces the possibility for higher order coupling reactions necessary for the creation of a large macromolecular cluster. Therefore, the chances for having a higher fraction of free or partially attached material within the network are higher for the first case as illustrated in Scheme 5. These results confirm the impact of the system co-functionality on the crosslinking density achieved after cure. One should note, however, that even in the worst case situation the fraction of unbound material is always lower than 25%. This indicates, at one instance, the importance of having good control over the composition mole ratio in the formulation of pure stoichiometric thiol–ene networks; and, in another instance, the properties of the final thermosets can be further modulated by adding small incremental amounts of thiol acting as a plasticizer.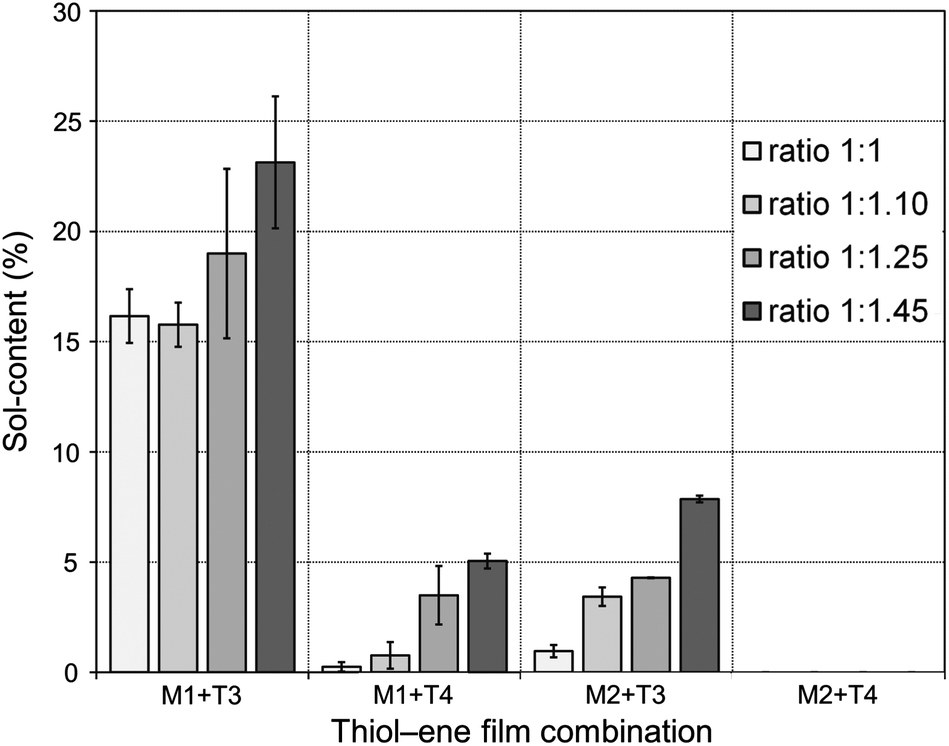 | ||
| Fig. 7 Sol-fraction of the thiol–ene films as determined gravimetrically. The inset ratios refer to proportional mole amounts of ene–thiol functional groups. | ||
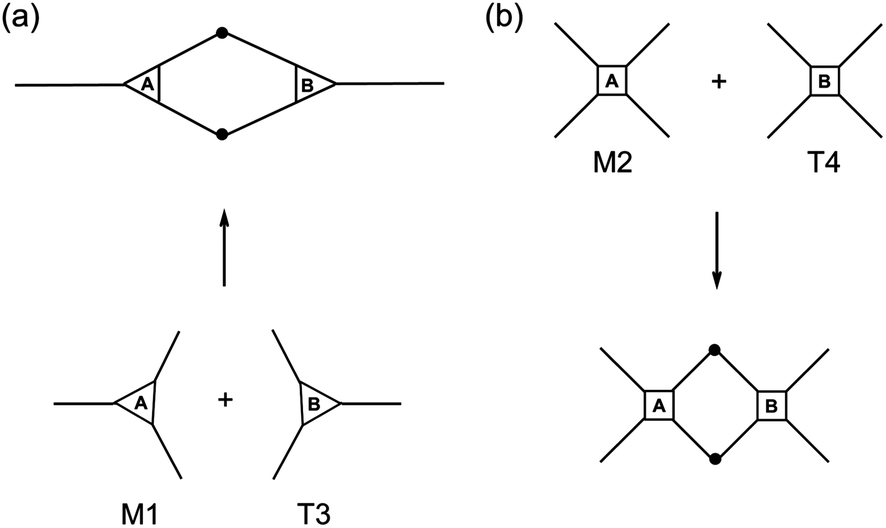 | ||
| Scheme 5 Theoretical effect of the monomer number functionality on the probability for higher order coupling reactions. | ||
FT-Raman spectroscopy was performed for the different formulations before and after UV-irradiation to determine the degree of curing which is correlated with the extent of crosslinking, since the two double bonds of limonene have shown to exhibit very low homopolymerization abilities.48,80 This is further supported by a recent study (2012) reporting on the free-radical homopolymerization of limonene using benzoyl peroxide (BPO) as an initiator at 85 °C in xylene with the reaction system following non-ideal kinetics and achieving low conversions due to both primary radical termination and degradative chain-transfer reactions.81 The results given in Fig. 8 illustrate well the extent of cure for the combination M1 + T3 which are also representative of other thiol–ene systems evaluated. The characteristic bands of thiol (∼2576 cm−1), exo-vinylidene (∼1645 cm−1) and endocyclic trisubstituted ene (∼1678 cm−1) chemical groups were all clearly visible before the cure. The ester carbonyl peak (1735 cm−1) remained unchanged throughout the reaction and was hence used as the internal standard. Practically full conversion of alkene functional groups was obtained for all cases in parallel with equivalent conversion of thiol groups. Residual thiol peaks correspond to unreacted fractions, as a consequence of an excess of thiol functional groups introduced within the system. Moving towards higher thiol–ene stoichiometries, increasingly more amounts of unreacted thiol become occluded onto the network resulting in higher peak heights. The extent and density of the chemical crosslinks achieved in combination with functionality and initial stoichiometry of the reacting system ultimately dictates the final thermal and mechanical properties of the fully cured thermosets. To determine more accurately the influence of these operational parameters, DSC and DMTA measurements were taken on the final films accounting for the representative thiol-ene stoichiometries of 1:1 and 1:1.45. The results presented in Table 1 and Fig. 9 provide a global unifying perspective of this effect.
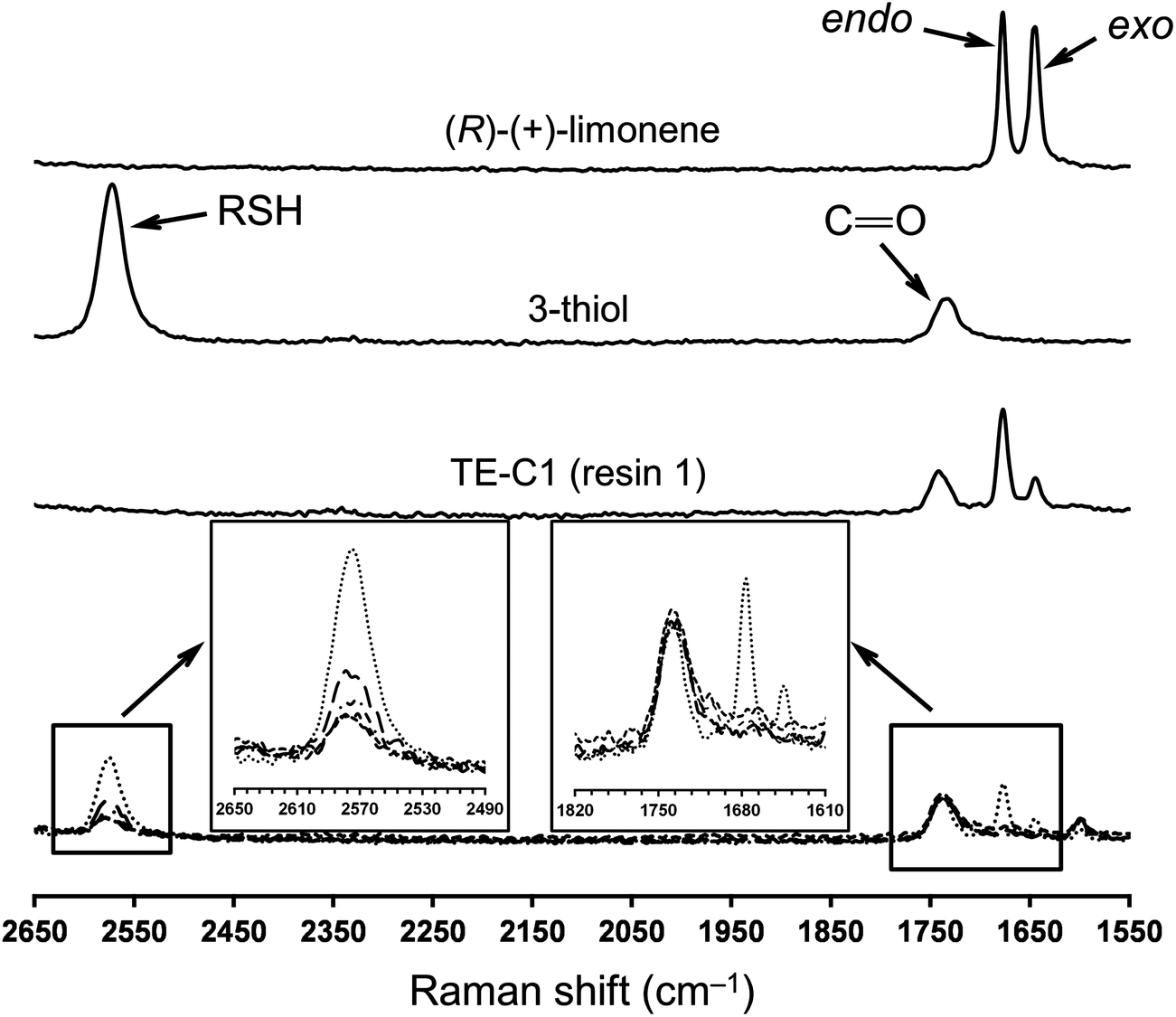 | ||
| Fig. 8 Spectral FT-Raman changes observed in thiol and alkene functional groups after UV-cure of the thiol–ene mixture comprised of M1 + T3. The dotted line refers to the initial thiol–ene mixture (before cure) with 45% relative excess of thiol functional groups. After the cure: long-dashed line (ratio, 1:1.45), dash-dotted line (ratio, 1:1.25), short-dashed line (ratio, 1:1.10), and mid-dashed line (ratio, 1:1). All spectra except limonene are normalized against the carbonyl ester band (CO, 1735 cm−1). | ||
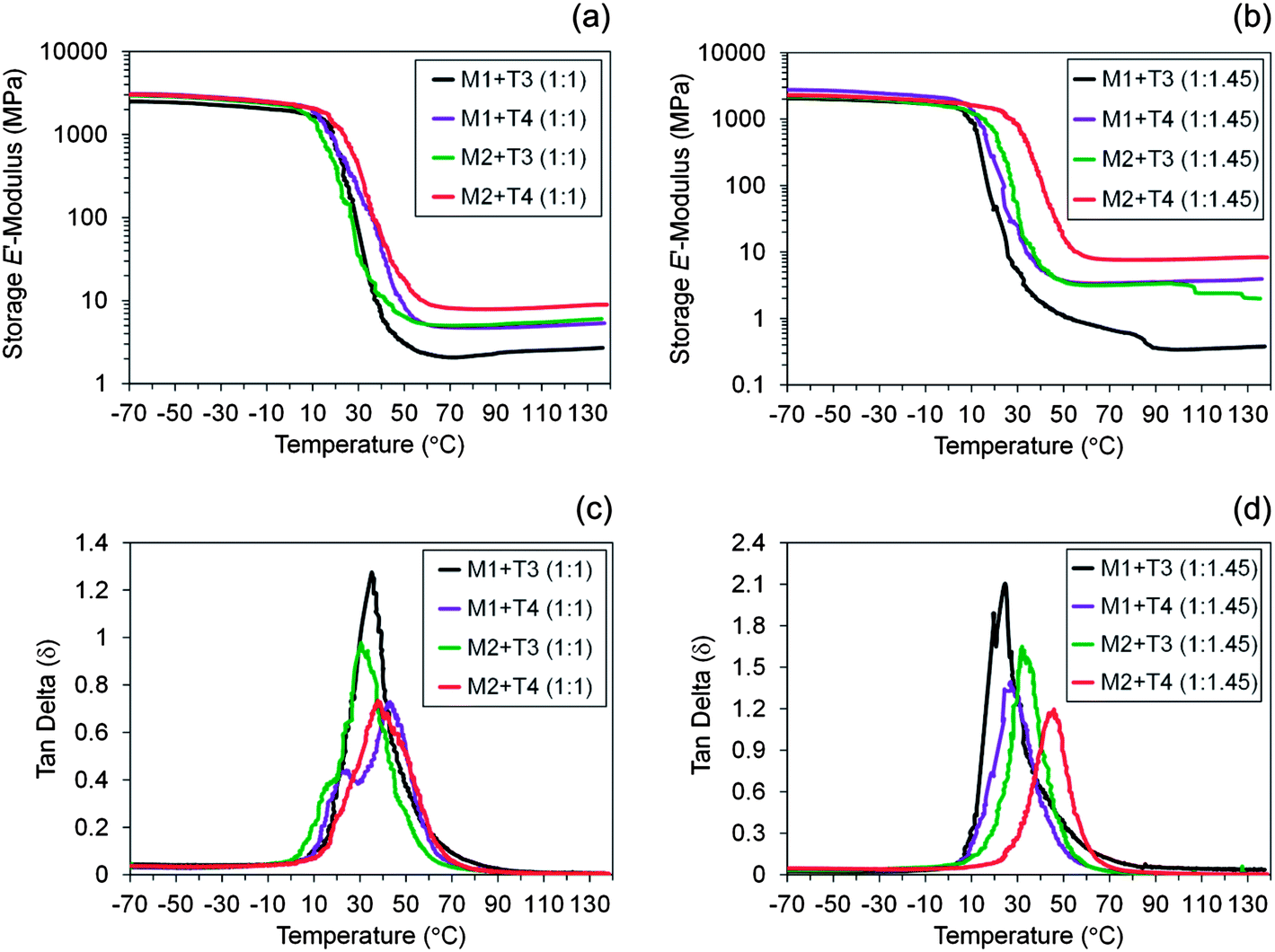 | ||
| Fig. 9 Influence of the type of thiol–ene formulation (combination and stoichiometry) on the thermo-viscoelastic properties of the final cured films. Plots (a) and (b) storage modulus versus temperature; and, plots (c) and (d) tanδ (E′′/E′) versus temperature. | ||
The thermal properties were first investigated by DSC. It can be observed that Tg increases when changing from low to high functionality materials due to an increase in crosslink density. The full range in different Tg values was 18.4 °C for pure stoichiometric films and 19.5 °C for off-stoichiometric materials, suggesting that an extra unreacted amount of thiol trapped within the networks works as a plasticizer softening the materials irrespective of network density. This is effectively confirmed by a decrease in Tg with excess of thiol which modifies the physical properties of the thermosets. To gain a better account of this effect DMTA measurements were performed because changes in modulus are usually much more pronounced than, for example, changes of heat capacity, cp, as in a DSC cycle.
DMTA results shown in plots (a) and (b) of Fig. 9 compare the evolution of the storage (elastic) modulus (E′) as a function of temperature, providing a complementary analysis to previous DSC results while allowing the thermal and viscoelastic properties of the final crosslinked networks to be determined. In DMTA, a sinusoidal mechanical stress is applied to the sample and the resulting sinusoidal strain is measured by the instrument. DMTA can be used to obtain the storage modulus (E′), glass-transition temperature (Tg) and determine the level of structural homogeneity. The crosslink density is normally determined by the storage E′-modulus at the rubbery plateau: the higher the relative level of this plateau, the higher the amount of crosslinks per unit volume or mass of material. The storage (elastic) E′-modulus gives a measure of the amount of energy stored by the material during one oscillating cycle, whereas the loss (viscous) E′′-modulus measures the amount of energy dissipated as heat during the same oscillating cycle. The ratio, of the loss and storage moduli, E′′/E′, affords the loss tangent (tanδ), a damping factor, which relates the energy dissipation relative to the energy stored within the material when the sample specimen is subjected to periodic deformation. The degree of homogeneity is usually quantified by the shape of the tanδ curve versus temperature determined by the full-width-at-half-maximum peak height (‘fwhm’), which also specifies the nominal range in Tg. A narrow and symmetrical (unimodal) curve characterizes a very homogeneous material with equal distribution of crosslinks throughout the entire network whereas a broad and/or asymmetrical (bimodal) curve describes a material with uneven distribution of crosslinks, disordered topology and even the presence of residual crystallinity. The intensity of the maximal tanδ value at the Tg is related to the degree of mobility of the chain segments between crosslinks at this particular temperature. Higher peak intensities reflect greater energy loss and, therefore, more viscous behavior; whereas lower intensities characterize more elastic behavior; i.e., more energy is stored within the material.
In both plots the storage modulus values depart from 2.6 ± 0.4 GPa (at −70 °C), which is typical of crosslinked polymers bellow Tg. As heating continues the modulus decreases gradually followed by a sudden glass-transition phase and then stabilizes at the rubbery plateau region attaining E′ values in the MPa range. The curves in plot (a) exhibit a narrow rubbery plateau when compared to film coatings cured with a higher amount of thiol depicted in plot (b). This was expected since an excess of free thiol intercalated throughout the networks reduces the crosslink density. In both cases the crosslink densities follow the order: M2 + T4 > M2 + T3 ≈ M1 + T4 > M1 + T3, which is remarkably well correlated with the previous sol-content results. The corresponding tanδ curves are displayed in plots (c) and (d). The shoulders observed at lower temperatures for the intermediary films M1 + T4 (1:1) and M2 + T3 (1:1) resulting in asymmetrical transition curves and larger widths at half-maximum peak heights are probably accredited to relaxation of chain segments for distinct crosslinked regions which indicates the presence of a non-uniform network structure. Intensities of the tanδ peaks also vary from each sample at the correspondent Tg values. Plot (d) shows a reduction in peak heights moving from very low functionality to very high functionality materials which is tightly connected to variations in crosslink density and Tg. A rise in Tg up to about 50 °C with increasing crosslink density results in lower energy loss during the thermal transition and, therefore, more elastic behavior (small heights) is observed; whereas, higher tanδ values indicate greater energy dissipation and increasing viscous behavior (large heights) as a consequence of the lower amount of crosslinks and reduced Tg. Therefore, a more dominating plasticizing (viscous) effect is observed in less densely crosslinked materials. A similar trend is observed in materials M1 + T3 (1:1) and M2 + T4 (1:1) represented in plot (c) but not for the intermediate networks for the reason pointed. Relatively equivalent tanδ curves of the two intermediate functionality materials, M1 + T4 (1:1.45) and M2 + T3 (1:1.45), with respect to height and Tg values were observed indicating that these networks exhibit comparable thermo-mechanical properties. In the former case the plasticizing effect can be excluded and the viscoelastic properties are determined exclusively by the crosslink density and regularity of the networks. The widths at half-maximum peak height give an account of the degree of homogeneity of the final networks with broader widths reflecting a material more heterogeneous. Interestingly, almost all materials with higher thiol content afforded narrower widths when compared to their stoichiometric counterparts although showing relatively higher tanδ heights. Possibly, this may be a consequence of the extra amount of thiol involved enabling a more ordered and regular build-up of the network.
It should be noted that the overall temperature difference between the different compositions is at the most in the range of 20 °C (Table 1). Using limonene as a cycloaliphatic monomer in comparison to conventional aliphatic systems demonstrates a more pronounced effect. The presence of the alicyclic ring in the final network increases the rigidity and significantly enhances the Tg in comparison to, for example, allyl ether based thiol–ene systems.82 This evidence further motivates the use of terpenes as renewable monomers in these types of organic systems as an ingenious green chemistry route to obtain higher Tg polymeric materials. This feature combined with off-stoichiometric manipulations in the thiol–ene composition and/or functionality, as well as the possibility to employ an assortment of thiol crosslinkers (e.g., thiol glycolates, triazine-based mercaptans and/or mercapto propionates) with different arm lengths and bulky core structures opens the door to tailor the thermo-mechanical properties of such organic thermoset systems without the need for resorting, for instance, to more complex hybrid inorganic–organic thiol–ene networks.83,84
3. Conclusions
The work outlined in this contribution offers a straightforward and efficient two-step synthesis procedure for the incorporation of D-limonene into thermoset polymeric materials by means of the free-radical thiol–ene coupling reaction. The first step takes advantage of the intrinsic difference in reactivity of the two unsaturations in limonene to form a branched oligomeric thermoset precursor. The choice of stoichiometry as well as thiol functionality allow for different resin structures to be formed. The thermoset precursor in the second step is crosslinked with a polythiol to form a final D-limonene-based poly(thioether) network with tunable properties depending on the initial stoichiometry and choice of thiol crosslinker. The efficiency of the thiol–ene coupling reaction enables high conversions without significant influence of side-reactions even for the highly substituted alkene present in the cycloaliphatic ring. The present study demonstrates how an appropriate synthesis strategy can be applied to terpene monomers allowing new bio-based thermoset polymers to be developed.Acknowledgements
We kindly acknowledge financial support from the Swedish Research Council (Vetenskapsrådet), grant # 621-2007-5723.References
- M. N. Belgacem and A. Gandini, in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2008, ch. 2, pp. 17–38 Search PubMed.
- A. Gandini, Green Chem., 2011, 13, 1061–1083 RSC.
- R. T. Mathers, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1–15 CrossRef CAS.
- P. A. Wilbon, F. Chu and C. Tang, Macromol. Rapid Commun., 2013, 34, 8–37 CrossRef CAS PubMed.
- K. Yao and C. Tang, Macromolecules, 2013, 46, 1689–1712 CrossRef CAS.
- R. T. Mathers and S. P. Lewis, Green Polym. Methods, 2011, 91–128 Search PubMed.
- P. S. Kulkarni, C. Brazinha, C. A. M. Afonso and J. G. Crespo, Green Chem., 2010, 12, 1990–1994 RSC.
- W. Schwab, C. Fuchs and F.-C. Huang, Eur. J. Lipid Sci. Technol., 2013, 115, 3–8 CrossRef CAS.
- M. Firdaus, L. Montero de Espinosa and M. A. R. Meier, Macromolecules, 2011, 44, 7253–7262 CrossRef CAS.
- M. Firdaus and M. A. R. Meier, Green Chem., 2013, 15, 370–380 RSC.
- F. M. Kerton and R. Marriot, in Alternative solvents for green chemistry, ed. J. H. Clark and G. A. Kraus, RSC Royal Society of Chemistry, Cambridge, UK, 1st edn, 2009, pp. 109–113 Search PubMed.
- E. A. Nonino, Perfum. Flavor., 1997, 22, 53–58 Search PubMed.
- W. J. Roberts and A. R. Day, J. Am. Chem. Soc., 1950, 72, 1226–1230 CrossRef CAS.
- M. Modena, R. B. Bates and C. S. Marvel, J. Polym. Sci., Part A: Gen. Pap., 1965, 3, 949–960 CrossRef CAS.
- T. Doiuchi, H. Yamaguchi and Y. Minoura, Eur. Polym. J., 1981, 17, 961–968 CrossRef CAS.
- J. Maslinska-Solich, T. Kupka, M. Kluczka and A. Solich, Macromol. Chem. Phys., 1994, 195, 1843–1850 CrossRef CAS.
- S. Sharma and A. K. Srivastava, Polym.-Plast. Technol. Eng., 2003, 42, 485–502 CrossRef CAS PubMed.
- S. Sharma and A. K. Srivastava, Des. Monomers Polym., 2006, 9, 503–516 CrossRef CAS PubMed.
- S. Sharma and A. K. Srivastava, J. Macromol. Sci., Pure Appl. Chem., 2003, A40, 593–603 CrossRef CAS PubMed.
- S. Sharma and A. K. Srivastava, Eur. Polym. J., 2004, 40, 2235–2240 CrossRef CAS PubMed.
- S. Sharma and A. K. Srivastava, Indian J. Chem. Technol., 2005, 12, 62–67 CAS.
- S. Sharma and A. K. Srivastava, J. Appl. Polym. Sci., 2007, 106, 2689–2695 CrossRef CAS.
- Y. Nakayama, Y. Sogo, Z. Cai and T. Shiono, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1223–1229 CrossRef CAS.
- F. J. B. Brum, F. N. Laux and M. M. C. Forte, Des. Monomers Polym., 2013, 16, 291–301 CrossRef CAS.
- H. Nakatani, T. Ichizyu, H. Miura and M. Terano, Polym. Int., 2010, 59, 1673–1682 CrossRef CAS.
- K. Xu, M. Chen, K. Zhang and J. Hu, Polymer, 2004, 45, 1133–1140 CrossRef CAS PubMed.
- R. T. Mathers, K. Damodaran, M. G. Rendos and M. S. Lavrich, Macromolecules, 2009, 42, 1512–1518 CrossRef CAS.
- R. T. Mathers, K. C. McMahon, K. Damodaran, C. J. Retarides and D. J. Kelley, Macromolecules, 2006, 39, 8982–8986 CrossRef CAS.
- R. T. Mathers and K. Damodaran, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3150–3165 CrossRef CAS.
- J. M. Delancey, M. D. Cavazza, M. G. Rendos, C. J. Ulisse, S. G. Palumbo and R. T. Mathers, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3719–3727 CrossRef CAS.
- K. Satoh, M. Matsuda, K. Nagai and M. Kamigaito, J. Am. Chem. Soc., 2010, 132, 10003–10005 CrossRef CAS PubMed.
- M. Matsuda, K. Satoh and M. Kamigaito, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1774–1785 CrossRef CAS.
- M. Matsuda, K. Satoh and M. Kamigaito, Macromolecules, 2013, 46, 5473–5482 CrossRef CAS.
- C. S. Marvel and L. E. Olson, J. Polym. Sci., 1957, 26, 23–28 CrossRef CAS.
- J. Yan, S. Ariyasivam, D. Weerasinghe, J. He, B. Chisholm, Z. Chen and D. Webster, Polym. Int., 2012, 61, 602–608 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 866–875 CrossRef CAS.
- J. Lalevee, N. Blanchard, M.-A. Tehfe, F. Morlet-Savary and J. P. Fouassier, Macromolecules, 2010, 43, 10191–10195 CrossRef CAS.
- M.-A. Tehfe, J. Lalevee, D. Gigmes and J. P. Fouassier, Macromolecules, 2010, 43, 1364–1370 CrossRef CAS.
- J. Lalevee, N. Blanchard, M.-A. Tehfe, M. Peter, F. Morlet-Savary and J. P. Fouassier, Macromol. Rapid Commun., 2011, 32, 917–920 CrossRef CAS PubMed.
- J. Lalevee, N. Blanchard, M.-A. Tehfe, M. Peter, F. Morlet-Savary, D. Gigmes and J. P. Fouassier, Polym. Chem., 2011, 2, 1986–1991 RSC.
- J. Lalevee and J. P. Fouassier, Polym. Chem., 2011, 2, 1107–1113 RSC.
- J. Lalevee, M.-A. Tehfe, F. Morlet-Savary, B. Graff, X. Allonas and J. P. Fouassier, Prog. Org. Coat., 2011, 70, 23–31 CrossRef CAS PubMed.
- M.-A. Tehfe, J. Lalevee, F. Morlet-Savary, B. Graff and J.-P. Fouassier, Macromolecules, 2011, 44, 8374–8379 CrossRef CAS.
- H. J. Park, C. Y. Ryu and J. V. Crivello, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 109–117 CrossRef CAS.
- J. A. Aikins and F. Williams, ACS Symp. Ser., 1985, 286, 335–359 CrossRef CAS PubMed.
- E. H. Nejad, A. Paoniasari, C. G. W. van Melis, C. E. Koning and R. Duchateau, Macromolecules, 2013, 46, 631–637 CrossRef CAS.
- H. Morinaga, Y. Kiyokawa, M. Kataoka, J. Masuda and D. Nagai, Polym. Bull., 2013, 70, 1113–1123 CrossRef CAS.
- M. Baehr, A. Bitto and R. Muelhaupt, Green Chem., 2012, 14, 1447–1454 RSC.
- L. Saikia, D. Srinivas and P. Ratnasamy, Appl. Catal., A, 2006, 309, 144–154 CrossRef CAS PubMed.
- L. Saikia, D. Srinivas and P. Ratnasamy, Microporous Mesoporous Mater., 2007, 104, 225–235 CrossRef CAS PubMed.
- D. Srinivas and P. Ratnasamy, Microporous Mesoporous Mater., 2007, 105, 170–180 CrossRef CAS PubMed.
- C. M. Byrne, S. D. Allen, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2004, 126, 11404–11405 CrossRef CAS PubMed.
- V. A. Morgunova, L. E. Nikitina, V. V. Plemenkov, M. G. Fazlyeva and Y. V. Chugunov, Chem. Nat. Compd., 1999, 35, 176–178 CrossRef CAS.
- K. Candela, R. Fellous, D. Joulain and R. Faure, Flavour Fragrance J., 2003, 18, 52–56 CrossRef CAS.
- J. F. Janes, I. M. Marr, N. Unwin, D. V. Banthorpe and A. Yusuf, Flavour Fragrance J., 1993, 8, 289–294 CrossRef CAS.
- O. Türünç, L. Montero de Espinosa, M. Firdaus and M. A. R. Meier, PMSE Prepr., 2010, 51, 724–725 Search PubMed.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- S. K. Kim and C. A. Guymon, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 465–475 CrossRef CAS.
- M. Uygun, M. A. Tasdelen and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 103–110 CrossRef CAS.
- H. Zheng, Y. Li, C. Zhou, Y. Li, W. Yang, W. Zhou, Z. Zuo and H. Liu, Chem.–Eur. J., 2011, 17, 2160–2167 CrossRef CAS PubMed.
- A. F. Jacobine, in Thiol–ene photopolymers, Elsevier, London, 1993, vol. 3, ch. 7, pp. 219–268 Search PubMed.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- M. Claudino, M. Johansson and M. Jonsson, Eur. Polym. J., 2010, 46, 2321–2332 CrossRef CAS PubMed.
- M. Claudino, I. van der Meulen, S. Trey, M. Jonsson, A. Heise and M. Johansson, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 16–24 CrossRef CAS.
- M. Shibata and M. Asano, J. Appl. Polym. Sci., 2013, 129, 301–309 CrossRef CAS.
- L. Lecamp, F. Houllier, B. Youssef and C. Bunel, Polymer, 2001, 42, 2727–2736 CrossRef CAS.
- C. Nilsson, E. Malmström, M. Johansson and S. M. Trey, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5815–5826 CrossRef CAS.
- C. Nilsson, E. Malmström, M. Johansson and S. M. Trey, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 589–601 CrossRef CAS.
- C. Nilsson, Ph. D. Thesis, KTH Royal Institute of Technology, 2008.
- M. Claudino, M. Jonsson and M. Johansson, RSC Adv., 2013, 3, 11021–11034 RSC.
- M. Claudino, M. Jonsson and M. Johansson, RSC Adv. Search PubMed , submitted.
- R. A. Clara, A. C. Gomez Marigliano and H. N. Solimo, J. Chem. Eng. Data, 2009, 54, 1087–1090 CrossRef CAS.
- R. F. T. Stepto, Polym. Int., 2010, 59, 23–24 CrossRef CAS.
- C. F. Carlborg, T. Haraldsson, K. Öberg, M. Malkoch and W. van der Wijngaart, Lab Chip, 2011, 11, 3136–3147 RSC.
- F. Saharil, C. F. Carlborg, T. Haraldsson and W. van der Wijngaart, Lab Chip, 2012, 12, 3032–3035 RSC.
- C. F. Carlborg, Ph. D. Thesis, KTH Royal Institute of Technology, 2011.
- J. Lange, Ph. D. Thesis, KTH Royal Institute of Technology, 1995.
- G. Odian, in Principles of Polymerization, ed. N. J. Boboken, John Wiley & Sons, Inc., New Jersey, 4th edn, 2004, ch. 3, pp. 275–279 Search PubMed.
- A. Singh and M. Kamal, J. Appl. Polym. Sci., 2012, 125, 1456–1459 CrossRef CAS.
- I. Carlsson, A. Harden, S. Lundmark, A. Manea, N. Rehnberg and L. Svensson, ACS Symp. Ser., 2003, 847, 65–75 CrossRef CAS PubMed.
- K. M. Schreck, D. Leung and C. N. Bowman, Macromolecules, 2011, 44, 7520–7529 CrossRef CAS PubMed.
- B. J. Sparks, T. J. Kuchera, M. J. Jungman, A. D. Richardson, D. A. Savin, S. Hait, J. Lichtenhan, M. F. Striegel and D. L. Patton, J. Mater. Chem., 2012, 22, 3817–3824 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3py01302b |
| This journal is © The Royal Society of Chemistry 2014 |