Synthesis of a phosphinate analogue of the anti-tumour phosphate di-ester perifosine via sequential radical processes†
Marios S.
Markoulides
and
Andrew C.
Regan
*
School of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: andrew.regan@manchester.ac.uk; Fax: +44 (0) 161 275 4617; Tel: +44 (0) 161 275 4617
First published on 3rd October 2012
Abstract
An efficient synthesis of a phosphinate analogue of the anti-tumour phosphate di-ester perifosine is described (6 steps and 50% overall yield). The two phosphorus–carbon bonds in the perifosine analogue were prepared by sequential double radical hydrophosphinylation processes. This is the first example of a phosphinate analogue of perifosine, designed to be resistant to hydrolysis by phospholipid-metabolizing enzymes.
Introduction
Alkyl phospholipids were synthesized in the late 1960s as metabolically stable analogues of lysophosphatidylcholine.1 Several of these synthetic analogues turned out to be potent immune modulators, but surprisingly some of these lipids also exhibited anti-tumour activities both in vitro and in vivo in a rather selective way.2 In the late 1980s, Eibl and Unger identified miltefosine 1 as the minimal structural requirement for the anti-tumour activity of synthetic analogues of phospholipids (Fig. 1).3 Remarkably, in addition to their initial anti-tumour action they have also been shown to be potentially useful for other therapeutic indications, which make synthetic analogues of phospholipids a fascinating family of compounds with several potential biomedical applications.4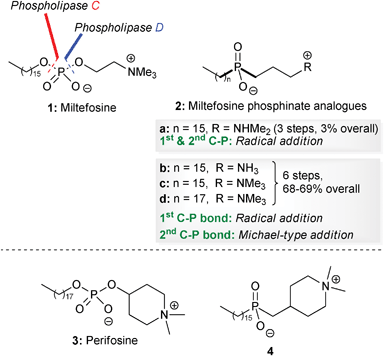 | ||
| Fig. 1 Structures of miltefosine 1, perifosine 3 and phosphinate analogues of miltefosine 2 and perifosine 4. | ||
Since then, new classes of alkyl phospholipids with promising biological activities have been synthesized and amongst these, the heterocyclic derivative perifosine 3 constitutes a second generation derivative of miltefosine 1, with potent anti-tumour activity (Fig. 1).5
Synthetic analogues of phospholipids display two important advantages: (a) they target the plasma membrane rather than directly interacting with cellular DNA, and (b) they reveal a strong apoptosis-inducing ability.4b,6 Whereas malignant cells are highly sensitive to the lethal action of synthetic phospholipids, normal cells may remain relatively unaffected, illustrating the potentially selective anti-tumour properties of these type of compounds.3
However, one disadvantage is that the presence of the phosphate diester group results in them being prone to biodegradation by phospholipid-metabolizing enzymes such as phospholipases C7 and D.8 Replacement of both of the O–P bonds of the phosphate diester in miltefosine 1 or perifosine 3 with two C–P bonds would result in phosphinate analogues 2 and 4, which should be resistant to hydrolysis by phospholipid-metabolizing enzymes (Fig. 1).
We have recently reported the synthesis of phosphinate analogues 2b–d of miltefosine 1, using a free-radical addition reaction to form the first phosphorus–carbon bond, and the conjugate addition reaction of a silyl phosphonite to form the second.9
During the synthesis of miltefosine analogues 2b–d, an alternative approach to compound 2a was also examined, involving two radical addition reactions, which had the attraction of requiring fewer steps overall. However the sequential radical approach proved to be low yielding, and no improvement was obtained upon modifying the conditions.9
In this paper we describe an efficient synthesis of the phosphinate analogue 4 of perifosine 3, involving two sequential radical hydrophosphinylation reactions to construct both of the carbon–phosphorus bonds. To the best of our knowledge, the phosphinate function has never been used in the synthesis of perifosine analogues, despite its close structural analogy. We have also carried out further investigations into the sequential radical approach to analogue 2a of miltefosine 1.
The retrosynthetic analysis of 4 is shown in Scheme 1. Disconnection of the first phosphorus–carbon bond suggested that this might be formed by regioselective free-radical addition of mono-alkylphosphinic ester 5 to the exo-methylene piperidine derivative 6. Phosphinate 5 is then a common intermediate for further analysis by two approaches, and our previous work on alkylation of trivalent silyl-phosphonite derivatives10 suggested that one possibility for preparation of 5 would be by the Arbuzov-type alkylation of a silyl derivative of hypophosphorous acid 8 with hexadecyl iodide 9.
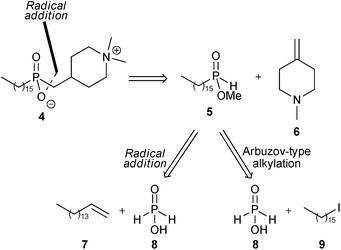 | ||
| Scheme 1 Retrosynthetic analyses of 4. | ||
An alternative approach to 4 envisioned the use of two sequential radical hydrophosphinylation addition reactions, where the carbon–phosphorus bond in intermediate 5 would also be constructed using a free-radical addition, here involving reaction of hypophosphorous acid 8 with 1-hexadecene 7.
The formation of mono-substituted phosphinic acids by free-radical addition of 8 or its salts to terminal alkenes has been known for some time.11 Nifant'ev12 developed the reaction using peroxide initiators; more recently AIBN has been employed with either the acid 813 or its esters,14 and the use of the combination of triethylborane and air is also effective.15 Changing the ratio of hypophosphorous acid 8 to the alkene can be used to influence the formation of either mono-substituted or symmetrical disubstituted phosphinic acids.16
There are relatively few examples of the formation of the second C–P bond in unsymmetrical disubstituted phosphinic acids by radical addition of mono-substituted H-phosphinates to alkenes.11,17 These reactions are often inefficient, probably due to the higher P–H bond dissociation energy, and often require a wastefully large excess of the alkene component.11 Phenyl H-phosphinates are more reactive than alkyl analogues,17a perhaps because the phenyl group can stabilise the adjacent phosphorus-centred radical.11
Nevertheless, there has been very little investigation of the use of sequential radical addition reactions to two different alkenes in order to form both phosphorus–carbon bonds in unsymmetrical disubstituted phosphinic acids.18 In the area of biological targets, there has been one report of this approach being employed to prepare a precursor for a phosphinate analogue of a dinucleotide.19
Results and discussion
As experienced in our previous work,9 attempts to alkylate bis(trimethylsilyl)phosphonite (BTSP) 10 with hexadecyl iodide 9 were not successful using our Arbuzov-type alkylation protocol (Scheme 2).10a In spite of extensive experimentation involving different solvents, including DCM, diglyme, benzene, and toluene, reaction without any solvent under neat conditions, and also modification of the conditions (changing the ratio of BTSP 10 to 9, and also temperature from room temperature to reflux), there was no improvement in yields. Although in some solvents (e.g. benzene or toluene) BTSP 10 was stable (thus allowing for longer reaction times), this did not improve the results. In more polar solvents (e.g. DCM) decomposition of BTSP 10 was observed to occur much more readily.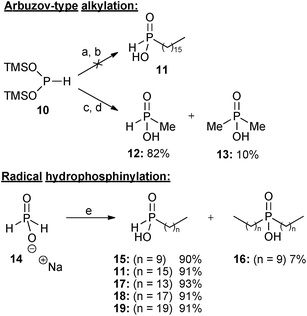 | ||
| Scheme 2 Reagents and conditions: (a) hexadecyl iodide, CH2Cl2, 0 °C to reflux, 2 days; (b) THF–H3O+, 0 °C to rt, 2 hours; (c) methyl iodide (0.33 eq), CH2Cl2, 0 °C, 2 hours, then rt, 12 hours; (d) THF–H3O+, 0 °C to rt, 2 hours; (e) terminal alkene, conc. H2SO4, AIBN or VAZO-88, EtOH, reflux, 1 day. | ||
The alkylation reaction was also repeated with greater success using methyl iodide10a and three equivalents of BTSP 10 (Scheme 2), resulting in formation of monomethyl phosphinic acid 12 in 82% yield together with a small amount of the corresponding dimethyl phosphinic acid 13. It appears that whereas alkylation of silyl phosphonite 10 with short-chain alkyl bromides and iodides is successful,10a long-chain iodides are problematic,10b even when reacted neat or in various solvents, and at elevated temperatures.
Having confirmed that the Arbuzov-type approach was unsuccessful, attention was then turned to free radical hydrophosphinylation reactions. Several long chain mono-alkyl phosphinic acids 11,915, 17, 189 and 19 were conveniently prepared in high yields by free radical addition of sodium hypophosphite 14 to the appropriate terminal alkenes (Scheme 2).9,13 Sodium hypophosphite 14 could be employed in either anhydrous form or as the monohydrate (depending on availability), and with essentially equivalent results. Because of changes in commercial availability VAZO-88 was sometimes used instead of AIBN as the radical initiator. In the case where 1-decene was used as the alkene, a small quantity (7%) of the corresponding disubstituted product 16 was also formed, and it may be that this is a result of less steric hindrance in the second radical addition than with the longer alkenes, where bis-addition was not observed.
Terminal alkenes with even numbers of carbon atoms were utilised in Scheme 2 because of their greater availability and lower cost. The consistently high yields obtained with alkenes ranging from 16 to 20 carbon atoms suggest that radical hydrophosphinylation reactions of alkenes with an odd number of carbons would be equally successful. It is not expected that changing the lengths of such long alkyl chains by one carbon atom would significantly alter the biological activities,1b however odd-numbered alkyl groups could be investigated in future work if desired.
In view of the very low yield (3%) obtained in our previous report9 for the radical hydrophosphinylation reaction between hexadecylphosphinic acid 11 and N,N-dimethylallylamine 22, further investigation of this methodology was undertaken by varying the phosphinate component between the acid forms (11 and 19), sodium salt 20, and methyl esters (5 and 21). The alkene component was also varied between the tertiary allyl amine 22 and the corresponding quaternary ammonium salt 23 (Scheme 3).
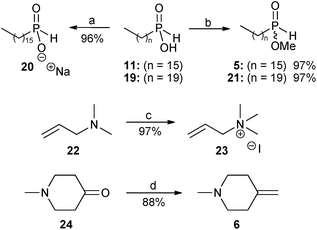 | ||
| Scheme 3 Reagents and conditions: (a) NaH, EtOH, 0 °C, 20 minutes, then rt, 30 minutes; (b) trimethyl orthoformate, reflux, 3.5 days; (c) methyl iodide, Et2O, rt, 3 hours; (d) NaH, dry DMSO, 75 °C, 45 minutes, then 0 °C, methyltriphenylphosphonium bromide in dry DMSO, then rt, 10 minutes, then 24, rt, 30 minutes. | ||
Sodium hexadecylphosphinate 20 was prepared from the acid 11 using sodium ethoxide. The coupling constant of the P–H doublet in the 1H NMR spectrum of sodium hexadecylphosphinate 20 was 486.5 Hz, reduced from the value found in hexadecylphosphinic acid 11, which was 539.7 Hz.9 The phosphorus NMR signal at δ 37.96 for 119 was also moved upfield to δ 27.97 for 20, as would be expected for a hypophosphite salt.
The mono-alkyl phosphinic acids 11 and 19 were then converted into their methyl esters 5 and 21 with trimethyl orthoformate.9,20 The 1H NMR spectrum of 5 displayed a new doublet at δ 3.72 (3JP–H = 11.9 Hz) attributable to the methyl ester group, and the signal for the corresponding carbon atom also appeared in the 13C NMR spectrum as a doublet at δ 52.68 (2JC–P = 6.4 Hz). The 31P NMR spectrum displayed a signal at δ 42.10, downfield from the corresponding signal in the starting material 11 (δ 37.96). For the longer chain alkylphosphinic acids 11 (C-16),918 (C-18)9 and 19 (C-20), the doublet attributable to the first carbon atom directly bonded to phosphorus could not be observed in the 13C NMR spectra, probably because it was hidden by other carbon signals of similar frequency. In the case of methyl hexadecylphosphinate 5 this doublet was separated and appeared at δ 28.47 (1JC–P = 92.7 Hz). Methyl icosylphosphinate 21 showed analogous spectroscopic data to 5.
Next, N,N,N-trimethylallylammonium iodide 23 was conveniently prepared in 97% yield by the reaction of N,N-dimethylallylamine 22 with methyl iodide in diethyl ether at room temperature.
Finally, for the preparation of the perifosine analogue 4, the alkene precursor required for the second radical hydrophosphinylation reaction was the exo-methylene piperidine 6, and this was prepared in 88% yield by a Wittig reaction on the piperidinone 24 using dimsyl sodium as a base21 (Scheme 3). The use of alternative conditions involving butyl lithium as a base22 gave unsatisfactory results in our hands.
The second radical hydrophosphinylation addition reaction was then investigated between the sodium phosphinate 20 and the tertiary amine 22, the phosphinic acid 11 and the quaternary ammonium salt 23, and the methyl phosphinate ester 5 with tertiary amine 22 (Scheme 4). However, all attempts to form the disubstituted products 25, 2c and 27 proved to be unsuccessful. Extensive repetition and variation of the procedures did not improve the results, and in all of the attempts the starting materials were substantially recovered at the end of the reaction.
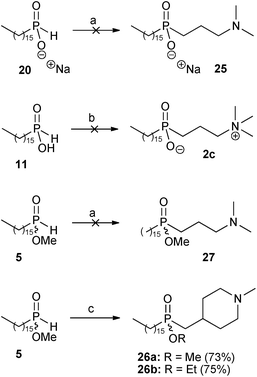 | ||
| Scheme 4 Reagents and conditions: (a) 22 (xs), MeOH, AIBN (xs), reflux, 6 days; (b) 23 (xs), EtOH, AIBN (xs), reflux, 6 days; (c) 6 (1 eq), AIBN (1.6 eq), MeOH (for 26a) or EtOH (for 26b), reflux, 3 days. | ||
However, much better results were obtained in the synthesis of the perifosine analogue 4. The radical hydrophosphinylation addition reaction between methyl hexadecylphosphinate 5 and 1-methyl-4-methylene-piperidine 6 afforded the disubstituted phosphinate 26a in 73% yield (Scheme 4). Remarkably, only one equivalent of the alkene 6 was required in this reaction, in contrast to the large excesses of alkenes often required for radical addition of mono-substituted H-phosphinates.11 This successful radical hydrophosphinylation involving methyl ester 5 to give 26a differed markedly from the very low-yielding radical addition of the corresponding unprotected phosphinic acid 11 to tertiary amine 22, which was reported in our previous work.9 The three unsuccessful radical hydrophosphinylation reactions in Scheme 4 were also originally aimed at the synthesis of the miltefosine analogues 2, and the failure of these reactions required us to resort to our previously-developed methodology of conjugate addition of silyl phosphonites in order complete that synthesis.9 Here, the successful radical reaction of the exo-methylene-piperidine 6, in contrast to those of the mono-substituted terminal alkenes 22 and 23, may be a result of alkene 6 being a disubstituted terminal alkene, so that the intermediate radical formed after the addition step is tertiary (and therefore more stable than the secondary radicals which would be formed from the mono-substituted terminal alkenes). An additional factor may be that in the alkenes 22 and 23 the electronegative nitrogen atom is only two bonds away from the radical centre formed during the addition step, whereas for alkene 6 it is more remote, being three bonds removed.
The successful hydrophosphinylation reaction between 5 and 6 was repeated in a higher boiling alcohol in an attempt to increase the yield. The only differences observed when ethanol (EtOH) was used as the solvent were that the product obtained was the corresponding transesterified ethyl ester 26b, and in a slightly improved yield of 75%.
The synthesis of phosphinate analogue 4 of perifosine was then completed as shown in Scheme 5. Quaternisation of the tertiary amine 26a with excess methyl iodide in the presence of anhydrous K2CO3 afforded the desired ammonium salt 28 in 95% yield. Finally, cleavage of the methyl ester group in 28 was achieved by treatment with iodotrimethylsilane (TMSI)9,23 followed by methanolysis, to afford the ammonium phosphinate inner salt 4 in 93% yield.
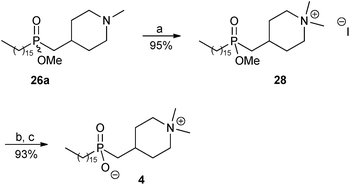 | ||
| Scheme 5 Reagents and conditions: (a) methyl iodide, anhydrous K2CO3, MeOH–CHCl3, reflux, 1 day; (b) TMSI, CH2Cl2, rt, overnight; (c) MeOH, rt, 30 minutes. | ||
The proton-decoupled 31P NMR spectrum of ester 28 displayed a signal at δ 57.06, whereas that of the zwitterion 4 displayed a signal which was shifted upfield to δ 52.85. Upon addition of one drop of concentrated hydrochloric acid to the same NMR tube containing 4 in CD3OD, the phosphorus signal moved downfield from δ 52.85 to 58.06, corresponding to protonation of P–O(−) to P–OH (see the ESI†). After the 31P NMR spectroscopic study the hydrochloride salt of 4 was converted back to the inner salt 4 by co-evaporation with MeOH, followed by pumping under high vacuum.
Conclusions
In summary, an efficient synthetic strategy has been developed for the synthesis of a phosphinate analogue 4 of the anti-tumour agent perifosine 3, making use of two radical hydrophosphinylation steps to form both of the carbon–phosphorus bonds. The second hydrophosphinylation in particular is unusual in that it is efficient and does not employ an excess of the alkene, which is important here because the alkene is a functionalised synthetic intermediate. Overall, the synthesis of the C16 alkyl phosphinate analogue 5 proceeded in six steps and 50% overall yield. By suitable modification of the two alkenes employed, further non-hydrolysable analogues may be designed in order to investigate biological structure-activity relationships.Experimental section
General information
All reagents and solvents were purchased from commercial sources and either used as supplied or purified using the appropriate standard procedures.24 Column chromatography was carried out using Merck silica gel 60H (40–60 nm, 230–300 mesh). Thin-layer chromatography (TLC) was carried out using glass, aluminum or plastic plates coated with Merck HF254/366 silica gel. Visualisation was achieved by viewing under a 254 nm ultraviolet (UV) light source and/or by immersion in solutions of potassium permanganate (KMnO4), anisaldehyde, phosphomolybdic acid (PMA) or ninhydrin, followed by heating. Electron impact/chemical ionisation (EI/CI) mass spectra were recorded on a Micromass Trio 2000 spectrometer. Electrospray (ES±) mass spectra were recorded on a Micromass Platform II spectrometer. High resolution mass spectra were recorded on a Thermo Finnigan MAT95XP spectrometer. Infrared (IR) spectra were recorded either on a PerkinElmer FT-IR RX1 as evaporated films (from deuteriochloroform CDCl3 or dichloromethane DCM) on sodium chloride (NaCl) plates or on a PerkinElmer FT-IR ATIR BX spectrometer, and bands are quoted in cm−1. NMR spectra were recorded using deuterated chloroform (CDCl3) as solvent unless otherwise stated. 1H NMR spectra were recorded either on a Bruker Avance II+ 500 (500 MHz), Bruker Avance III 400 (400 MHz), Varian INOVA Unity 300 (300 MHz) or on a Bruker Avance 200 (200 MHz) spectrometer. Residual non-deuterated solvent was used as the internal standard. Chemical shifts (δH) are quoted in parts per million (ppm) downfield from tetramethylsilane (TMS). 13C NMR spectra were recorded either on a Bruker Avance II+ 500 at 125 MHz, Bruker Avance III 400 at 100 MHz or on a Varian INOVA Unity 300 at 75 MHz spectrometer, using a carbon signal of the solvent as the internal standard. Chemical shifts (δC) are quoted in parts per million (ppm) downfield from tetramethylsilane (TMS). 31P NMR spectra were recorded either on a Bruker Avance III 400 at 162 MHz or on a Bruker Avance 200 spectrometer at 81 MHz. Chemical shifts for phosphorus (δP) are reported in parts per million (ppm) on the δ scale relative to phosphoric acid as the external standard. Peak assignments were aided by 1H–1H COSY, 1H–13C HMQC, 1H–13C HMBC, DEPT-135 and/or DEPT-90, whenever necessary. The resonance multiplicity patterns are described as singlet (s), broad singlet (br s), doublet (d), triplet (t), quartet (q), quintet (quin.), multiplet (m), or combinations of these. Coupling constants (J) are quoted in hertz (Hz). Microanalyses were carried out by the staff of the Microanalytical Laboratory at the School of Chemistry, University of Manchester. Reagents: Copper stabilisers were removed from methyl iodide (MeI) and trimethylsilyl iodide (TMSI) prior to use. 1-Adamantanamine was purified by dissolving it in diethyl ether (Et2O) and washing the organic layer (three times) with a dilute solution of sodium hydroxide (1 M NaOH). The organic layer was then dried over anhydrous sodium sulfate (anh. Na2SO4), and concentrated under reduced pressure to give the compound as a bright white powder. Melting points (mp) were determined either on a Stuart Scientific SMP10 apparatus or on a Reichert (Shandon Scientific) hot stage microscope, and were uncorrected.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 3 cm3), acidified with a few drops of concentrated HCl, were added and the solution was stirred for two hours at room temperature. The solvent was removed under reduced pressure and the crude material was washed with hot hexane to afford the product 12 as a yellow oil (0.15 g, 82%). The compound was not purified further. 1H NMR (300 MHz, CDCl3): δH 12.22 (1H, br s, OH), 7.34 (1H, dq, 1JP–H = 562.4 Hz, 3JH–H = 2.2 Hz, CH3PH), 1.65 (3H, dd, 2JP–H = 15.2 Hz, 3JH–H = 2.3 Hz, CH3PH). 13C NMR (75 MHz, CDCl3): δC 15.58 (d, 1JC–P = 94.4 Hz, CH3PH). 31P NMR (162 MHz, CDCl3): δP 34.36 (CH3PH).
:2.5:0.05, CHCl3–MeOH–TFA, stain: PMA). HRMS (ESI−): calculated for C2H6O2P [M − H]− requires 93.0111; found: 93.0113. IR, νmax (NaCl, evap. film)/cm−1: 2984 (m, CH3), 2626 (br, PO–H), 1429 (w, CH3), 1306 (w, CH3), 1149 (m, P
:1, 3 cm3), acidified with a few drops of concentrated HCl, were added and the solution was stirred for two hours at room temperature. The solvent was removed under reduced pressure and the crude material was washed with hot hexane to afford the product 12 as a yellow oil (0.15 g, 82%). The compound was not purified further. 1H NMR (300 MHz, CDCl3): δH 12.22 (1H, br s, OH), 7.34 (1H, dq, 1JP–H = 562.4 Hz, 3JH–H = 2.2 Hz, CH3PH), 1.65 (3H, dd, 2JP–H = 15.2 Hz, 3JH–H = 2.3 Hz, CH3PH). 13C NMR (75 MHz, CDCl3): δC 15.58 (d, 1JC–P = 94.4 Hz, CH3PH). 31P NMR (162 MHz, CDCl3): δP 34.36 (CH3PH).
:2.5:0.05, CHCl3–MeOH–TFA, stain: PMA). HRMS (ESI−): calculated for C2H6O2P [M − H]− requires 93.0111; found: 93.0113. IR, νmax (NaCl, evap. film)/cm−1: 2984 (m, CH3), 2626 (br, PO–H), 1429 (w, CH3), 1306 (w, CH3), 1149 (m, P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1062 (br, P–OH), 877 (w, P–O), 646 (w, P–C). 1H NMR (400 MHz, CDCl3): δH 11.25 (1H, br s, OH), 1.22 (6H, d, 2JP–H = 14.4 Hz, CH3PCH3). 13C NMR (100 MHz, CDCl3): δC 16.42 (d, 1JC–P = 96.0 Hz, CH3PCH3). 31P NMR (162 MHz, CDCl3): δP 52.42 (CH3PCH3). MS (ESI−), m/z (rel. intensity %): 93 (M − H−, 30%).
O), 1127 (s, C–N), 1039 (s, P–O), 843 (m, P–O), 725 (w, CH2), 645 (w, P–C). 1H NMR (400 MHz, CD3OD): δH 2.07 (3H, br s, 3 × CH), 1.79 (6H, d, J = 2.3 Hz, 6 × CH), 1.70 (3H, d, J = 12.7 Hz, 3 × CH), 1.62 (3H, d, J = 12.1 Hz, 3 × CH), 1.14 (6H, d, 2JP–H = 13.6 Hz, CH3PCH3). 13C NMR (100 MHz, CD3OD): δC 52.65 (C–N), 41.58 (CH2), 36.52 (CH2), 30.46 (CH), 18.47 (d, 1JC–P = 93.8 Hz, CH3PCH3). 31P NMR (162 MHz, CD3OD, proton-coupled): δP 36.66 (sp, 2JP–H = 13.7 Hz, CH3PCH3).
O), 1079 and 1051 (m, P–OH), 973 and 946 (s, P–H), 721 (m, CH2), 675 (w, P–C). 1H NMR (400 MHz, CDCl3): δH 12.23 (1H, br s, OH), 7.1 (1H, dt, 1JP–H = 539.5 Hz, 3JH–H = 1.8 Hz, PH), 1.78–1.70 (2H, m, PCH2), 1.63–1.52 (2H, m, PCH2CH2), 1.38 (2H, quin., J = 6.6 Hz, PCH2CH2CH2), 1.25 (12H, br s, 6 × CH2), 0.87 (3H, t, J = 7.1 Hz, CH3). 13C NMR (100 MHz, CDCl3): δC 31.88, 30.43 (d, 3JC–P = 16.0 Hz, PCH2CH2CH2), 29.54, 29.36, 29.30, 29.24 (d, 1JC–P = 93.7 Hz, PCH2), 29.15, 22.68, 20.68 (d, 2JC–P = 2.9 Hz, PCH2CH2CH2), 14.10 (CH3). 31P NMR (162 MHz, CDCl3, proton-coupled): δP 37.29 (d quin., 1JP–H = 538.0 Hz, 2JP–H = 3JP–H = 28.4 Hz, CH2CH2PH). MS (ESI−), m/z (rel. intensity %): 205 (M − H−, 100%).
O), 1126 (w, C–N), 1039 (s, P–O−), 990 (w, P–H), 720 (w, CH2), 690 (w, P–C). 1H NMR (400 MHz, CDCl3): δH 8.83 (3H, br s, NH3), 7.1 (1H, d, 1JP–H = 479.2 Hz, PH), 2.11 (3H, br s), 1.94 (6H, br s), 1.66 (6H, br s), 1.52 (4H, br s), 1.35 (2H, br s), 1.24 (12H, br s), 0.87 (3H, t, J = 7.1 Hz, CH3). 13C NMR (100 MHz, CDCl3): δC 50.60 (CNH3), 40.63, 35.78, 33.22 (d, 1JC–P = 90.8 Hz, PCH2), 31.90, 31.05 (d, 3JC–P = 16.0 Hz, PCH2CH2CH2), 29.64, 29.52, 29.47, 29.33, 29.02, 22.84 (d, 2JC–P = 1.4 Hz, PCH2CH2), 22.68, 14.11 (CH3). 31P NMR (162 MHz, CDCl3): δP 25.56 (CH2PH). MS (ESI−), m/z (rel. intensity %): 205 (M−, 100%). MS (ESI+), m/z (rel. intensity %): 152 (M+, 100%).
O), 1062 (br, P–OH), 877 (w, P–O), 646 (w, P–C). 1H NMR (400 MHz, CDCl3): δH 11.25 (1H, br s, OH), 1.22 (6H, d, 2JP–H = 14.4 Hz, CH3PCH3). 13C NMR (100 MHz, CDCl3): δC 16.42 (d, 1JC–P = 96.0 Hz, CH3PCH3). 31P NMR (162 MHz, CDCl3): δP 52.42 (CH3PCH3). MS (ESI−), m/z (rel. intensity %): 93 (M − H−, 30%).
O), 1127 (s, C–N), 1039 (s, P–O), 843 (m, P–O), 725 (w, CH2), 645 (w, P–C). 1H NMR (400 MHz, CD3OD): δH 2.07 (3H, br s, 3 × CH), 1.79 (6H, d, J = 2.3 Hz, 6 × CH), 1.70 (3H, d, J = 12.7 Hz, 3 × CH), 1.62 (3H, d, J = 12.1 Hz, 3 × CH), 1.14 (6H, d, 2JP–H = 13.6 Hz, CH3PCH3). 13C NMR (100 MHz, CD3OD): δC 52.65 (C–N), 41.58 (CH2), 36.52 (CH2), 30.46 (CH), 18.47 (d, 1JC–P = 93.8 Hz, CH3PCH3). 31P NMR (162 MHz, CD3OD, proton-coupled): δP 36.66 (sp, 2JP–H = 13.7 Hz, CH3PCH3).
O), 1079 and 1051 (m, P–OH), 973 and 946 (s, P–H), 721 (m, CH2), 675 (w, P–C). 1H NMR (400 MHz, CDCl3): δH 12.23 (1H, br s, OH), 7.1 (1H, dt, 1JP–H = 539.5 Hz, 3JH–H = 1.8 Hz, PH), 1.78–1.70 (2H, m, PCH2), 1.63–1.52 (2H, m, PCH2CH2), 1.38 (2H, quin., J = 6.6 Hz, PCH2CH2CH2), 1.25 (12H, br s, 6 × CH2), 0.87 (3H, t, J = 7.1 Hz, CH3). 13C NMR (100 MHz, CDCl3): δC 31.88, 30.43 (d, 3JC–P = 16.0 Hz, PCH2CH2CH2), 29.54, 29.36, 29.30, 29.24 (d, 1JC–P = 93.7 Hz, PCH2), 29.15, 22.68, 20.68 (d, 2JC–P = 2.9 Hz, PCH2CH2CH2), 14.10 (CH3). 31P NMR (162 MHz, CDCl3, proton-coupled): δP 37.29 (d quin., 1JP–H = 538.0 Hz, 2JP–H = 3JP–H = 28.4 Hz, CH2CH2PH). MS (ESI−), m/z (rel. intensity %): 205 (M − H−, 100%).
O), 1126 (w, C–N), 1039 (s, P–O−), 990 (w, P–H), 720 (w, CH2), 690 (w, P–C). 1H NMR (400 MHz, CDCl3): δH 8.83 (3H, br s, NH3), 7.1 (1H, d, 1JP–H = 479.2 Hz, PH), 2.11 (3H, br s), 1.94 (6H, br s), 1.66 (6H, br s), 1.52 (4H, br s), 1.35 (2H, br s), 1.24 (12H, br s), 0.87 (3H, t, J = 7.1 Hz, CH3). 13C NMR (100 MHz, CDCl3): δC 50.60 (CNH3), 40.63, 35.78, 33.22 (d, 1JC–P = 90.8 Hz, PCH2), 31.90, 31.05 (d, 3JC–P = 16.0 Hz, PCH2CH2CH2), 29.64, 29.52, 29.47, 29.33, 29.02, 22.84 (d, 2JC–P = 1.4 Hz, PCH2CH2), 22.68, 14.11 (CH3). 31P NMR (162 MHz, CDCl3): δP 25.56 (CH2PH). MS (ESI−), m/z (rel. intensity %): 205 (M−, 100%). MS (ESI+), m/z (rel. intensity %): 152 (M+, 100%).
Elem. Anal. (%): calculated for C20H43O2P requires C, 69.32; H, 12.51; P, 8.94%; found: C, 68.96; H, 12.54; P, 8.77%. IR, νmax (NaCl, evap. film)/cm−1: 2951 (m, C–H), 2915 (s, C–H), 2848 (s, C–H), 2600 (br, PO–H), 1470 (m, C–H), 1290 (w, C–H), 1137 (m, PO), 995 (w, P–OH), 719 (m, CH2). 1H NMR (400 MHz, CDCl3): δH 11.11 (1H, br s, OH), 1.71–1.54 (8H, m, CH2CH2PCH2CH2), 1.37 (4H, quin., J = 7.5 Hz, CH2CH2CH2PCH2CH2CH2), 1.26 (24H, br s, 12 × CH2), 0.88 (6H, t, J = 7.0 Hz, 2 × CH3). 13C NMR (100 MHz, CDCl3): δC 31.92, 30.94 (d, 3JC–P = 15.3 Hz, PCH2CH2CH2), 29.62, 29.49, 29.34, 29.23, 28.95 (d, 1JC–P = 91.6 Hz, PCH2), 22.70, 21.60 (d, 2JC–P = 3.6 Hz, PCH2CH2CH2), 14.13 (CH3). 31P NMR (162 MHz, CDCl3): δP 59.51 (CH2PCH2). MS (ESI−), m/z (rel. intensity %): 345 (M − H−, 100%).
O), 1121 (s, C–N), 1033 (s, P–O−), 734 (m, CH2), 645 (w, P–C). 1H NMR (400 MHz, CDCl3): δH 8.10 (3H, br s, NH3), 2.02 (3H, br s), 1.87 (6H, br s), 1.60 (6H, br s), 1.44 (8H, br s), 1.18 (28H, br s), 0.81 (6H, t, J = 7.0 Hz, CH3). 13C NMR (100 MHz, CDCl3): δC 50.26 (CNH3), 41.11, 35.88, 31.94, 31.56 (d, 3JC–P = 15.2 Hz, PCH2CH2CH2), 30.71 (d, 1JC–P = 88.6 Hz, PCH2), 29.71, 29.63, 29.57, 29.38, 29.15, 24.13, 22.71, 14.13 (CH3). 31P NMR (162 MHz, CDCl3): δP 41.76 (CH2PCH2).
O), 1075 and 1037 (m, P–OH), 975 and 959 (m, P–H), 720 and 709 (m, CH2). 1H NMR (400 MHz, CDCl3): δH 10.37 (1H, br s, OH), 7.1 (1H, dt, 1JP–H = 540.3 Hz, 3JH–H = 1.7 Hz, PH), 1.80–1.72 (2H, m, PCH2), 1.65–1.54 (2H, m, PCH2CH2), 1.40 (2H, quin., J = 6.8 Hz, PCH2CH2CH2), 1.26 (20H, br s, 10 × CH2), 0.89 (3H, t, J = 7.1 Hz, CH3). 13C NMR (100 MHz, CDCl3): δC 31.95, 30.44 (d, 3JC–P = 16.0 Hz, PCH2CH2CH2), 29.71, 29.69, 29.67, 29.65, 29.61, 29.38, 29.26 (d, 1JC–P = 93.7 Hz, PCH2), 29.16, 22.71, 20.63 (d, 2JC–P = 2.9 Hz, PCH2CH2CH2), 14.14 (CH3). 31P NMR (162 MHz, CDCl3, proton-coupled): δP 38.11 (d quin., 1JP–H = 539.9 Hz, 2JP–H = 3JP–H = 27.4 Hz, CH2CH2PH). MS (ESI−), m/z (rel. intensity %): 261 (M − H−, 100%).
O), 1127 (m, C–N), 1042 (s, P–O−), 976 (m, P–H), 719 (m, CH2), 690 (m, P–C). 1H NMR (400 MHz, CDCl3): δH 8.78 (3H, br s, NH3), 7.0 (1H, d, 1JP–H = 478.7 Hz, PH), 2.04 (3H, br s), 1.88 (6H, br s), 1.60 (6H, br s), 1.46 (4H, br s), 1.29 (2H, br s), 1.17 (20H, br s), 0.81 (3H, t, J = 7.1 Hz, CH3). 13C NMR (100 MHz, CDCl3): δC 50.57 (CNH3), 40.63, 35.79, 33.27 (d, 1JC–P = 90.8 Hz, PCH2), 31.95, 31.07 (d, 3JC–P = 16.0 Hz, PCH2CH2CH2), 29.72, 29.69, 29.67, 29.54, 29.49, 29.39, 29.02, 22.88, 22.71, 14.14 (CH3). 31P NMR (162 MHz, CDCl3): δP 25.47 (CH2PH).
O), 1086 and 1047 (s, P–OH), 945 (w, P–H), 736 (m, CH2), 699 (m, P–C). 1H NMR (500 MHz, CDCl3): δH 10.73 (1H, br s, OH), 7.1 (1H, dt, 1JP–H = 540.4 Hz, 3JH–H = 1.9 Hz, PH), 1.80–1.73 (2H, m, PCH2), 1.65–1.56 (2H, m, PCH2CH2), 1.41 (2H, quin., J = 7.0 Hz, PCH2CH2CH2), 1.27 (32H, br s, 16 × CH2), 0.90 (3H, t, J = 6.9 Hz, CH3). 13C NMR (125 MHz, CDCl3): δC 31.95, 30.44 (d, 3JC–P = 16.4 Hz, PCH2CH2CH2), 29.72, 29.70, 29.68, 29.66, 29.61, 29.38, 29.16, 28.96, 22.71, 20.59 (d, 2JC–P = 3.7 Hz, PCH2CH2CH2), 14.13 (CH3). 31P NMR (162 MHz, CDCl3, proton-coupled): δP 38.39 (d quin., 1JP–H = 539.9 Hz, 2JP–H = 3JP–H = 27.4 Hz, CH2CH2PH). MS (ESI−), m/z (rel. intensity %): 345 (M − H−, 100%).
O), 1127 (w, C–N), 1040 (s, P–O−), 985 (w, P–H), 720 (w, CH2), 690 (w, P–C). 1H NMR (400 MHz, CDCl3): δH 8.82 (3H, br s, NH3), 7.0 (1H, d, 1JP–H = 479.5 Hz, PH), 2.04 (3H, br s), 1.88 (6H, br s), 1.60 (6H, br s), 1.46 (4H, br s), 1.29 (2H, br s), 1.18 (32H, br s), 0.81 (3H, t, J = 7.1 Hz, CH3). 13C NMR (75 MHz, CDCl3): δC 50.48 (CNH3), 40.53, 35.81, 33.23 (d, 1JC–P = 90.4 Hz, PCH2), 31.95, 31.09 (d, 3JC–P = 15.8 Hz, PCH2CH2CH2), 29.74, 29.69, 29.56, 29.51, 29.39, 29.02, 22.84, 22.71, 14.14 (CH3). 31P NMR (162 MHz, CDCl3): δP 25.69 (CH2PH).
O), 1086 and 1049 (s, P–O), 879 (m, P–H), 721 (w, CH2), 680 (w, P–H). 1H NMR (400 MHz, CD3OD): δH 6.98 (1H, d, 1JP–H = 486.5 Hz, PH), 1.57–1.37 (6H, m, 3 × CH2), 1.30 (24H, br s, 12 × CH2), 0.91 (3H, t, J = 7.0 Hz, CH3). 13C NMR (100 MHz, CD3OD): δC 34.22, 33.32, 33.14, 32.18 (d, 3JC–P = 16.0 Hz, CH2CH2CH2P), 30.86, 30.84, 30.69, 30.56, 30.54, 23.80, 23.12 (d, 2JC–P = 2.2 Hz, CH2CH2CH2P), 14.53 (CH3). 31P NMR (162 MHz, CD3OD, proton-decoupled): δP 27.95 (CH2PH). MS (ESI+), m/z (rel. intensity %): 335 (M + Na+, 100%).
:0.5, CHCl3–MeOH, stain: PMA) to afford the product 5 as a white solid (12.14 g, 97%). mp 41–43 °C. Rf = 0.44 (9.5:0.5, CHCl3–MeOH, stain: PMA). HRMS (ESI+): calculated for C17H41O2NP [M + NH4+]+ requires 322.2869; found: 322.2869. IR, νmax (NaCl, evap. film)/cm−1: 2949 (w, C–H), 2917, 2849 and 1467 (m, C–H), 1234 (w, C–H), 1217 (w, P–OC), 1194 (w, PO), 1035 (w, P–O–C), 951 (w, P–H), 722 (w, CH2). 1H NMR (400 MHz, CDCl3): δH 6.98 (1H, dt, 1JP–H = 527.9 Hz, 3JH–H = 2.0 Hz, PH), 3.72 (3H, d, 3JP–H = 11.9 Hz, POCH3), 1.75–1.67 (2H, m, CH2P), 1.57–1.47 (2H, m, CH2CH2P), 1.32 (2H, quin., J = 6.8 Hz, CH2CH2CH2P), 1.18 (24H, br s, 12 × CH2), 0.81 (3H, t, J = 6.8 Hz, CH3). 13C NMR (125 MHz, CDCl3): δC 52.68 (d, 2JC–P = 6.4 Hz, POCH3), 31.90, 30.42 (d, 3JC–P = 15.5 Hz, CH2CH2CH2P), 29.66, 29.64, 29.59, 29.54, 29.35, 29.29, 29.08, 28.47 (d, 1JC–P = 92.7 Hz, CH2CH2CH2P), 22.67, 20.63 (d, 2JC–P = 1.8 Hz, CH2CH2CH2P), 14.10 (CH3). 31P NMR (162 MHz, CDCl3, proton-coupled): δP 42.10 (dm, 1JP–H = 528.2 Hz, CH2PH). MS (ESI+), m/z (rel. intensity %): 322 (M + NH4+, 100%).
:0.5, CHCl3–MeOH, stain: PMA) to afford the product 21 as a white solid (9.47 g, 97%). mp 56–58 °C.
R
f = 0.44 (9.5:0.5, CHCl3–MeOH, stain: PMA). HRMS (ESI+): calculated for C21H45O2NaP [M + Na+]+ requires 383.3049; found: 383.3054. IR, νmax (NaCl, evap. film)/cm−1: 2935 (m, C–H), 2917 and 2850 (s, C–H), 2346 (w, P–H), 1467 and 1236 (m, C–H), 1208 (m, P–OC), 1175 (w, PO), 1037 (m, P–O–C), 954 (m, P–H), 802 and 789 (w, P–O–C), 721 (w, CH2). 1H NMR (400 MHz, CDCl3): δH 6.72 (1H, dt, 1JP–H = 528.1 Hz, 3JH–H = 1.8 Hz, PH), 3.46 (3H, d, 3JP–H = 11.6 Hz, POCH3), 1.49–1.41 (2H, m, CH2P), 1.32–1.21 (2H, m, CH2CH2P), 1.07 (2H, quin., J = 7.0 Hz, CH2CH2CH2P), 0.93 (32H, br s, 16 × CH2), 0.55 (3H, t, J = 7.0 Hz, CH3). 13C NMR (100 MHz, CDCl3): δC 52.64 (d, 2JC–P = 6.5 Hz, POCH3), 31.88, 30.38 (d, 3JC–P = 16.0 Hz, CH2CH2CH2P), 29.66, 29.64, 29.62, 29.57, 29.51, 29.33, 29.27, 29.06, 28.45 (d, 1JC–P = 93.0 Hz, CH2CH2CH2P), 22.63, 20.60 (d, 2JC–P = 2.9 Hz, CH2CH2CH2P), 14.05 (CH3). 31P NMR (162 MHz, CDCl3, proton-coupled): δP 41.94 (dm, 1JP–H = 528.1 Hz, CH2PH). MS (ESI+), m/z (rel. intensity %): 361 (M + H+, 60%), 383 (M + Na+, 100%).
C–H), 2944 (w, C–H), 1639 (w, CC), 1473 (s, C–H), 1405 (m,C–H), 1369 (w, C–H), 1126 (w, C–N), 1008 (m,C–H), 956 and 890 (s,C–H). 1H NMR (500 MHz, CD3OD): δH 5.75 (1H, ddt, J = 17.1, 9.8, 7.6 Hz, –CH), 5.38 (1H, d, J = 16.7 Hz,CH2), 5.34 (1H, d, J = 10.1 Hz,CH2), 3.71 (2H, d, J = 7.2 Hz, NCH2), 2.80 (9H, s, 3 × NCH3). 13C NMR (100 MHz, CD3OD): δC 129.8 (CH2), 126.7 (–CH), 69.44 (NCH2), 53.59 (NCH3). MS (ESI+), m/z (rel. intensity %): 100 (M+, 20%).
:3, CHCl3–MeOH, stain: ninhydrin). HRMS (ESI+): calculated for C7H14N [M + H]+ requires 112.1121; found: 112.1111. IR, νmax (ATIR)/cm−1: 3079 (w,C–H), 2939 and 2848 (m, C–H), 2781 (m, N–CH2), 1653 (m, CC), 1436, 1371, 1282, 1268 and 1222 (m, C–H), 1135 (m, C–N), 1014 and 887 (s,C–H). 1H NMR (400 MHz, CDCl3): δH 4.58 (2H, t, 4JH–H = 1.0 Hz,CH2), 2.32 (4H, t, J = 6.3 Hz, 2 × CH2N), 2.19 (3H, s, NCH3), 2.17 (4H, t, J = 5.5 Hz, 2 × CCH2). 13C NMR (100 MHz, CDCl3): δC 145.6 (>C), 107.9 (CH2), 57.01 (CH2N), 46.04 (NCH3), 34.49 (CCH2). MS (ESI+), m/z (rel. intensity %): 112 (M + H+, 100%).
:1:0.05, CHCl3–MeOH–conc. NH4OH, stain: ninhydrin) to afford the product 26a as a colourless oil (1.00 g, 73%). Rf = 0.28 (8:1:0.05, CHCl3–MeOH–conc. NH4OH, stain: ninhydrin). HRMS (ESI+): calculated for C24H51O2NP [M + H]+ requires 416.3652; found: 416.3649. IR, νmax (NaCl, evap. film)/cm−1: 2925 and 2853 (s, C–H), 2780 (m, N–CH2), 1463 (m, C–H), 1279 (w, C–H), 1207 (m, P–OC), 1194 (m, PO), 1142 (w, C–N), 1039 (m, P–O–C), 822 and 804 (w, P–O–C), 719 (w, CH2). 1H NMR (400 MHz, CDCl3): δH 3.62 (3H, d, 3JP–H = 10.4 Hz, POCH3), 2.84 (2H, apparent d, J = 11.1 Hz, 2 × CHN), 2.25 (3H, s, NCH3), 1.99 (2H, apparent dd, J = 11.7, 1.6 Hz, 2 × CHN), 1.86–1.78 (2H, m), 1.67–1.55 (5H, m), 1.50–1.39 (4H, m), 1.34–1.25 (2H, m), 1.19 (24H, br s, 12 × CH2), 0.81 (3H, t, J = 7.1 Hz, CH3). 13C NMR (100 MHz, CDCl3): δC 54.43 (CH2N), 49.70 (d, 2JC–P = 6.6 Hz, POCH3), 44.98 (NCH3), 33.01 (d, 1JC–P = 89.5 Hz, PCH2), 32.38, 30.93, 29.90 (d, 3JC–P = 15.4 Hz), 28.69, 28.67, 28.63, 28.59, 28.38, 28.36, 28.18, 28.12, 27.30, 21.69, 21.11, 13.13 (CH3). 31P NMR (162 MHz, CDCl3): δP 58.02 (CH2PCH2). MS (ESI+), m/z (rel. intensity %): 416 (M + H+, 10%).
:1:0.05, CHCl3–MeOH–conc. NH4OH, stain: ninhydrin) to afford the product 26b as a colourless oil (0.21 g, 75%). Rf = 0.28 (8:1:0.05, CHCl3–MeOH–conc. NH4OH, stain: ninhydrin). HRMS (ESI+): calculated for C25H53O2NP [M + H]+ requires 430.3808; found: 430.3806. IR, νmax (NaCl, evap. film)/cm−1: 2927 and 2852 (m, C–H), 2782 (w, N–CH2), 1465, 1460 and 1277 (w, C–H), 1218 (w, P–OC), 1193 (m, PO), 1143 (w, C–N), 1040 (m, P–O–C), 823 (w, P–O–C), 718 (w, CH2). 1H NMR (400 MHz, CDCl3): δH 4.02–3.92 (2H, m, POCH2), 2.75 (2H, d, J = 11.4 Hz, 2 × CHN), 2.18 (3H, s, NCH3), 1.89 (2H, dd, J = 11.8, 1.8 Hz, 2 × CHN), 1.83–1.77 (2H, m), 1.69–1.55 (5H, m), 1.53–1.42 (2H, m), 1.37–1.28 (4H, m), 1.24 (3H, t, J = 7.1 Hz, POCH2CH3), 1.19 (24H, br s, 12 × CH2), 0.81 (3H, t, J = 6.9 Hz, CH3). 13C NMR (100 MHz, CDCl3): δC 59.83 (d, 2JC–P = 6.5 Hz, POCH2), 55.62 (CH2N), 46.36 (NCH3), 34.62 (d, 1JC–P = 89.4 Hz, PCH2), 33.89, 31.92, 30.90 (d, 3JC–P = 15.3 Hz), 29.79 (d, 2JC–P = 3.6 Hz), 29.69, 29.66, 29.65, 29.63, 29.58, 29.38, 29.36, 29.12, 28.80, 22.69, 22.12 (d, 2JC–P = 3.6 Hz), 16.71 (d, 3JC–P = 5.8 Hz, POCH2CH3), 14.13 (CH3). 31P NMR (162 MHz, CDCl3): δP 56.58 (CH2PCH2). MS (ESI+), m/z (rel. intensity %): 430 (M + H+, 100%), 452 (M + Na+, 95%).
:1, 10 cm3). The mixture was refluxed under a dry nitrogen atmosphere and monitored by means of thin-layer chromatography (10:2:0.2, CHCl3–MeOH–conc. NH4OH, stain: ninhydrin) and 1H NMR spectroscopy. After one day of refluxing the reaction mixture cooled to room temperature and the solvent evaporated under reduced pressure. The residue was then re-dissolved in chloroform and filtered. The filtrate was concentrated under reduced pressure to give a yellow solid which was purified by recrystallization from tetrahydrofuran–ethyl acetate to afford the product 28 as a bright white powder (0.15 g, 95%). mp 233–234 °C (dec.) (from THF–EtOAc). Rf = 0.32 (6.5:2.5:0.3, CHCl3–MeOH–conc. NH4OH, stain: ninhydrin). HRMS (ESI+): calculated for C25H53O2NP [M]+ requires 430.3808; found: 430.3827. IR, νmax (NaCl, evap. film)/cm−1: 2916, 2852 and 1468 (m, C–H), 1409 (w, +NMe2), 1304 (w, C–H), 1207 (m, P–OC), 1191 (m, PO), 1040 (m, P–O–C), 954 (w, +NMe2), 823 and 814 (w, P–O–C), 718 (w, CH2). 1H NMR (400 MHz, CDCl3): δH 3.76 (2H, d, J = 12.6 Hz, 2 × CHN), 3.64 (3H, d, 3JP–H = 10.6 Hz, POCH3), 3.60 (2H, dd, J = 12.4, 2.5 Hz, 2 × CHN), 3.49 (3H, s, NCH3), 3.37 (3H, s, NCH3), 2.30–2.15 (1H, m), 2.12–2.06 (2H, m), 1.98–1.87 (2H, m), 1.85–1.75 (2H, m), 1.71–1.61 (2H, m), 1.54–1.42 (2H, m), 1.37–1.27 (2H, m), 1.19 (24H, br s, 12 × CH2), 0.81 (3H, t, J = 7.0 Hz, CH3). 13C NMR (100 MHz, CDCl3): δC 61.56 (CH2N), 55.23 (NCH3), 50.20 (d, 2JC–P = 6.6 Hz, POCH3), 47.57 (NCH3), 31.41 (d, 1JC–P = 90.2 Hz, PCH2), 30.93, 29.87 (d, 3JC–P = 14.7 Hz), 28.71, 28.70, 28.67, 28.62, 28.44, 28.37, 28.19, 27.78 (d, 1JC–P = 88.8 Hz, PCH2), 26.63 (d, 2JC–P = 4.4 Hz), 26.48, 26.39, 26.33, 26.25, 21.70, 21.08 (d, 2JC–P = 4.4 Hz), 13.15 (CH3). 31P NMR (162 MHz, CDCl3): δP 57.05 (CH2PCH2). MS (ESI+), m/z (rel. intensity %): 430 (M+, 100%).
O), 1134 (w, C–N), 1035 (w, P–O), 954 (w, +NMe2), 720 (w, CH2). 1H NMR (400 MHz, CD3OD): δH 3.39 (2H, d, J = 12.6 Hz, 2 × CHN), 3.29 (2H, dd, J = 12.7, 2.5 Hz, 2 × CHN), 3.07 (3H, s, NCH3), 3.03 (3H, s, NCH3), 2.00–1.97 (3H, m), 1.80–1.69 (4H, m), 1.67–1.59 (2H, m), 1.55–1.44 (2H, m), 1.33 (2H, quin., J = 6.5 Hz, CH2CH2CH2P), 1.19 (24H, br s, 12 × CH2), 0.81 (3H, t, J = 7.0 Hz, CH3). 13C NMR (100 MHz, 328.1 K, CD3OD): δC 63.69 (CH2N), 56.79 (NCH3), 48.81 (NCH3), 35.00 (d, 1JC–P = 89.5 Hz, PCH2), 33.02, 31.96 (d, 3JC–P = 14.7 Hz), 30.74, 30.70, 30.54, 30.39, 30.29, 29.12, 28.80, 28.71, 23.67, 22.96 (d, 2JC–P = 3.7 Hz), 14.41 (CH3). 31P NMR (162 MHz, CD3OD): δP 52.85 (CH2PCH2). MS (ESI+), m/z (rel. intensity %): 416 (M + H+, 100%), 438 (M + Na+, 90%).
Acknowledgements
We gratefully acknowledge the University of Manchester and the A. G. Leventis Foundation (Cyprus) for financial support and Dr C. I. F. Watt at Manchester for valuable assistance and helpful discussions.Notes and references
- (a) H. Brachwitz and C. Vollgraf, Pharmacol. Ther., 1995, 66, 39–82 CrossRef CAS; (b) W. J. Houlihan, M. Lohmeyer, P. Workman and S. H. Cheon, Med. Res. Rev., 1995, 15, 157–223 CrossRef CAS.
- (a) G. S. Tarnowski, I. M. Mountain, C. C. Stock, P. G. Munder, H. U. Weltzien and O. Westphal, Cancer Res., 1978, 38, 339–344 CAS; (b) P. G. Munder, M. Modolell, R. Andreesen, H. U. Weltzien and O. Westphal, Springer Semin. Immunopathol., 1979, 2, 187–203 CrossRef CAS; (c) D. Berkovic, Gen. Pharmacol., 1998, 31, 511–517 CrossRef CAS.
- H. Eibl and C. Unger, Cancer Treat. Rev., 1990, 17, 233–242 CrossRef CAS.
- (a) F. Mollinedo, Expert Opin. Ther. Pat., 2007, 17, 385–405 CrossRef CAS; (b) S. R. Vink, W. J. van Blitterswijk, J. H. M. Schellens and M. Verheij, Cancer Treat. Rev., 2007, 33, 191–202 CrossRef CAS.
- (a) J. Stekar, P. Hilgard, R. Voegeli, H. R. Maurer, J. Engel, B. Kutscher, G. Nößner and W. Schumacher, Cancer Chemother. Pharmacol., 1993, 32, 437–444 CrossRef CAS; (b) P. Hilgard, T. Klenner, J. Stekar, G. Nössner, B. Kutscher and J. Engel, Eur. J. Cancer, 1997, 33, 442–446 CrossRef CAS.
- C. Oberle, U. Massing and H. F. Krug, Biol. Chem., 2005, 386, 237–245 CrossRef CAS.
- V. Jendrossek and R. Handrick, Curr. Med. Chem.: Anti-Cancer Agents, 2003, 3, 343–353 CrossRef CAS.
- R. Ulbrich-Hofmann, A. Lerchner, M. Oblozinsky and L. Bezakova, Biotechnol. Lett., 2005, 27, 535–5439 CrossRef CAS.
- M. S. Markoulides and A. C. Regan, Tetrahedron Lett., 2011, 52, 2954–2956 CrossRef CAS.
- (a) E. A. Boyd, K. James and A. C. Regan, Tetrahedron Lett., 1994, 35, 4223 CrossRef CAS; (b) E. L. Ravaschino, R. Docampo and J. B. Rodriguez, J. Med. Chem., 2006, 49, 426–435 CrossRef CAS.
- L. Coudray and J.-L. Montchamp, Eur. J. Org. Chem., 2008, 3601–3613 CrossRef CAS.
- (a) E. E. Nifant'ev and M. P. Koroteev, J. Gen. Chem. USSR (Engl. Trans.), 1967, 37, 1293 Search PubMed; (b) E. E. Nifant'ev, R. K. Magdeeva and N. P. Shchepet'eva, J. Gen. Chem. USSR (Engl. Trans.), 1980, 50, 1416 Search PubMed.
- (a) D. S. Karanewsky, M. C. Badia, D. W. Cushman, J. M. DeForrest, T. Dejneka, M. J. Loots, M. G. Perri, E. W. Petrillo Jr. and J. R. Powell, J. Med. Chem., 1988, 31, 204–212 CrossRef CAS; (b) N. G. Anderson, M. L. Coradetti, J. A. Cronin, M. L. Davies, M. B. Gardineer, A. S. Kotnis, D. A. Lust and V. A. Palaniswamy, Org. Process Res. Dev., 1997, 1, 315–319 CrossRef CAS; (c) Y. Feng and J. K. Coward, J. Med. Chem., 2006, 49, 770–778 CrossRef CAS; (d) C. Selvam, N. Oueslati, I. A. Lemasson, I. Brabet, D. Rigault, T. Courtiol, S. Cesarini, N. Triballeau, H.-O. Bertrand, C. Goudet, J.-P. Pin and F. C. Acher, J. Med. Chem., 2010, 53, 2797–2813 CrossRef CAS.
- M. I. Antczak and J.-L. Montchamp, Synthesis, 2006, 3080–3084 CAS.
- S. Deprèle and J.-L. Montchamp, J. Org. Chem., 2001, 66, 6745–6755 CrossRef.
- For a recent example see: A. F. Parsons and A. Wright, Synlett, 2008, 2142–2146 CrossRef CAS.
- For examples, see: (a) L.-B. Han and C.-Q. Zhao, J. Org. Chem., 2005, 70, 10121–10123 CrossRef CAS; (b) S. R. Piettre, Tetrahedron Lett., 1996, 37, 2233–2236 CrossRef CAS; (c) H.-J. Zeiss, Tetrahedron, 1992, 48, 8263–8270 CrossRef CAS; (d) J. G. Dingwall and B. Tuck, J. Chem. Soc., Perkin Trans. 1, 1986, 2081–2090 RSC; (e) M. Finke and H.-J. Kleiner, Liebigs Ann. Chem., 1974, 741–750 CrossRef CAS; (f) L. P. Reiff and H. S. Aaron, J. Am. Chem. Soc., 1970, 92, 5275–5276 CrossRef CAS.
- For an example, see: A. Gautier, G. Garipova, C. Salcedo, S. Balieu and S. R. Piettre, Angew. Chem., Int. Ed., 2004, 43, 5963–5967 CrossRef CAS.
- O. Dubert, A. Gautier, E. Condamine and S. R. Piettre, Org. Lett., 2002, 4, 359–362 CrossRef CAS.
- S. J. Fitch, J. Am. Chem. Soc., 1964, 86, 61–64 CrossRef CAS.
- A. M. Schmidt and P. Eilbracht, J. Org. Chem., 2005, 70, 5528–5535 CrossRef CAS.
- E. W. Della and P. A. Smith, J. Org. Chem., 1999, 64, 1798–1806 CrossRef CAS.
- (a) J. Zygmunt, P. Kafarski and P. Mastalerz, Synthesis, 1978, 609–612 CrossRef CAS; (b) S. A. Biller, C. Forster, E. M. Gordon, T. Harrity, L. C. Rich, J. C. Marretta and C. P. Ciosek Jr., J. Med. Chem., 1991, 34, 1912–1914 CrossRef CAS.
- (a) D. D. Perrin and W. L. F. Armarego, Purification of Laboratory Chemicals, Butterworth-Heinemann, Oxford, UK, 4th edn, 1996 Search PubMed; (b) B. S. Furniss, A. J. Hannaford, P. W. G. Smith and A. R. Tatchell, Vogel's Textbook of Practical Organic Chemistry, Pearson Education, Harlow, Essex, UK, 5th edn, 1989 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H, 13C and 31P NMR spectra. See DOI: 10.1039/c2ob26395e |
| This journal is © The Royal Society of Chemistry 2013 |