UVA1 is skin deep: molecular and clinical implications†
Angela
Tewari
*a,
Mette M. L.
Grage
b,
Graham I.
Harrison
a,
Robert
Sarkany
a and
Antony R.
Young
a
aKing's College London (KCL), King's College London School of Medicine, Division of Genetics and Molecular Medicine, St John's Institute of Dermatology, London, UK. E-mail: angela.tewari@kcl.ac.uk
bDanish Meteorological Institute, Lyngbyvej 100-2100, Copenhagen, Denmark
First published on 22nd November 2012
Abstract
Long wavelength UVA1 (340–400 nm) is the main component of terrestrial UVR and is increasingly used in skin phototherapy. Its damage to critical biomolecules such as DNA has been widely attributed to its ability to generate reactive oxygen species (ROS) via other chromophores. However recent studies in vitro and in vivo have shown that UVA1 has a specific ability to generate cyclobutane pyrimidine dimers (CPD), especially thymine dimers (T<>T), and that this is probably due to direct absorption of UVR. The CPD has been implicated in many aspects of skin cancer. Measuring UVB-induced CPD in the epidermis and dermis in vivo shows that, as expected, the skin attenuates UVB. In contrast, our data show that this is not the case with UVA1: in fact there is more damage with increased skin depth. This suggests that the basal layer, which contains keratinocyte stem cells and melanocytes, is more vulnerable to the carcinogenic effects of UVA1 than would be predicted by mouse models. These data support the continuing trend for better UVA1 protection by sunscreens.
Erythema as a biological measure of exposure to solar UVR
Ultraviolet radiation (UVR) is electromagnetic radiation spanning 100–400 nm and is officially divided into UVC (100–280 nm), UVB (280–315 nm) and UVA (315–400 nm), with UVA being sub-classified into UVA2 (315–340 nm) and UVA1 (340–400 nm). However, a UVB–UVA cutoff at 320 nm is widely used in photodermatology. Since stratospheric ozone absorbs all UVR below ∼295 nm, terrestrial sunlight contains only UVB (5–10% of total UVR) and UVA (90–95% of total UVR), the latter being primarily UVA1 (∼75%). The precise ratio of UVB to UVA varies with the solar zenith angle that is determined by latitude, season and time of day. This is demonstrated by Fig. 1a, which shows the monthly variation of UVA and UVB irradiance at Chilton, UK (51.6°N), in which a radiative transfer model (SMARTS)1 generated the data taking total ozone into account.2 Similarly, Fig. 1b shows the daily variation of UVA and UVB taken in mid-summer (21st June) and mid-winter (21st December) at the same location.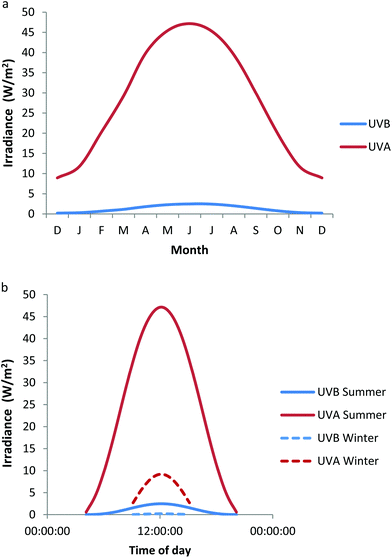 | ||
| Fig. 1 Variation of UVB and UVA irradiance at Chilton, UK (a) monthly at noon on the 21st day of each month (b) daily at summer and winter solstices. These data have been modeled taking total ozone into account.2 | ||
Erythema is widely used as a clinical measure of UVR exposure. Thus, the data in Fig. 1a and 1b have been weighted with the CIE erythema action spectrum3 to produce erythemally effective energy (EEE) as shown in Fig. 2a and 2b respectively. Fig. 2a shows that in mid-summer, UVA at temperate latitudes contributes about 25% of EEE of solar UVR. However, this decreases either side of this period such that the ratio is approximately 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 from December through to January. Fig. 2b shows that UVA contributes to a maximum of about 25% of EEE at noon in mid-summer, but that in mid-winter the relative contributions of UVB and UVA to erythema are independent of time of day, i.e. erythemal exposure is achieved equally by UVB and UVA. In this context, it must be remembered that considerable molecular and cellular damage can be done in human skin in vivo by single and repeated sub-erythemal exposure,4 irrespective of spectrum. Furthermore, suppression of the skin's immune function can occur with about 25% of the minimal erythema dose (MED).5
:50 from December through to January. Fig. 2b shows that UVA contributes to a maximum of about 25% of EEE at noon in mid-summer, but that in mid-winter the relative contributions of UVB and UVA to erythema are independent of time of day, i.e. erythemal exposure is achieved equally by UVB and UVA. In this context, it must be remembered that considerable molecular and cellular damage can be done in human skin in vivo by single and repeated sub-erythemal exposure,4 irrespective of spectrum. Furthermore, suppression of the skin's immune function can occur with about 25% of the minimal erythema dose (MED).5
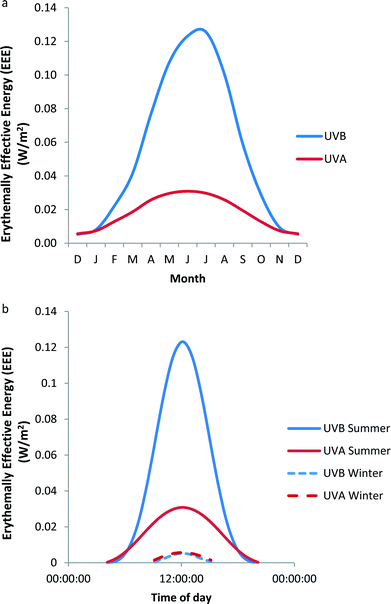 | ||
| Fig. 2 Variation of UVB and UVA EEE at Chilton, UK (a) monthly at noon on the 21st day of each month (b) daily at summer and winter solstices. These data are based on the data in Fig. 1. | ||
The standard erythema dose (SED)6 is increasingly used as a measure of human exposure, especially in epidemiological studies. Unlike the MED, the SED is independent of individual UVR sensitivity. During mid-summer in Chilton at midday it would take approximately 9.5 minutes to receive a full UVR spectrum SED (unpublished data) which is approximately 1/3 of a minimal erythema dose (MED) in a skin type I/II person.7 It would take approximately 4 times longer to get an SED from the UVA component alone. These data show considerable potential exposure to UVA whether determined by physical or erythemal exposure parameters.
UVR and skin cancer
There has been a dramatic increase in incidence of skin cancers in Europe and the USA, especially malignant melanoma (MM), in the past 25 years.8,9 This has increased the urgency of identifying and protecting against the causative factors of which UVR has emerged as the most important. This conclusion has resulted from classic epidemiology and more recently, by molecular epidemiology that identifies UVR-specific mutations in skin cancers. The focus has been largely on UVB because of a combination of action spectrum data from a mouse model of squamous cell carcinoma (SCC)10,11 and the well-established finding that UVB readily induces the production of DNA photoproducts (see section below) associated with skin cancer.12–14 However, a combination of factors has arisen in recent years that implies that UVA plays a more significant role than was previously thought. First, epidemiological data have shown that MM shows a better latitude correlation with UVA than with UVB15,16 and that the use of artificial tanning devices (which mainly emit UVA) increases the risk of skin cancer, especially MM.17 UVA artificial tanning devices are widely used,18 especially by the young, and use under the age of 35 confers the greatest risk.19 Second, it has become increasingly clear that UVA1 is not photochemically or biologically inactive, and that UVA1 is a potent inducer of reactive oxygen species (ROS),20 which are thought to play a role in skin cancer.21,22 The question of whether UVA1 is a significant carcinogen has become more important as exposure of the human population to UVA has significantly increased. Until relatively recently, sunscreens were primarily UVB absorbing products that enabled prolonged sunbathing without much protection against UVA, paradoxically increasing exposure to UVA in sunbathers.23,24 This can occur to the extent that erythema from repeated sub-erythemal exposure can be caused by UVA.25 Finally UVA1 phototherapy has been developed in the last 20 years and is used to treat a variety of dermatological diseases26 with comparable efficacy to other types of phototherapy (TLO1 or narrowband UVB) for some conditions. Although the action spectrum for induction of SCC in mice is predominantly in the UVB waveband, the action spectrum for induction of MM in mammalian skin is still unknown. Mechanistically, the proven mutagenicity and carcinogenicity of UVB has led to the dogma that UVB is the only significant cause of skin cancer.The rest of this review will focus on what is currently known about UVA1 photodamage of DNA, the significance of this damage in terms of mutagenicity and carcinogenicity, and the clinical and public health implications of these new findings.
UVR-induced DNA damage
DNA is susceptible to modification by UVR which may result in mutation if not successfully repaired. There are many different types of DNA photolesions, which show wavelength dependence in their formation. Some are formed by the direct absorption of UVR and others are formed indirectly as described below.(i) Di-pyrimidine photolesions
UVR is absorbed by skin chromophores27 and DNA pyrimidine bases are primarily chromophores for UVB (thymine, cytosine and the minor 5 methylcytosine),28 but also weakly absorb UVA. The direct excitation of these nucleobases by UVB in an oxygen independent manner induces two main classes of photolesions in the DNA of skin fibroblasts and keratinocytes.29 These are cyclobutane pyrimidine dimers (CPD) and 6-4 pyrimidine pyrimidone adducts (6-4PP) (which make up 65% and 35% of the UVB induced DNA lesions respectively.30,31 The CPD arises by linkage of two adjacent pyrimidine bases of which the T<>T type is the most common.32Until recently the presence of CPD-induced “fingerprint” mutations (e.g. C→T) in skin cancers was taken as a priori evidence for a causal role for UVB, even though it has been known for some time that UVA, including UVA1, induces CPD in human skin in vivo.33,34 More recent studies have confirmed that UVA1 induces CPD in vitro, ex vivo and in vivo32,35 but not 6-4PP,32,35 these being predominantly (but not exclusively) T<>T at frequencies comparable to or exceeding that of oxidative damage to DNA (see below) but at several orders of magnitude lower efficiency than UVB. The dogma that C→T mutations are only caused by UVB has been recently challenged.36
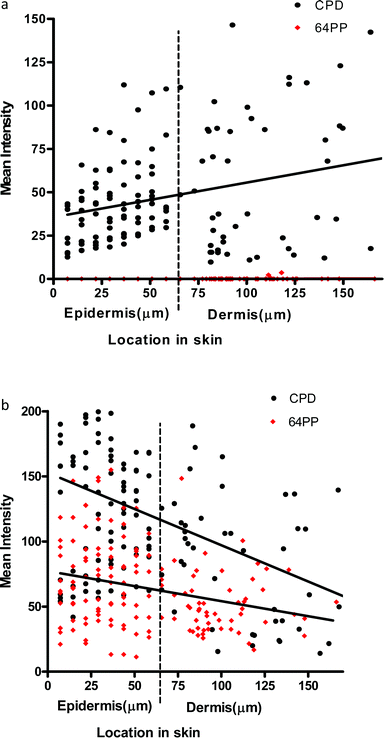 | ||
| Fig. 3 (a) Linear regression for UVA1-induced CPD (p = 0.0006 for slope) and 6-4PP (p = 0.56 for slope) in the epidermis and dermis in vivo immediately after a 3MED exposure. More CPD appear to be induced as depth increases through the epidermis and dermis. Note the lack of UVA1 induced 6-4PP. (b) Linear regression for UVB (300 nm)-induced CPD (p = <0.0001 for slope) and 6-4PP (p = 0.0006 for slope) in the epidermis and dermis in vivo immediately after a 3MED exposure. | ||
UVA1 is less well absorbed by the upper layers of the epidermis than UVB and hence can penetrate deeper into tissue, but the reasons for the “reverse attenuation” with UVA1 are not known. One possibility is that UVA1 photons are preferentially scattered by the epidermis and dermis, in a forward or backward direction, rather than absorbed by chromophores.37,38 Back scattering results in remittance (a type of “reflection”), which provides additional opportunity for chromophore absorption during the return pathway. Dermal remittance increases between 300 and 400 nm.37 Thus, it is possible that the higher number of UVA1-induced CPD seen in the basal layer is due to dermal back scatter (e.g., from collagen), as well as epidermal forward scatter.39
Although we have only measured CPD and 6-4PP, these can be used as a surrogate for UVR penetration into skin and can inform about the possibility of generating other types of DNA and bio-molecule damage. Overall, our data demonstrate that the basal layer, which contains melanocytes and proliferating keratinocytes, is especially sensitive to UVA1 induced DNA damage, and they challenge the paradigm that UVB is the only important spectral region for skin cancer. This is supported by studies on engineered human skin show that, in contrast to UVB, UVA-induced mutations were mainly located in the basal layer.40
(ii) Oxidative damage
DNA damage also occurs indirectly through the formation of ROS.44,47 These are generated by the absorption of UVA by non-DNA endogenous chromophores (photosensitizers) such as porphyrins, flavins and NADH/NADPH44,48 which can be excited to a triplet state. Damage can occur via a type 1 photosensitization reaction which can cause DNA strand breaks by the generation of superoxide anions, hydrogen peroxide and hydroxyl radicals, the latter being very reactive with all DNA bases.47 There are also type 2 photosensitization reactions where energy is transferred to molecular oxygen giving rise to singlet O2 (1O2). This species specifically reacts with guanine residues49 producing 8-oxo-7,8-dihydro-2′deoxyguanosine (8oxodG) and is thought to be important in UVA induced cell death.50 Few studies have reported the detection of UVR-induced oxidative damage to DNA in vivo, but this has been reported in mice51 and in human skin.22,52 8oxodG has also been reported in urine after the exposure of human skin to UVA.53When UVB (300 nm) and UVA1 doses are given to produce comparable levels of erythema in human skin (1MED ∼ 30 mJ cm−2 and ∼50 J cm−2 respectively), UVA1 induces 3–4 fold fewer T<>T than UVB implying other non-DNA chromophores have an important role in UVA1 erythema, and it is possible that this is mediated via oxidative damage to DNA. Unlike UVB erythema, UVA erythema is oxygen dependent,54 implying a role for ROS in UVA erythema. Different mechanisms for UVB and UVA1 erythema are supported by a lack of correlation between UVB and UVA1 induced MED on the same individuals (Fig. 4). A detailed laser action spectrum for human erythema showed a peak at 300 nm but also identified a minor UVA1 peak (at approximately 360 nm)55 which also suggests that more than one chromophore is important (Fig. 5).
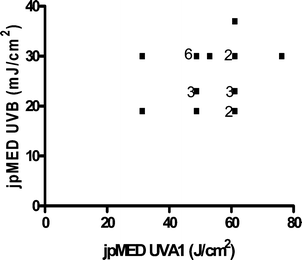 | ||
| Fig. 4 Lack of relationship between a given individual's UVB and UVA1 MED (n = 22 with some data points overlapping). jp = just perceptible. | ||
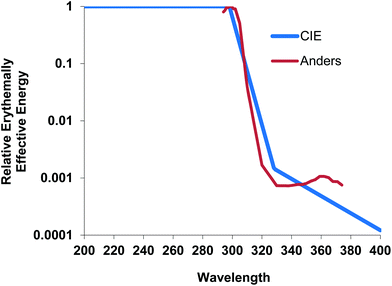 | ||
| Fig. 5 Erythema action spectrum55 showing small peak at approximately 360 nm (raw data obtained from the late Dr Anders) normalized at 300 nm with the CIE action spectrum.3 | ||
Consequences of UVR induced DNA damage in human skin
(i) Non-genetic
The action spectra for CPD and erythema (inflammation) are similar (especially between 280–340 nm) suggesting that DNA is a chromophore for erythema33 which is also supported in studies in the marsupial Monodelphis domestica in which the MED was reduced by a factor of approximately four after enhanced CPD repair induced by UVA activation of endogenous photolyase which is not present in placental mammals.56 In other words the CPD triggers inflammation of the skin. Inflammation plays a role in many types of cancer57,58 by fostering tumour progression, and UVR-induced erythema is a risk factor for MM and possibly BCC.59 Persistent erythema is also found in xeroderma pigmentosum patients who are unable to repair CPD and who are also 2000–10000 times more likely to present with melanomas and non-melanoma type skin cancers.12,60
Apart from erythema, the CPD has been shown to trigger pro-inflammatory cytokine release and immunosuppression that are thought to play a role in skin cancer.61,62 We have recently shown that 3 SED, irrespective of spectrum (including UVA1), is predictive of the suppression of the sensitization phase of the contact hypersensitivity (CHS) response, which is regarded as a model for the immunological events in non-melanoma skin cancer (manuscript in preparation). In conclusion, there is considerable evidence that DNA damage, especially the CPD, causes inflammation/immunosuppression that may play a role in skin cancer. Recently published work also suggested that erythema is predictive of photoageing induced by matrix metalloproteinase 1 (MMP1) via the CPD.63
Oxidative damage and immunosuppression
The CPD and urocanic acid have been identified as possible chromophores for UVR-induced immunosuppression.64,65 However several studies in the mouse66,67 have shown that antioxidants can inhibit UVR-induced immunosuppression, which suggests other possibly UVA absorbing chromophores. In addition, antioxidants have been shown to inhibit mouse photocarcinogenesis.68,69 However, there is no direct evidence that oxidative damage to DNA is a trigger for UVA-induced immunosuppression.(ii) Genetic
Faithful NER of CPD is crucial for the prevention of skin cancer. One study on ex vivo skin suggested poorer repair of UVA1-induced CPD compared with those induced by UVB32 but our data did not support this when the epidermis was assessed in total.35 However, a subsequent analysis of repair in the basal layer alone suggests that repair of UVA induced CPD is less efficient than that with UVB (unpublished). UVA can induce a G2/S block in melanocytes and melanoma cells suggesting that the DNA damage induced by UVA can have potent effects as they may not be easily repaired at the basal epidermis.75 Recent work76,77 suggests that UVB initiates melanoma and that both UVB and UVA are involved in the progression of the disease.
Oxidative damage to DNA results in what have been termed “UVA fingerprint mutations” via G:C→A:T transversions which have been detected in human SCC and actinic keratosis, especially in the basal layer.22 These were found to be more prominent than C→T, transitions21 but p53 gene mutations do not contain UVA specific T and G transversions implying that although UVA causes oxidative DNA damage, such lesions may not contribute to the generation of p53 mutations.82 Also, the UVA mutation spectrum in mammalian cells does not exhibit a predominance of G:C→T:A transversions83 which, combined with the lack of increase in mutation rate in cells deficient in repair of 8oxodG,84 suggests that other lesions are involved in UVA mutagenesis. New work in mice85 examined UVB and UVA induced DNA damage and their contribution to melanoma. UVB induced melanoma was independent of melanin, but UVA-induced melanoma required melanin. UVA, but not UVB, induced oxidative DNA damage in melanocytes of pigmented mice. These data support other work that suggests that melanocytes are particularly sensitive to UVA.86
Clinical implications of UVA1 DNA photodamage in the skin
As discussed earlier, potential exposure to solar UVA1 is substantial, even in temperate UK climates. Further south, on a summer's day in the south of France the maximum ambient daily UVR dose is ∼22MED and the maximum UVA dose is ∼137 J cm−2,23 of which the majority is UVA1. The accumulation of high UVA1 doses also occurs during the treatment of dermatological skin disorders such as morphea87,88 and atopic eczema89 where on average patients receive cumulative doses of approximately 900 J cm−2 UVA1 (for example 50 J cm−2 3 times a week for 6–8 weeks) and some conditions such as atopic eczema of the hands or cutaneous T-cell lymphoma receiving single doses of up to 100 J cm−2 UVA1.90 The long-term consequences of such exposures are not known. We have shown that repeated daily exposure to sub-erythemal doses of solar simulated radiation (SSR) results in an accumulation of CPD4,25 probably because repair of this lesion is relatively slow. To the best of our knowledge, the effects of repeated UVA1 sub-erythemal exposure on CPD has not been studied.A recent study91 demonstrated that uncontrolled activation of a major mediator of the Hedgehog pathway signalling can lead to the development of nodular BCC from hair follicle stem cells and superficial BCC-like tumours from the basal cell compartment of the epidermis. Such cells may also thus be vulnerable to UVA1 because of its skin penetration properties.
Increasing recognition of the possible long-term harmful effects of solar UVA exposure and development of better UVA sunscreens is now required by regulatory bodies in Europe and the USA.23 Recent government regulation in the UK and other countries and prohibits the use of sunbeds for those under 18 years of age.
Conclusions
Our data, and those of others, show that the basal epidermis of human skin is especially vulnerable to UVA1-induced CPD, and ROS-induced mutations and therefore suggests that the carcinogenic potential of this spectral region, which is the major UVR component of sunlight, may be much greater than has been estimated for SCC in the albino mouse, which has a very thin epidermis and unlikely to show much evidence of differential UVA1 induced damage in the epidermis.The UVA1 sensitive basal layer is the location of proliferating keratinocytes and melanocytes. The action spectrum for MM in mammalian skin is unknown, but there are epidemiological data to suggest a greater role for UVA for MM compared to non-melanoma skin cancer, furthermore recent mouse data suggest that melanocyte DNA is especially sensitive to oxidative damage.85 Overall, the data support the use of broad-spectrum sunscreen protection.
Acknowledgements
The research was funded/supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Guy's and St Thomas’ NHS Foundation Trust and King's College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. Angela Tewari was awarded a medical research council (MRC)/British skin foundation (BSF)/British association of dermatology (BAD) clinical training fellowship during which the above research was completed. This research has also received funding from the European Community's Seventh Framework Programme (FP7/2007–2013) under grant agreement n° 227020.References
- C. A. Gueymard, Parameterized transmittance model for direct beam and circumsolar spectral irradiance, Sol. Energy, 1995, 71(5), 325–346 CrossRef; C. A. Gueymard, SMARTS, A Simple Model of the Atmospheric Radiative Transfer of Sunshine: Algorithms and Performance Assessment, Technical Paper FSEC-PF-270-95, Florida Solar Energy Center, Cocoa, FL, 1995 Search PubMed.
- A. R. D. Smedley, et al., Total ozone and surface UV trends in the United Kingdom: 1979–2008, Int. J. Climatol., 2012, 32(3), 338–346 CrossRef.
- CIE Standard. Erythema reference action spectrum and standard erythema dose. 1998(CIE S 007/E-1998, Vienna: Commision Internationale de l'Eclairage).
- S. Seite, et al., Photodamage to human skin by suberythemal exposure to solar ultraviolet radiation can be attenuated by sunscreens: a review, Br. J. Dermatol., 2010, 163(5), 903–914 CrossRef CAS.
- D. A. Kelly, et al., Sensitivity to sunburn is associated with susceptibility to ultraviolet radiation-induced suppression of cutaneous cell-mediated immunity, J. Exp. Med., 2000, 191(3), 561–566 CrossRef CAS.
- B. L. Diffey, et al., The standard erythema dose: a new photobiological concept, Photodermatol. Photoimmunol. Photomed., 1997, 13(1–2), 64–66 CrossRef CAS.
- G. I. Harrison and A. R. Young, Ultraviolet radiation-induced erythema in human skin, Methods, 2002, 28(1), 14–19 CrossRef CAS.
- D. E. Godar, Worldwide increasing incidences of cutaneous malignant melanoma, J. Skin Cancer, 2011, 2011, 858425 Search PubMed.
- CRUK Malignant Melanoma Trends in European Age-Standardised Incident Rates 1975–2010, http://www.cancerresearchuk.org/cancer-info/cancerstats/types/skin/incidence/uk-skin-cancer-incidence-statistics.
- F. R. de Gruijl, et al., Wavelength dependence of skin cancer induction by ultraviolet irradiation of albino hairless mice, Cancer Res., 1993, 53(1), 53–60 CAS.
- A. de Laat, J. C. van der Leun and F. R. de Gruijl, Carcinogenesis induced by UVA (365 nm) radiation: the dose–time dependence of tumor formation in hairless mice, Carcinogenesis, 1997, 18(5), 1013–1020 CrossRef CAS.
- J. J. DiGiovanna and K. H. Kraemer, Shining a light on xeroderma pigmentosum, J. Invest. Dermatol., 2012, 132(3 Pt 2), 785–796 CrossRef CAS.
- P. T. Bradford, et al., Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair, J. Med. Genet., 2011, 48(3), 168–176 CrossRef.
- N. M. Wikonkal and D. E. Brash, Ultraviolet radiation induced signature mutations in photocarcinogenesis, J. Invest. Dermatol. Symp. Proc., 1999, 4(1), 6–10 CrossRef CAS.
- J. Moan, A. C. Porojnicu and A. Dahlback, Ultraviolet radiation and malignant melanoma, Adv. Exp. Med. Biol., 2008, 624, 104–116 CrossRef.
- J. Moan, et al., UVA, UVB and incidence of cutaneous malignant melanoma in Norway and Sweden, Photochem. Photobiol. Sci., 2012, 11(1), 191–198 CAS.
- F. El Ghissassi, et al., A review of human carcinogens – part D: radiation, Lancet Oncol., 2009, 10(8), 751–752 CrossRef.
- J. F. Dore and M. C. Chignol, Tanning salons and skin cancer, Photochem. Photobiol. Sci., 2012, 11(1), 30–37 CAS.
- International Agency for Research on Cancer Working Group on artificial ultraviolet (UV) light and skin cancer, The association of use of sunbeds with cutaneous malignant melanoma and other skin cancers: a systematic review, Int. J. Cancer, 2007, 120(5), 1116–1122 Search PubMed.
- M. D. Evans, M. Dizdaroglu and M. S. Cooke, Oxidative DNA damage and disease: induction, repair and significance, Mutat. Res., 2004, 567(1), 1–61 CrossRef CAS.
- G. M. Halliday, et al., UV-A fingerprint mutations in human skin cancer, Photochem. Photobiol., 2005, 81(1), 3–8 CrossRef CAS.
- N. S. Agar, et al., The basal layer in human squamous tumors harbors more UVA than UVB fingerprint mutations: a role for UVA in human skin carcinogenesis, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(14), 4954–4959 CrossRef CAS.
- A. Fourtanier, D. Moyal and S. Seite, UVA filters in sun-protection products: regulatory and biological aspects, Photochem. Photobiol. Sci., 2012, 11(1), 81–89 CAS.
- F. P. Gasparro, Sunscreens, skin photobiology, and skin cancer: the need for UVA protection and evaluation of efficacy, Environ. Health Perspect., 2000, 108(Suppl 1), 71–78 CAS.
- A. R. Young, et al., A sunscreen's labeled sun protection factor may overestimate protection at temperate latitudes: a human in vivo study, J. Invest. Dermatol., 2010, 130(10), 2457–2462 CrossRef CAS.
- A. C. Kerr, et al., Ultraviolet A1 phototherapy: a British photodermatology group workshop report, Clin. Exp. Dermatol., 2012, 37, 219–226 CrossRef CAS.
- A. R. Young, Chromophores in human skin, Phys. Med. Biol., 1997, 42(5), 789–802 CrossRef CAS.
- J. L. Ravanat, T. Douki and J. Cadet, Direct and indirect effects of UV radiation on DNA and its components, J. Photochem. Photobiol., B, 2001, 63(1–3), 88–102 CrossRef CAS.
- J. Cadet, et al., Sensitized formation of oxidatively generated damage to cellular DNA by UVA radiation, Photochem. Photobiol. Sci., 2009, 8(7), 903–911 CAS.
- J. Cadet, E. Sage and T. Douki, Ultraviolet radiation-mediated damage to cellular DNA, Mutat. Res., 2005, 571(1–2), 3–17 CrossRef CAS.
- G. P. Pfeifer and A. Besaratinia, UV wavelength-dependent DNA damage and human non-melanoma and melanoma skin cancer, Photochem. Photobiol. Sci., 2012, 11(1), 90–97 CAS.
- S. Mouret, et al., Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(37), 13765–13770 CrossRef CAS.
- A. R. Young, et al., The similarity of action spectra for thymine dimers in human epidermis and erythema suggests that DNA is the chromophore for erythema, J. Invest. Dermatol., 1998, 111(6), 982–988 CrossRef CAS.
- E. Sage, P. M. Girard and S. Francesconi, Unravelling UVA-induced mutagenesis, Photochem. Photobiol. Sci., 2012, 11(1), 74–80 CAS.
- A. Tewari, R. P. Sarkany and A. R. Young, UVA1 induces cyclobutane pyrimidine dimers but not 6-4 photoproducts in human skin in vivo, J. Invest. Dermatol., 2011, 132, 394–400 CrossRef.
- T. M. Runger and U. P. Kappes, Mechanisms of mutation formation with long-wave ultraviolet light (UVA), Photodermatol. Photoimmunol. Photomed., 2008, 24(1), 2–10 CrossRef CAS.
- R. R. Anderson and J. A. Parrish, The optics of human skin, J. Invest. Dermatol., 1981, 77(1), 13–19 CAS.
- M. J. van Gemert, et al., Skin optics, IEEE Trans. Biomed. Eng., 1989, 36(12), 1146–1154 CrossRef CAS.
- W. A. Bruls, et al., Transmission of human epidermis and stratum corneum as a function of thickness in the ultraviolet and visible wavelengths, Photochem. Photobiol., 1984, 40(4), 485–494 CrossRef CAS.
- X. X. Huang, F. Bernerd and G. M. Halliday, Ultraviolet A within sunlight induces mutations in the epidermal basal layer of engineered human skin, Am. J. Pathol., 2009, 174(4), 1534–1543 CrossRef CAS.
- V. Lhiaubet-Vallet, et al., Triplet excited fluoroquinolones as mediators for thymine cyclobutane dimer formation in DNA, J. Phys. Chem. B, 2007, 111(25), 7409–7414 CrossRef CAS.
- K. S. Robinson, et al., Cyclobutane pyrimidine dimers are photosensitised by carprofen plus UVA in human HaCaT cells, Toxicol. in Vitro, 2010, 24(4), 1126–1132 CrossRef CAS.
- T. Douki, et al., Bipyrimidine photoproducts rather than oxidative lesions are the main type of DNA damage involved in the genotoxic effect of solar UVA radiation, Biochemistry, 2003, 42(30), 9221–9226 CrossRef CAS.
- B. Epe, DNA damage spectra induced by photosensitization, Photochem. Photobiol. Sci., 2012, 11(1), 98–106 CAS.
- Y. Jiang, et al., UVA generates pyrimidine dimers in DNA directly, Biophys. J., 2009, 96(3), 1151–1158 CrossRef CAS.
- S. Mouret, et al., UVA-induced cyclobutane pyrimidine dimers in DNA: a direct photochemical mechanism?, Org. Biomol. Chem., 2010, 8(7), 1706–1711 CAS.
- C. Kielbassa, L. Roza and B. Epe, Wavelength dependence of oxidative DNA damage induced by UV and visible light, Carcinogenesis, 1997, 18(4), 811–816 CrossRef CAS.
- W. Baumler, et al., UVA and endogenous photosensitizers–the detection of singlet oxygen by its luminescence, Photochem. Photobiol. Sci., 2012, 11(1), 107–117 Search PubMed.
- J. L. Ravanat, et al., Singlet oxygen induces oxidation of cellular DNA, J. Biol. Chem., 2000, 275(51), 40601–4 CrossRef CAS.
- S. Basu-Modak and R. M. Tyrrell, Singlet oxygen: a primary effector in the ultraviolet A/near-visible light induction of the human heme oxygenase gene, Cancer Res., 1993, 53(19), 4505–4510 CAS.
- Y. Hattori, et al., 8-Hydroxy-2′-deoxyguanosine is increased in epidermal cells of hairless mice after chronic ultraviolet B exposure, J. Invest. Dermatol., 1996, 107(5), 733–737 CAS.
- S. Liardet, et al., Protection against pyrimidine dimers, p53, and 8-hydroxy-2′-deoxyguanosine expression in ultraviolet-irradiated human skin by sunscreens: difference between UVB + UVA and UVB alone sunscreens, J. Invest. Dermatol., 2001, 117(6), 1437–1441 CrossRef CAS.
- M. S. Cooke, et al., Induction and excretion of ultraviolet-induced 8-oxo-2′-deoxyguanosine and thymine dimers in vivo: implications for PUVA, J. Invest. Dermatol., 2001, 116(2), 281–285 CrossRef CAS.
- M. Auletta, et al., Effect of cutaneous hypoxia upon erythema and pigment responses to UVA, UVB, and PUVA (8-MOP + UVA) in human skin, J. Invest. Dermatol., 1986, 86(6), 649–652 CAS.
- A. Anders, et al., Action spectrum for erythema in humans investigated with dye lasers, Photochem. Photobiol., 1995, 61(2), 200–205 CrossRef CAS.
- R. D. Ley, Photoreactivation of UV-induced pyrimidine dimers and erythema in the marsupial Monodelphis domestica, Proc. Natl. Acad. Sci. U. S. A., 1985, 82(8), 2409–2411 CrossRef CAS.
- L. M. Coussens and Z. Werb, Inflammation and cancer, Nature, 2002, 420(6917), 860–867 CrossRef CAS.
- A. Mantovani, et al., Cancer-related inflammation, Nature, 2008, 454(7203), 436–444 CrossRef CAS.
- R. P. Gallagher, et al., Sunlight exposure, pigmentary factors, and risk of nonmelanocytic skin cancer, I. Basal cell carcinoma, Arch. Dermatol., 1995, 131(2), 157–163 CAS.
- H. Fassihi, Spotlight on ‘xeroderma pigmentosum’, Photochem. Photobiol. Sci., 2013 10.1039/C2PP25267H.
- D. B. Yarosh, DNA repair, immunosuppression, and skin cancer, Cutis, 2004, 74(5 Suppl), 10–13 Search PubMed.
- J. Kibitel, et al., UV-DNA damage in mouse and human cells induces the expression of tumor necrosis factor alpha, Photochem. Photobiol., 1998, 67(5), 541–546 CrossRef CAS.
- A. Tewari, et al., Human erythema and matrix metalloproteinase-1 mRNA induction, in vivo, share an action spectrum which suggests common chromophores, Photochem. Photobiol. Sci., 2012, 11(1), 216–223 CAS.
- A. A. Vink, D. B. Yarosh and M. L. Kripke, Chromophore for UV-induced immunosuppression: DNA, Photochem. Photobiol., 1996, 63(4), 383–386 CrossRef CAS.
- M. Norval, Chromophore for UV-induced immunosuppression: urocanic acid, Photochem. Photobiol., 1996, 63(4), 386–390 CrossRef CAS.
- D. P. Steenvoorden and G. Beijersbergen van Henegouwen, Protection against UV-induced systemic immunosuppression in mice by a single topical application of the antioxidant vitamins C and E, Int. J. Radiat. Biol., 1999, 75(6), 747–755 CrossRef CAS.
- S. Widyarini, et al., Photoimmune protective effect of the phytoestrogenic isoflavonoid equol is partially due to its antioxidant activities, Photochem. Photobiol. Sci., 2012, 11(7), 1186–1192 CAS.
- R. D. Ley and V. E. Reeve, Chemoprevention of ultraviolet radiation-induced skin cancer, Environ. Health Perspect., 1997, 105(Suppl 4), 981–984 CrossRef CAS.
- J. F. Zhao, et al., Green tea protects against psoralen plus ultraviolet A-induced photochemical damage to skin, J. Invest. Dermatol., 1999, 113(6), 1070–1075 CrossRef CAS.
- E. D. Pleasance, et al., A comprehensive catalogue of somatic mutations from a human cancer genome, Nature, 2010, 463(7278), 191–196 CrossRef CAS.
- D. L. Mitchell and R. S. Nairn, The biology of the (6–4) photoproduct, Photochem. Photobiol., 1989, 49(6), 805–819 CrossRef CAS.
- V. J. Bykov, et al., In situ repair of cyclobutane pyrimidine dimers and 6–4 photoproducts in human skin exposed to solar simulating radiation, J. Invest. Dermatol., 1999, 112(3), 326–331 CrossRef CAS.
- H. Ikehata and T. Ono, The mechanisms of UV mutagenesis, J. Radiat. Res., 2011, 52(2), 115–125 CrossRef CAS.
- J. Jans, et al., Powerful skin cancer protection by a CPD-photolyase transgene, Curr. Biol., 2005, 15(2), 105–115 CrossRef CAS.
- C. I. Kowalczuk, et al., Wavelength dependence of cellular responses in human melanocytes and melanoma cells following exposure to ultraviolet radiation, Int. J. Radiat. Biol., 2006, 82(11), 781–792 CrossRef CAS.
- D. Mitchell and A. Fernandez, The photobiology of melanocytes modulates the impact of UVA on sunlight-induced melanoma, Photochem. Photobiol. Sci., 2012, 11(1), 69–73 CAS.
- D. E. Godar, R. J. Landry and A. D. Lucas, Increased UVA exposures and decreased cutaneous Vitamin D(3) levels may be responsible for the increasing incidence of melanoma, Med. Hypotheses, 2009, 72(4), 434–443 CrossRef CAS.
- A. Besaratinia, et al., DNA lesions induced by UV A1 and B radiation in human cells: comparative analyses in the overall genome and in the p53 tumor suppressor gene, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(29), 10058–10063 CrossRef CAS.
- A. Besaratinia, et al., G-to-T transversions and small tandem base deletions are the hallmark of mutations induced by ultraviolet a radiation in mammalian cells, Biochemistry, 2004, 43(25), 8169–8177 CrossRef CAS.
- A. Besaratinia, S. I. Kim and G. P. Pfeifer, Rapid repair of UVA-induced oxidized purines and persistence of UVB-induced dipyrimidine lesions determine the mutagenicity of sunlight in mouse cells, FASEB J., 2008, 22(7), 2379–2392 CrossRef CAS.
- P. J. Rochette, et al., UVA-induced cyclobutane pyrimidine dimers form predominantly at thymine–thymine dipyrimidines and correlate with the mutation spectrum in rodent cells, Nucleic Acids Res., 2003, 31(11), 2786–2794 CrossRef CAS.
- E. A. Drobetsky, J. Turcotte and A. Chateauneuf, A role for ultraviolet A in solar mutagenesis, Proc. Natl. Acad. Sci. U. S. A., 1995, 92(6), 2350–2354 CrossRef CAS.
- U. P. Kappes, et al., Short- and long-wave UV light (UVB and UVA) induce similar mutations in human skin cells, J. Invest. Dermatol., 2006, 126(3), 667–675 CrossRef CAS.
- U. P. Kappes and T. M. Runger, No major role for 7,8-dihydro-8-oxoguanine in ultraviolet light-induced mutagenesis, Radiat. Res., 2005, 164(4 Pt 1), 440–445 CrossRef CAS.
- F. P. Noonan, et al., Melanoma induction by ultraviolet A but not ultraviolet B radiation requires melanin pigment, Nat. Commun., 2012, 3, 884 CrossRef.
- S. Mouret, A. Forestier and T. Douki, The specificity of UVA-induced DNA damage in human melanocytes, Photochem. Photobiol. Sci., 2012, 11(1), 155–162 CAS.
- O. Su, et al., Effectiveness of medium-dose ultraviolet A1 phototherapy in localized scleroderma, Int. J. Dermatol., 2011, 50(8), 1006–1013 CrossRef.
- C. Andres, et al., Successful ultraviolet A1 phototherapy in the treatment of localized scleroderma: a retrospective and prospective study, Br. J. Dermatol., 2010, 162(2), 445–447 CrossRef CAS.
- T. Gambichler, et al., Medium-dose ultraviolet (UV) A1 vs. narrowband UVB phototherapy in atopic eczema: a randomized crossover study, Br. J. Dermatol., 2009, 160(3), 652–658 CrossRef CAS.
- A. C. Kerr, et al., Ultraviolet A1 phototherapy: a British Photodermatology Group workshop report, Clin. Exp. Dermatol., 2012, 37(3), 219–226 CrossRef CAS.
- M. Grachtchouk, et al., Basal cell carcinomas in mice arise from hair follicle stem cells and multiple epithelial progenitor populations, J. Clin. Invest., 2011, 121(5), 1768–1781 CAS.
Footnote |
| † This article is published as part of a themed issue on current topics in photodermatology. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2013 |