Syntheses and photophysical properties of 5′–6-locked fluorescent nucleosides†
Kristmann
Gislason
,
Dnyaneshwar B.
Gophane
and
Snorri Th.
Sigurdsson
*
University of Iceland, Department of Chemistry, Science Institute, Dunhagi 3, 107 Reykjavik, Iceland. E-mail: snorrisi@hi.is; Fax: +354 5528911; Tel: +354 5254801
First published on 31st October 2012
Abstract
Nine fluorescent 5′–6-locked nucleosides were synthesized by condensation of various 1,2-diketones with 5-amino-2′-deoxycytidine. The nucleosides have different substituents on the pyrazine core structure, ranging from two methyl groups to polyaromatic rings. The photophysical properties of each nucleoside were determined, with the nucleosides displaying diverse absorption and emission maxima, extinction coefficients and quantum yields. The nucleoside with the highest fluorescence brightness was phosphitylated and incorporated into an oligonucleotide by means of automated oligonucleotide synthesis. The labelled oligonucleotide in aqueous buffer exhibited a substantially lowered extinction coefficient and quantum yield compared to the nucleoside in THF. The photophysical properties of the nucleoside were also compared in different DNA structural contexts, a single strand, a 14-mer duplex, a 14-mer duplex with an 11-mer overhang, and a 25-mer nicked duplex labelled at the nick site. Circular dichroism and melting temperature studies verified that the nucleoside did not perturb or destabilize the DNA helixes. In fact, when incorporated at the nick site, the nucleoside was found to stabilize the nicked duplex notably compared to its unmodified counterpart. The brightness of the fluorescent nucleoside in DNA increased as the polarity of its surroundings decreased, being highest in the 25-mer nicked duplex where exposure to the polar solvent is minimized by stacking to the adjacent bases on both the 3′- and 5′-side. The nucleosides brightness in the nicked duplex was also found to increase with lowered temperature, in accordance with expected temperature-dependent changes in the stacked–unstacked equilibrium at the nick site.
Introduction
Fluorescence spectroscopy is one of the most widely used spectroscopic techniques in nucleic acid research.1–4 Applications range from visualizing the biomolecules, both in vitro and in vivo, to the study of intra- and intermolecular interactions. Due to the lack of emission from the natural nucleotides, fluorescence studies of nucleic acids require incorporation of fluorescent labels (fluorophores). For many applications, it is sufficient to tether a fluorophore to an appropriate position in the biomolecule with a flexible linker spanning a few atoms. However, the flexibility of such linkers limits the applicability for structural studies.3 To circumvent the limitation associated with flexible linkers, semi-rigid alkyne linkers have been used for conjugation to the 5-position of pyrimidines or the 7-position of 7-deazapurines.5,6 However, this approach still allows movements of the fluorophore independent of the nucleic acid, thus reducing the sensitivity of such a label for detection of changes in the secondary or tertiary structure of the nucleic acid.An alternate approach for conjugating the fluorophore to the natural nucleotide is to replace a nucleotide base with a fluorescent polycyclic aromatic hydrocarbon.3 Despite being bulkier than the natural bases and lacking hydrogen bonding capability, they have been applied successfully in a number of biophysical studies.7–10 Nevertheless, structural studies of native nucleic acids require less intrusive labels; labels that have larger bases than the natural nucleosides they replace, and/or do not base-pair to opposite natural bases, can distort the natural secondary structure of the biomolecule.
In recent years, a number of fluorescent nucleoside analogs that contain nucleobases in which the aromatic ring system has been extended to provide a fluorescent base, while retaining the hydrogen bonding face of the base, have been synthesized and applied in various biophysical studies.2,11 For example, our group has previously reported the detection and identification of single nucleotide mismatches employing a cytidine analog (Çf), where a tetramethylisoindoline is fused to the cytosine base to form a phenoxazine analog.12–14 Additionally, two nucleosides with similar structures were recently applied as the first nucleoside analog fluorescence resonance energy transfer (FRET)-pair.15 Although a number of different nucleoside analogs containing extended fluorescent bases have been synthesized and applied in different nucleic acid studies, the search for new nucleosides with advantageous properties is still an active area of research. It is of interest to discover fluorescent probes that have increased brightness (increased absorption coefficient and/or quantum yield), increased Stokes shift, emission at higher wavelengths (in the near infrared range) and increased sensitivity to its microenvironment (base pairing, stacking, etc.).1
An important aspect in the design of new fluorescent analogs, like other spectroscopic probes, is their ease of synthesis. We have previously reported the 1,10-phenanthroline-containing 5′–6-locked nucleoside 1 (Fig. 1) which was readily accommodated at the end of DNA duplexes.16 Nucleoside 1 is a T-analog, forming a base-pair with A, where one of the nitrogens in the pyrazine ring replaces the 4-carboxy oxygen of T as a hydrogen bond acceptor (Fig. 1). Nucleoside 1 was simply prepared by the reaction of a 1,2-diketone with 5-amino-2′-deoxycytidine (2) and was directly phosphitylated for incorporation of 1 into nucleic acids by solid-phase synthesis. The phenanthroline nucleoside displayed fluorescence both before and after incorporation into oligonucleotides.
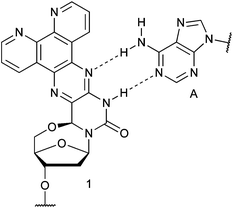 | ||
| Fig. 1 Previously reported 5′–6-locked, 1,10-phenanthroline-containing nucleoside 1, shown base-paired to A. | ||
To investigate if the readily-prepared 5′–6 ring-closed structural scaffold could yield fluorescent nucleosides with improved spectroscopic properties and to demonstrate the generality of the synthetic approach, we have prepared nine fluorescent 5′–6-locked nucleosides. The structure of the pyrazine ring extension ranges from a dimethyl substituted pyrazine ring to pyrazine rings conjugated to polyaromatic moieties. The nucleosides exhibited a wide range of absorption maxima, emission maxima, extinction coefficients and quantum yields. Additionally, one nucleoside was incorporated into an oligonucleotide and its photophysical properties studied, both at the end of a DNA double helix and at an internal nick site in a DNA duplex.
Results and discussion
Synthesis and characterization of fluorescent nucleosides
An advantage of the synthetic approach is that a variety of 1,2-diketones can be condensed with the diamino nucleoside 2 (Scheme 1). Eight of the nine diketones used here are commercially available while one (9a, Fig. S1†) was obtained in a single step from a commercially available precursor (8a).17 Condensation of the diketones used here (3a–11a, Fig. S1†) yielded the 5′–6-locked nucleosides 3–11 (Fig. 2), that can be cast into four categories. Nucleoside 3 contains two methyl substituents on the core base structure, while the second group (4–7) contains a pair of aromatic rings. The third group contains the acenaphthylene nucleosides 8 and 9, and the fourth group is comprised of nucleosides where the aromatic system of the core base structure has been extended with three and four benzene rings (nucleosides 10 and 11).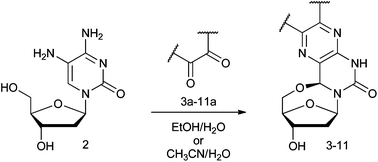 | ||
| Scheme 1 General reaction scheme for synthesis of 5′–6-locked nucleosides. Detailed structures of diketones are shown in ESI (Fig. S1†) and of the product in Fig. 2. The yields of the reaction are listed in Table 1. | ||
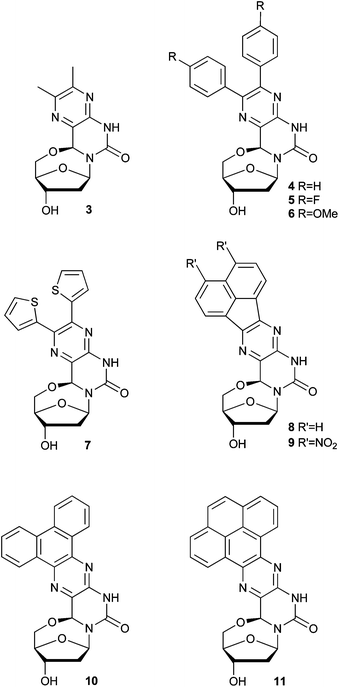 | ||
| Fig. 2 Structures of 5′–6-locked nucleosides 3–11. | ||
Nucleosides 3, 8, 10 and 11 were prepared in excellent yields (Table 1) by simply heating the corresponding 1,2-diketone with 218–20 in a mixture of ethanol and water.16 However, nucleosides 4–7 and 9 could not be synthesized by this approach, even after prolonged heating, presumably due to the limited solubility of the corresponding 1,2-diketones in the polar solvent system. Indeed, replacing ethanol with acetonitrile yielded the remaining nucleosides in moderate yields (Table 1). The stereochemistry of the chiral centers formed by the condensation was assumed to be the same as determined by Kalman and co-workers using NOE-experiments.19 UV-Vis and fluorescence spectra were recorded for all nucleosides in tetrahydrofuran (Fig. 3). The excitation wavelength for fluorescence measurements was chosen just below the maximum of the highest absorption peak to avoid scattering of the excitation light interfering with the measurements. The photophysical properties for nucleosides 3–11 are listed in Table 1. Nucleoside 3 was fluorescent with an absorption maximum at 319 nm and a Stokes shift of 36 nm. Its moderate extinction coefficient and quantum yield results in a fluorescence brightness (FB) of 3240. Thus, the core structure is moderately fluorescent and its photophysical properties might be tunable by substituents.
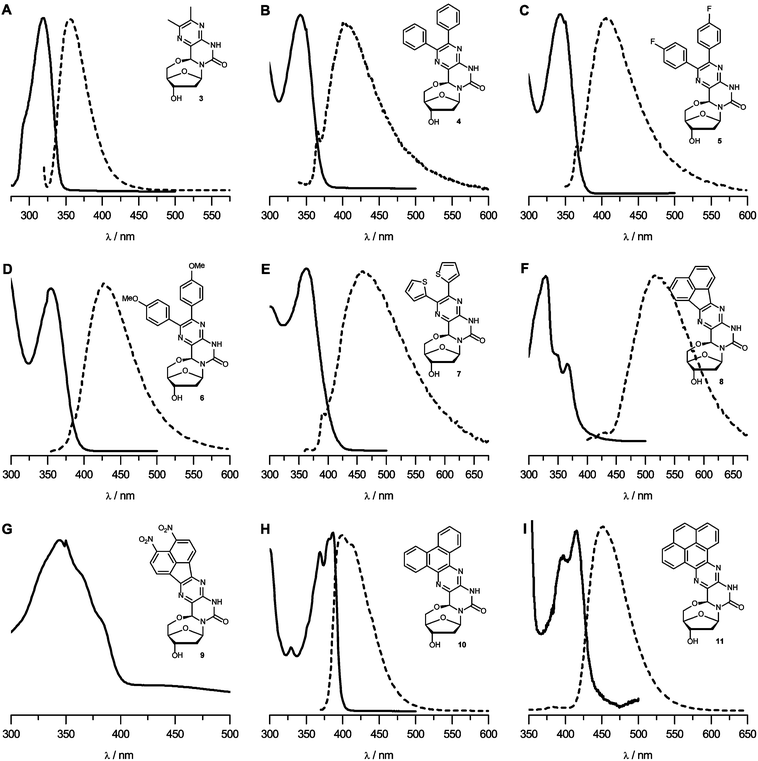 | ||
| Fig. 3 (A–I) UV-Vis and fluorescence spectra for nucleosides 3–11 in THF, shown as solid and broken lines, respectively. Y-axes are omitted for clarity, spectra with y-axes can be seen in Fig. S6.† | ||
| Compound | Yield | λ ab | ε | λ em |
Φ
F![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a a |
FBb |
|---|---|---|---|---|---|---|
| a Fluorescence quantum yield. b Fluorescence brightness in M−1 cm−1, calculated as FB = ΦF × ε. | ||||||
| 3 | 84% | 319 | 8000 | 355 | 0.405 | 3240 |
| 4 | 47% | 340 | 11000 |
403 | 0.005 | 52.3 |
| 5 | 50% | 342 | 11200 |
406 | 0.007 | 73.3 |
| 6 | 20% | 355 | 10200 |
428 | 0.019 | 191 |
| 7 | 28% | 363 | 11200 |
459 | 0.012 | 133 |
| 8 | 70% | 329, 366 | 17000, 50900 |
517 | 0.054, 0.047 | 920, 2410 |
| 9 | 10% | 344, 383 | — | — | — | — |
| 10 | 95% | 386 | 21800 |
399 | 0.358 | 7790 |
| 11 | 91% | 415 | 8380 | 452 | 0.771 | 6460 |
In compounds 4–7, both the methyl groups of nucleoside 3 have been substituted with phenyl, p-fluorophenyl, p-methoxyphenyl or thiophene rings, respectively. The ortho-terphenyl-like structure of these nucleosides allows only partial conjugation of the aromatic rings with the central pyrazine ring due to steric hindrance. Not unexpectedly, extending the aromatic system yielded higher extinction coefficients but the quantum yields for 4–7 were considerably lower than that of 3. Nucleosides 4–7 had absorption and emission maxima that are red-shifted compared to those of nucleoside 3 and increased Stokes shifts. Although nucleosides 4–7 had similar photophysical properties, the thiophene-containing nucleoside 7 is the most notable with the highest emission wavelength and Stokes shift.
Nucleosides 8 and 9 both contain naphthalene moieties connected to the pyrazine ring through a five membered ring. Nucleoside 9 contains two nitro substituents on the naphthalene ring that increase the π-electron delocalization in the aromatic ring and thereby affect the absorption and emission of the fluorophores. For this reason, nitro groups are often incorporated into fluorophores in an attempt to obtain beneficial changes in photophysical properties.21 In nucleoside 9, however, the effect was a dramatic quenching of its emission with a maximum at 590 nm. Nucleoside 8, on the other hand, exhibited quantifiable emission although the quantum yield was low. The absorption spectrum of 8 (Fig. 3F) has an interesting profile; it has a weak absorption peak at 366 nm and an intense one at 329 nm (a similar absorption structure was seen for nucleoside 9). Excitation at either maximum resulted in emission with a maximum at 517 nm. Excitation at both bands leads to similar quantum yields but the much higher extinction coefficient at the lower wavelength resulted in considerably higher FB. Nucleoside 8 displayed the highest Stokes shift of the nucleosides in this study (∼150 nm) and the highest emission maximum wavelength (517 nm).
In contrast to the other nucleosides in this study, nucleosides 10 and 11 contain fully fused six-membered aromatic rings where the π-electrons can resonate throughout the whole structure. Nucleosides 10 and 11 displayed red shifts in both absorption (67 nm and 96 nm, respectively) and emission (44 nm and 97 nm, respectively) compared to nucleoside 3. While nucleoside 11 has a similar extinction coefficient as 3 and a higher quantum yield, nucleoside 10 has a similar quantum yield but a much higher extinction coefficient, yielding the brightest nucleoside in this study. Nucleosides 10 and 11 have rather small Stokes shifts, similar to nucleoside 3.
Photophysical studies of DNA constructs containing nucleoside 10
The brightest nucleoside in this study, nucleoside 10, was chosen to investigate the photophysical properties of fluorescent 5′–6-locked nucleosides in DNA. Thus, phosphoramidite 12 was synthesized in one step from nucleoside 10 using standard reaction conditions, except that a few drops of dry DMSO were added to the reaction mixture due to limited solubility of 10 in CH2Cl2 (Scheme 2). Although the lack of a 5′-OH group restricts the use of 5′–6-locked nucleosides to 5′-end labelling of nucleic acids, the 5′–6-locked nucleosides have the advantage that preparation of their phosphoramidites for oligonucleotide synthesis does not require the usual 5′-trityl protection.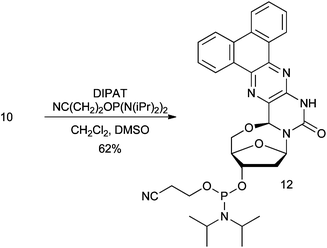 | ||
| Scheme 2 Synthesis of phosphoramidite 12 from nucleoside 10 (DIPAT: diisopropylammonium tetrazolide). | ||
Nucleoside 10 was incorporated into a 14-mer oligonucleotide (I, Table 2) using standard solid-phase DNA synthesis and its incorporation was verified by mass spectrometry. The photophysical properties of DNA I were subsequently studied in an aqueous solution. DNA I absorbs at approximately the same wavelength as nucleoside 10 in THF (385 nm). The emission maximum is, however, red-shifted by ∼20 nm (420 nm). The extinction coefficient and quantum yield of DNA I were lowered, when compared to 10 in THF, leading to a considerably lower fluorescence brightness (Table 2). It is not uncommon for the photophysical properties of fluorescent nucleosides to change upon incorporation into oligonucleotides. Such changes can be attributed to several factors, such as base-stacking and the polarity of the solvent.2,22
| DNA | Structure | T m (°C)a | Solventb | ε (M−1 cm−1) | Φ F | FBc |
|---|---|---|---|---|---|---|
| a Values in parentheses are for unmodified duplexes where 10 is replaced by T. b A = phosphate buffer, B = 30% ethylene glycol in phosphate buffer. c Fluorescence brightness in M−1 cm−1, calculated as FB = ΦF × ε. | ||||||
| I | 3′-GTGCTACGCTCCG10-5′ | A | 7760 | 0.008 | 62.8 | |
| B | 7220 | 0.018 | 131 | |||
| II | 5′-CACGATGCGAGGCA-3′, 3′-GTGCTACGCTCCG10-5′ | 66.2 (65.5) | A | 6830 | 0.010 | 65.9 |
| B | 7580 | 0.017 | 125 | |||
| III | 5′-CACGATGCGAGGCA-AGAGGACTCGC-3′, 3′-GTGCTACGCTCCG10-5′ | A | 7770 | 0.013 | 99.4 | |
| B | 6850 | 0.023 | 160 | |||
| IV | 5′-CACGATGCGAGGCA–AGAGGACTCGC-3′, 3′-GTGCTACGCTCCG10 TCTCCTGAGCG-5′ | 58.6 (53.3), 69.7 (66.9) | A | 5770 | 0.031 | 179 |
| B | 5720 | 0.044 | 251 | |||
To further investigate the effect of the solvent exposure on the emission of I, we designed and prepared DNA structures II–IV: the fully complementary 14-mer duplex (II), a 14-mer duplex containing an 11-mer overhang (III) and a 25-mer nicked duplex (IV) to examine how the fluoroside behaves at a nick site in duplex DNA (Table 2). To verify that the nucleosides did not significantly perturb or destabilize the DNA duplexes, circular dichroism (CD) spectra and melting temperatures (Tms) were recorded for duplexes II and IV along with their unmodified counterparts, where 10 was replaced with T. CD-spectra of modified and unmodified duplexes displayed the same structures with the spectra showing the characteristic B-DNA molar ellipticities at ca. 250 nm (negative) and 280 nm (positive) (ESI†). DNA II displayed a minor increase in stability over its unmodified duplex (ΔTm = 0.7 °C). The three strand duplexes (DNA IV) displayed two Tms, with the lower Tms presumably reflecting the melting of the 11-mer duplex region. The stabilizing effect of 10 was more pronounced in the three strand system IV when compared to its unmodified counterpart (ΔTm1 = 5.3 °C and ΔTm2 = 2.8 °C), presumably due to stronger stacking interactions of 10 to the flanking thymine base at the 3′-end of the 11-mer oligonucleotide.
Having established that nucleoside 10 does not significantly distort B-form DNA duplexes, the photophysical properties of 10 in structures II–IV were determined. While the 14-mer duplex II displayed approximately the same brightness as the single strand I, the FB increased by ∼35% by addition of the 11-mer overhang in III and by ∼65% in the nicked duplex IV, the similar FBs obtained for I and II indicate that stacking interactions and base-pairing have little effect on the brightness of 10.22 The increase in brightness for III and IV also indicates that exposure of nucleoside 10 to the solvent has a significant effect on its brightness. In duplex II, one side of the nucleoside is still exposed to the solvent, while the overhang in III could, in principle, provide stacking interactions. In the 25-mer nicked duplex IV, the shielding effect is most pronounced, indicating that the 5′-flanking T–A base-pair is stacking on top of the 10-A pair despite the nick in the duplex. Crystal structures of nicked DNAs have shown that the helix adopts a B-DNA conformation over the nick site indicating that base-pairing and stacking interactions overcome the loss of the covalent phosphodiester connection.23 Additionally, nicked duplexes have been shown to have mobilities similar to their un-nicked counterparts in polyacrylamide gels.24 Thus, our data indicate that 5′–6-locked nucleosides can be incorporated into internal positions in duplex DNAs for spectrophotometric studies or to investigate electron transport through a three component system as in IV.
The equilibrium between un-stacked and stacked conformations in nicked DNAs follows the Boltzmann distribution,24 resulting in an increasing population of the un-stacked conformation with rising temperature. Thus, if the brightness of nucleoside 10 in duplex IV increased due to stacking with the base-pair on the other side of the nick, the brightness should also be temperature-dependent. Therefore, the photophysical properties of duplexes II and IV were compared at three different temperatures (Fig. 4). Indeed, while the fluorescence brightness of II showed virtually no temperature dependence, the brightness of IV showed a marked decrease as the temperature was raised from 4 °C to 30 °C. Hence, the enhancement in the brightness of 10 in IV most likely stems from stacking at the nick site, which shields the nucleoside from the solvent.
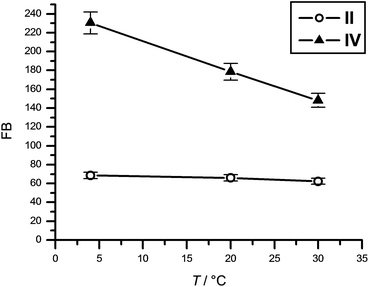 | ||
| Fig. 4 Temperature dependence of the fluorescence brightness of nucleoside 10 in II and IV. | ||
Further evidence for the sensitivity of nucleoside 10 to the polarity of its surroundings was obtained by repeating the aforementioned experiments in 30% aqueous ethylene glycol, commonly used as a cryoprotectant in biological samples (Table 2, Fig. S1†). The lower polarity of the solvent resulted in an increase in the brightness of 10 in all DNA structures. The difference in brightness between DNAs where the nucleoside is exposed to (I and II) or shielded from (III and IV) the solvent was lower for the ethylene glycol solution (two-fold) than water (three-fold). This was expected, because of a smaller change in polarity of the fluorophore's microenvironment upon shielding from a less polar solvent.
Conclusions
Nine 5′–6-locked nucleosides have been prepared and their photophysical properties determined. The core structure of nucleoside 3 had a moderate extinction coefficient and a quantum yield with absorption and emission maxima at rather low wavelengths. While additions to the core structure in all cases red-shifted both maxima, the effect on fluorescence brightness varied from almost quantitative quenching in 9 to a significant increase in nucleosides 10 and 11. The absorption and emission of nucleoside 10 were significantly quenched upon incorporation into DNA, but to a smaller extent when placed at a nick in a DNA duplex. The emissive strength of 10 in the nicked helix also showed correlation with temperature in agreement with the expected changes in the ratio between stacking–unstacking at the nick site. Tm measurements showed that nucleoside 10 stabilized the nicked duplex when compared to thymidine. Since previous work has shown that natural helixes mostly retain their structure despite a single nick in one strand, the Tm results indicate that 5′–6-locked nucleosides might be applicable for internal labelling of duplexes through three strand systems as in DNA IV. It should be mentioned that, although all the diketones in this study are symmetrical, the reactivity of the amino groups of 2 differs enough for unsymmetrical diketones to be condensed, yielding one product, as long as one of the ketone groups is more reactive.19 A clear advantage of the strategy described here is that the preparation of labelled oligonucleotides only requires three synthetic steps from the corresponding diketone, which should facilitate the preparation of DNAs modified with new 5′–6-locked nucleotides. Thus, nucleosides within this 5′–6-locked family that have higher fluorescence brightness should be readily accessible, for example by incorporating chromophores that contain both electron-withdrawing and electron-donating groups, which is known to polarize the π-system and thereby increase the brightness.25,26Experimental section
Chemicals were purchased from Sigma Aldrich, Acros and Apollo Scientific Ltd and used without further purification. Thin layer chromatography (TLC) was carried out using glass plates pre-coated with silica gel (0.25 mm, F-254) from Silicycle. Nucleosides were identified using staining with p-anisaldehyde. Flash column chromatography was performed using ultra pure flash silica gel from Silicycle (40–63 μm, 60 Å). Acetonitrile, dichloromethane and pyridine were freshly distilled from calcium hydride prior to use. DMSO was dried by vacuum distillation and stored over activated 3 Å molecular sieves under argon until used. All moisture- and air sensitive reactions were carried out in oven- or flame-dried glassware under an inert argon atmosphere. The phosphate buffer used contained 10 mM phosphate, 100 mM NaCl, 0.1 mM EDTA, pH 7. NMR spectra were recorded on a Bruker Advance 400 spectrometer. 1H NMR chemical shifts are reported in reference to undeuterated residual solvent (D2O (4.60 ppm), CDCl3 (7.26 ppm), d6-DMSO (2.50 ppm)). 13C NMR chemical shifts are reported in reference to undeuterated residual solvent (CDCl3 (77.0 ppm), d6-DMSO (39.43 ppm)). 31P NMR chemical shifts were reported relative to 85% H3PO4 as an external standard. UV-Vis spectra were recorded on a Varian Cary 4000 UV-Vis Spectrometer. Fluorescence spectra were recorded on a JY Horiba SPEX Fluorolog τ-3 spectrofluorimeter. CD and Tm measurements were performed on a Jasco J-810 spectropolarimeter. MALDI-ToF-MS measurements were performed on a Bruker Autoflex III and ESI-MS measurements were performed on a Bruker MicroTOF Q.Nucleoside 3
5-Amino-2′-deoxycytidine 2 (100 mg, 0.413 mmol) and biacetyl 3a (36 mg, 0.413 mmol) were suspended in 75% aq. ethanol (2 mL) and heated at 83 °C for 4 hours. The reaction mixture was allowed to cool, the solvent removed under vacuum. The compound was purified on preparative TLC (MeOH:CH2Cl2, 10:90) to yield 3 as a white solid (102 mg or 84%). 1H NMR (CDCl3) δ 2.40–2.46 (1H, m, H2′), 2.48 (3H, s, CH3), 2.50 (3H, s, CH3), 2.76–2.82 (1H, m, H2′′), 4.04 (1H, d, J = 12.6, H5′), 4.10 (1H, dd, J1 = 12.7, J2 = 2.3, H5′′), 4.48 (1H, d, J = 2.6, H4′), 4.50 (1H, d, J = 3.7, H3′), 6.09 (1H, s, H6), 6.13 (1H, d, J = 6.4, H1′), 8.26 (1H, s, H3). 13C NMR (CDCl3) δ 21.18, 46.43, 50.78, 72.35, 73.26, 85.22, 88.77, 90.35, 128.08, 141.43, 146.87, 149.48, 152.68. ESI-MS: [M + H]+: calc. 293.12, found 293.12.
Nucleoside 4
5-Amino-2′-deoxycytidine 2 (100 mg, 0.413 mmol) and benzil 4a (104 mg, 0.496 mmol) were suspended in acetonitrile (5.5 mL) and water (1.5 mL) and heated at 45–50 °C for 16 h. The reaction mixture was allowed to cool, the solvent evaporated and the residue purified by column chromatography (CH2Cl2:MeOH; 98:2 to 95:5) to yield 4 as a white solid (80 mg, 47%). 1H-NMR (400 MHz, DMSO-d6): δ 2.20 (1H, ddd, J = 14.2, 6.2, 3.7, C2′H), 2.47 (1H, d, J = 7.8, C2′H), 3.95 (2H, dd, J = 17.4, 6.9, C5′H), 4.22–4.31 (1H, m, C4′H), 4.35 (1H, s, OH), 5.17 (1H, d, J = 4.2, C3′H), 5.95 (1H, d, J = 6.2 Hz, C1′H), 6.09 (1H, s, C6H), 7.31 (5H, dd, J = 7.0, 2.0 Hz, ArH), 7.34 (5H, d, J = 3.9 Hz, ArH), 10.81 (1H, s, NH). 13C-NMR (400 MHz, DMSO-d6): δ 45.70, 71.28, 71.86, 84.52, 87.92, 90.02, 127.97, 128.66, 129.28, 129.38, 130.10, 137.69, 138.00, 142.75, 145.54, 148.85, 151.13. ESI-MS: [M + H]+: calc. 417.15, found 417.16.
Nucleoside 5
5-Amino-2′-deoxycytidine 2 (150 mg, 0.620 mmol) and 1,2-bis(4-fluorophenyl)ethane-1,2-dione 5a (183 mg, 0.743 mmol) were suspended in acetonitrile (8 mL) and water (2 mL) and heated at 40–45 °C for 16 h. The reaction mixture was allowed to cool, the solvent evaporated and the residue purified by column chromatography (CH2Cl2:MeOH; 98:2 to 95:5) to yield 5 as a white solid (140 mg, 50%). 1H-NMR (400 MHz, DMSO-d6): δ 2.19 (1H, ddd, J = 14.1, 6.5, 3.7 Hz, C2′H), 2.47 (1H, d, J = 7.8, C2′H), 3.94 (2H, dd, J = 16.8, 7.3, C5′H), 4.19–4.30 (1H, m, C4′H), 4.34 (1H, s, OH), 5.16 (1H, d, J = 4.3, C3′H), 5.94 (1H, d, J = 5.8, C1′H), 6.07 (1H, s, C6H), 7.18 (4H, q, J = 9.1, ArH), 7.35 (4H, ddd, J = 21.4, 8.8, 5.5, ArH), 10.82 (1H, s, NH). 13C-NMR (400 MHz, DMSO-d6): δ 45.70, 71.28, 71.88, 84.45, 87.92, 90.01, 114.95, 115.01, 115.17, 115.22, 130.23, 131.40, 131.48, 131.63, 131.72, 133.98, 134.01, 134.31, 134.34, 144.51, 148.81, 150.13, 160.57, 160.97, 163.01, 163.42. ESI-MS: [M + H]+: calc. 453.14, found 453.14.
Nucleoside 6
5-Amino-2′-deoxycytidine 2 (150 mg, 0.619 mmol) and diketone 6a (200 mg, 0.743 mmol) were suspended in acetonitrile (7.5 mL) and water (1.5 mL) and heated at 50 °C for 16 h. The reaction mixture was allowed to cool, the solvent evaporated and the residue purified by column chromatography (CH2Cl2:MeOH; 98:2 to 95:5) to yield 6 as a white solid (60 mg, 20%). 1H-NMR (400 MHz, DMSO-d6): δ 2.19 (1H, ddd, J = 14.0, 6.4, 3.7, C2′H), 2.47 (1H, d, J = 7.6, C2′H), δ 3.75 (6H, s, OCH3), 3.84–4.03 (2H, m, C5′H), 4.20–4.28 (1H, m, C4′H), 4.34 (1H, s, OH), 5.15 (1H, d, J = 4.2, C3′H), 5.94 (1H, d, J = 6.2, C1′H), 6.04 (1H, s, C6H), 6.88 (4H, dd, J = 8.8, 3.4, ArH), 7.26 (4H, dd, J = 25.1, 8.7, ArH), 10.69 (1H, s, NH). 13C-NMR (400 MHz, DMSO-d6): δ 45.73, 55.01, 55.05, 71.30, 71.80, 84.57, 87.91, 90.02, 113.45, 113.48, 129.23, 130.02, 130.47, 130.57, 130.78, 142.24, 145.09, 148.92, 150.42, 158.92, 159.51. ESI-MS: [M + H]+: calc. 477.17, found 477.17.
Nucleoside 7
5-Amino-2′-deoxycytidine 2 (100 mg, 0.413 mmol) and 1,2-di(thiophen-2-yl)ethane-1,2-dione 7a (111 mg, 0.496 mmol) were suspended in acetonitrile (5.5 mL) and water (1.5 mL) and heated at 45–50 °C for 16 h. The reaction mixture was allowed to cool, the solvent evaporated and the residue purified by column chromatography (CH2Cl2:MeOH; 98:2 to 95:5) to yield 7 as a white solid (50 mg, 28%). 1H-NMR (400 MHz, DMSO-d6): δ 2.18 (1H, ddd, J = 14.1, 6.5, 3.8, C2′H), 2.47 (1H, dd, J = 10.2, 4.4, C2′H), 3.90 (2H, dt, J = 12.5, 7.4, C5′H), 4.20 (1H, m, C4′H), 4.33 (1H, s, OH), 5.16 (1H, d, J = 4.3, C3′H), 5.93 (1H, d, J = 5.7, C1′H), 6.00 (1H, s, C6H), 7.00–7.08 (2H, m, ArH), 7.08–7.14 (2H, m, ArH), 7.71 (2H, ddd, J = 9.1, 4.1, 2.2, ArH), 10.83 (1H, s, NH). 13C-NMR (400 MHz, DMSO-d6): δ 45.67, 71.27, 71.83, 84.24, 87.89, 89.98, 127.53, 127.76, 128.01, 128.16, 128.88, 129.89, 129.93, 138.12, 139.64, 139.82, 142.65, 144.68, 148.67. ESI-MS: [M + H]+: calc. 429.06, found 429.07.
Nucleoside 8
5-Amino-2′-deoxycytidine 2 (100 mg, 0.413 mmol) and diketone 8a (75 mg, 0.413 mmol) were suspended in 75% ethanol (2 mL) and heated at 95 °C overnight. The reaction mixture was allowed to cool and the solid formed collected by filtration, washed with ethanol and ether and dried. Column chromatography (CH2Cl2:MeOH; 98:1 to 90:10) yielded 8 as a pale yellow solid (113 mg, 70%). 1H NMR (DMSO-d6) δ 2.22–2.28 (1H, m, H2′), 2.54–2.60 (1H, m, H2′′), 4.10 (2H, s, H5′), 4.31–4.33 (1H, m, H4′), 4.43 (1H, s, H3′), 6.05 (1H, d, J = 6.2, H1′), 6.25 (1H, s, H6), 8.13–8.19 (4H, m, Ar-H), 8.37 (1H, d, J = 7.4, Ar-H), 8.44 (1H, d, J = 7.5 Hz, Ar-H), 9.11 (1H, d, J = 7.6, Ar-H), 9.14 (1H, d, J = 7.5, Ar-H). 13C NMR (DMSO-d6) δ 152.33, 148.88, 146.51, 143.22, 131.58, 131.13, 130.59, 130.42, 129.19, 128.85, 128.78, 128.67, 122.97, 121.64, 90.19, 88.01, 85.02, 71.87, 71.36, 45.79. ESI-MS: [M + H]+: calc. 439.12, found 439.12.
Nucleoside 9
5-Amino-2′-deoxycytidine 2 (100 mg, 0.413 mmol) and 5,6-dinitroacenaphthylene-1,2-dione 9a (90 mg, 0.496 mmol) were suspended in acetonitrile (8 mL) and water (2 mL) and heated at 45 °C for 16 h. The reaction mixture was allowed to cool, the solvent evaporated and the residue purified by preparative TLC (CH2Cl2:MeOH, 90:10) to yield 9 as a yellow solid (18 mg, 10%). 1H-NMR (400 MHz, DMSO-d6): δ 2.20 (1H, ddd, J = 14.1, 6.5, 3.7, 1H, C2′H), 2.51–2.60 (1H, m, C2′H), 3.99 (2H, dd, J = 19.0, 7.2, C5′H), 4.24–4.32 (1H, m, C4′H), 4.38 (1H, s, OH), 5.20 (1H, d, J = 3.8, C3′H), 5.95 (1H, d, J = 6.0, C1′H), 6.05 (1H, s, C6H), 8.23 (2H, dd, J = 7.5, 4.5, ArH), 8.51 (2H, dd, J = 7.2, 4.5, ArH), 11.12 (1H, s, NH). 13C-NMR (400 MHz, DMSO-d6): δ 45.69, 71.33, 72.05, 84.60, 88.06, 90.16, 113.11, 121.96, 123.80, 129.50, 130.08, 131.65, 132.59, 136.06, 139.92, 144.49, 144.89, 145.46, 145.76, 148.44, 152.22. ESI-MS: [M + H]+: calc. 479.09, found 479.09.
Nucleoside 10
5-Amino-2′-deoxycytidine 2 (100 mg, 0.413 mmol) and phenanthrene-9,10-dione 10a (103 mg, 0.496 mmol) were suspended in 75% ethanol (2 mL) and heated at 83 °C for 16 h. The reaction mixture was allowed to cool and the solid formed collected by filtration, washed with ethanol and ether and dried under suction. Crude product was purified by trituration from THF with ether to yield 10 as a pale yellow solid (162 mg, 95%). 1H NMR (d6-DMSO) δ 2.19–2.26 (1H, m, H2′), 2.52–2.58 (1H, m, H2′′), 4.07 (2H, s, H5′), 4.29–4.32 (1H, m, H4′), 4.41 (1H, s, H3′), 6.02 (1H, d, J = 6.0, H1′), 6.24 (1H, s, H6), 7.76–7.82 (4H, m, ArH), 8.80–8.85 (2H, m, ArH), 8.94–8.98 (2H, m, ArH), 11.12(1H, s, NH). 13C NMR (DMSO-d6) δ 149.04, 143.71, 139.38, 135.08, 133.89, 131.38, 130.06, 129.34, 128.95, 128.67, 128.45, 127.95, 127.74, 124.99, 123.82, 123.44, 123.35, 90.16, 88.02, 84.82, 72.20, 71.45, 45.81. ESI-MS: [M + H]+: calc. 415.13, found 415.13.Nucleoside 11
5-Amino-2′-deoxycytidine 2 (100 mg, 0.413 mmol) and pyrene-4,5-dione 11a (96 mg, 0.413 mmol) were suspended in 95% ethanol (4 mL) and heated at 83 °C overnight. The reaction mixture was allowed to cool and the solid formed collected by filtration, washed with ethanol and ether. Crude product was purified by trituration from THF with ether to yield 11 as a yellow solid (165 mg or 91%). 1H NMR (DMSO-d6) δ 2.22–2.28 (1H, m, H2′), 2.54–2.60 (1H, m, H2′′), 4.10 (2H, s, H5′), 4.31–4.33 (1H, m, H4′), 4.43 (1H, s, H3′), 6.05 (1H, d, J = 6.2, H1′), 6.25 (1H, s, H6), 8.13–8.19 (4H, m, Ar-H), 8.37 (1H, d, J = 7.4, Ar-H), 8.44 (1H, d, J = 7.5, Ar-H), 9.11 (1H, d, J = 7.6, Ar-H), 9.14 (1H, d, J = 7.5, Ar-H), 11.12(1H, s, NH). 13C NMR (DMSO-d6) δ 149.03, 134.84, 130.85, 128.54, 128.19, 127.85, 127.77, 127.71, 127.41, 127.15, 127.07, 126.96, 126.73, 124.85, 122.65, 121.32, 90.17, 88.05, 84.84, 72.24, 71.46, 45.82. ESI-MS: [M + H]+: calc. 439.13, found 439.15.Phosphoramidite 12
Nucleoside 10 (25 mg, 0.060 mmol) and diisopropyl ammonium tetrazolide (25 mg, 0.144 mmol) were suspended in CH2Cl2 (2 mL) and anhydrous DMSO added dropwise until the suspension went into solution. NC(CH2)2OP[N(iPr)2]2 (47 mg, 0.156 mmol) was then added and the solution stirred for 2 hours. CH2Cl2 (3 mL) was added and the organic layer washed with sat. NaHCO3(aq) (3 × 5 mL) and brine (5 mL). The organic phase was dried with anhydrous Na2SO4 and the solvent removed in vacuo. The residue was dissolved in a minimum amount of dry CH2Cl2 and precipitated with petroleum ether. Precipitation was repeated twice and the residue dried under vacuum to yield 12 as a pale yellow solid (23 mg, 62%). 1H NMR (400 MHz, CDCl3) δ 0.86 (2H, t, J = 6.9), 1.33–1.08 (14H, m), 2.60–2.50 (1H, m), 2.62 (2H, t, J = 6.6), 2.81–2.69 (1H, m), 2.95–2.83 (1H, m), 3.66–3.54 (1H, m), 3.90–3.70 (1H, m), 4.29–4.08 (2H, m), 4.74–4.53 (1H, m), 6.21–6.15 (1H, m), 6.29 (1H, s), 7.80–7.61 (3H, m), 8.60–8.52 (1H, m), 8.94 (1H, d, J = 7.9), 9.10–9.02 (1H, m). 31P NMR (CDCl3) δ 148.78, 148.52.HPLC analysis
Analytical HPLC chromatograms were obtained on a Beckman Coulter Gold HPLC system, using a GL Sciences Inertsustain C18 4.6 × 150 mm analytical column with UV detection at 260 nm. The method was a linear gradient from 96:4, H2O:CH3CN to 100% CH3CN.
Oligonucleotide synthesis
Oligonucleotides were synthesized on a 1000 nmol scale by using an ASM 800 DNA/RNA synthesizer with the manufacturer's standard protocols. The modified nucleotide was introduced by pausing the synthesizer program after completion of the prior cycle, removing the column from the synthesizer and running 200 μL of standard activator solution and 200 μL of a 0.05 M solution of 12 in CH3CN back and forth through the column for 15 min. Then the column was re-mounted on the synthesizer to complete the cycle. The oligonucleotides were cleaved from the solid support, deprotected by using standard conditions (conc. aq. NH3, 55 °C, 12 h) and purified by 20% denaturing polyacrylamide gel electrophoresis. The modified oligonucleotide was characterized by MALDI-ToF mass spectrometry: 5′-d(10GC CTC GCA TCG TG) (I) calc. 4404, found 4404.UV-Vis and fluorescence studies
Solutions of nucleosides were prepared by dissolving a known amount of each nucleoside in 10 mL of THF. Solutions of oligonucleotides were prepared by dissolving 3 nmol of each strand in 100 μL phosphate buffer, annealing and diluting to 1 mL with phosphate buffer. UV-Vis and fluorescence spectra were then recorded of each sample at three different concentrations by diluting the original sample with phosphate buffer. Quantum yields were determined using the equation Φx = Φst(Gradx/Gradst)(ηx/ηst), where the subscripts st and x denote standard and sample respectively, Φ is the fluorescence quantum yield, Grad the gradient from the plot of integrated fluorescence intensity vs. absorbance and η the refractive index of the solvent. Anthracene in ethanol was used as a standard (ΦF = 0.27).27,28T m and CD measurements
Solutions of oligonucleotides were prepared by dissolving 3 nmol of each strand in 100 μL phosphate buffer, annealing and diluting to 0.5 mL with phosphate buffer. Samples for Tm studies were heated at a rate of 5 °C min−1.Acknowledgements
We thank the Icelandic Research Fund (070600007) and the Science Institute, University of Iceland for financial support, Prof. Fredrik Westerlund for assistance with UV-Vis and fluorescence measurements and members of the Sigurdsson research group for helpful discussions.Notes and references
- R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2010, 110, 2579–2619 CrossRef CAS.
- L. M. Wilhelmsson, Q Rev Biophys, 2010, 43, 159–183 CrossRef CAS.
- J. N. Wilson and E. T. Kool, Org. Biomol. Chem., 2006, 4, 4265–4274 CAS.
- A. Haller, M. F. Souliere and R. Micura, Acc. Chem. Res., 2011, 44, 1339–1348 CrossRef CAS.
- A. Okamoto, K. Tainaka, K. Nishiza and I. Saito, J. Am. Chem. Soc., 2005, 127, 13128–13129 CrossRef CAS.
- M. Vrabel, R. Pohl, I. Votruba, M. Sajadi, S. A. Kovalenko, N. P. Ernsting and M. Hocek, Org. Biomol. Chem., 2008, 6, 2852–2860 CAS.
- J. M. Gao, C. Strassler, D. Tahmassebi and E. T. Kool, J. Am. Chem. Soc., 2002, 124, 11590–11591 CrossRef CAS.
- F. Garo and R. Haner, Angew. Chem., Int. Ed., 2012, 51, 916–919 CrossRef CAS.
- F. Samain, S. Ghosh, Y. N. Teo and E. T. Kool, Angew. Chem., Int. Ed., 2010, 49, 7025–7029 CrossRef CAS.
- J. Guo, S. L. Wang, N. Dai, Y. N. Teo and E. T. Kool, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3493–3498 CrossRef CAS.
- J. Riedl, R. Pohl, L. Rulisek and M. Hocek, J. Org. Chem., 2012, 77, 1026–1044 CrossRef CAS.
- N. Barhate, P. Cekan, A. P. Massey and S. T. Sigurdsson, Ang. Chem., Int. Ed., 2007, 46, 2655–2658 CrossRef CAS.
- H. Gardarsson, A. S. Kale and S. T. Sigurdsson, Chembiochem, 2011, 12, 567–575 CrossRef CAS.
- H. Gardarsson and S. T. Sigurdsson, Bioorg. Med. Chem., 2010, 18, 6121–6126 CrossRef CAS.
- K. Borjesson, S. Preus, A. H. El-Sagheer, T. Brown, B. Albinsson and L. M. Wilhelmsson, J. Am. Chem. Soc., 2009, 131, 4288–4293 CrossRef.
- K. Gislason and S. T. Sigurdsson, Eur. J. Org. Chem., 2010, 4713–4718 CrossRef CAS.
- P. A. Blair, S. J. Chang and H. Shechter, J. Org. Chem., 2004, 69, 7123–7133 CrossRef CAS.
- P. Ge and T. I. Kalman, Bioorg. Med. Chem. Lett., 1997, 7, 3023–3026 CrossRef CAS.
- P. Ge and T. I. Kalman, J. Heterocycl. Chem., 1998, 35, 257–260 CrossRef CAS.
- P. Ge, G. O. Voronin and T. I. Kalman, Nucleosides Nucleotides, 1996, 15, 1701–1710 CAS.
- V. Madhura, M. V. Kulkarni, S. Badami, J. Yenagi and J. Tonannavar, Spectrochim. Acta, Part A, 2011, 84, 137–143 CrossRef CAS.
- J. N. Wilson, Y. J. Cho, S. Tan, A. Cuppoletti and E. T. Kool, Chembiochem, 2008, 9, 279–285 CrossRef CAS.
- J. Aymami, M. Coll, G. A. Vandermarel, J. H. Vanboom, A. H. J. Wang and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 2526–2530 CrossRef CAS.
- E. Protozanova, P. Yakovchuk and M. D. Frank-Kamenetskii, J. Mol. Biol., 2004, 342, 775–785 CrossRef CAS.
- C. A. B. Rodrigues, I. F. A. Mariz, E. M. S. Macoas, C. A. M. Afonso and J. M. G. Martinho, Dyes Pigm., 2012, 95, 713–722 CrossRef CAS.
- R. S. Butler, P. Cohn, P. Tenzel, K. A. Abboud and R. K. Castellano, J. Am. Chem. Soc., 2009, 131(2), 623–633 CrossRef CAS.
- W. R. Dawson and M. W. Windsor, J. Phys. Chem., 1968, 72, 3251–3260 CrossRef CAS.
- D. F. Eaton, Pure Appl. Chem., 1988, 60, 1107–1114 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Structures for diketones 3a–11a. Temperature dependence spectra for II and IV in 30% ethylene glycol in aqueous buffer. CD spectra for DNAs II and IV and their unmodified counterparts. HPLC traces for nucleosides 3–11. NMR spectra for nucleosides 3–11 and phosphoramidite 12. See DOI: 10.1039/c2ob26536b |
| This journal is © The Royal Society of Chemistry 2013 |