 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotolabile plasmonic vesicles assembled from amphiphilic gold nanoparticles for remote-controlled traceable drug delivery†
Jibin
Song
,
Zheng
Fang
,
Chenxu
Wang
,
Jiajing
Zhou
,
Bo
Duan
,
Lu
Pu
and
Hongwei
Duan
*
School of Chemical and Biomedical Engineering, Nanyang Technological University, 70 Nanyang Drive, 637457, Singapore. E-mail: hduan@ntu.edu.sg
First published on 23rd April 2013
Abstract
We have developed a new type of photo-responsive plasmonic vesicles that allow for active delivery of anticancer payloads to specific cancer cells and personalized drug release regulated by external photo-irradiation. Our results show that amphiphilic gold nanoparticles carrying hydrophilic poly(ethylene glycol) (PEG) and photo-responsive hydrophobic poly(2-nitrobenzyl acrylate) (PNBA) can assemble into plasmonic vesicles with gold nanoparticles embedded in the hydrophobic shell of PNBA, which can be converted into hydrophilic poly(acrylic acid) upon photo exposure. Benefiting from the interparticle plasmonic coupling of gold nanoparticles in close proximity, the plasmonic vesicles assembled from amphiphilic gold nanoparticles exhibit distinctively different optical properties from single nanoparticle units, which offer the opportunity to track the photo-triggered disassembly of the vesicles and the associated cargo release by plasmonic imaging. We have shown the dense layer of PEG grafts on the vesicles not only endow plasmonic vesicles with excellent colloidal stability, but also serve as flexible spacers for bioconjugation of targeting ligands to facilitate the specific recognition of cancer cells. The targeted delivery of model anticancer drug doxorubicin, investigated by dual-modality plasmonic and fluorescence imaging and toxicity studies, clearly demonstrated the potential of photolabile plasmonic vesicles as multi-functional drug carriers.
1 Introduction
Drug delivery systems (DDS) are actively explored to address major challenges associated with conventional small molecular anticancer therapeutics such as non-specific exposure to healthy cells/tissues, limited solubility and stability in physiological conditions, and transient blood circulation.1–3 Active targeting of nanoscale DDS to cancer cells can be achieved by tagging their surfaces with ligands recognizing overexpressed biomarkers on cancer cell surfaces.1–3 In parallel, nanocarriers that release their payloads in response to external stimuli offer new possibilities for targeted drug delivery by taking advantage of the disease-specific biochemical microenvironment such as pH and enzymes or bioorthogonal exogenous triggers such as photo-irradiation.4–22 Particularly, photo-regulated cargo release affords flexible spatiotemporal control and personalized kinetics, emerging as a universal remote-control mechanism for targeted drug delivery in a wide range of cell types.5,10–16,23–27 Recently, growing research efforts have led to the development of nanocarriers based on polymeric micelles, mesoporous silica, and inorganic nanocrystals with photo-responsive mechanisms for the controlled release of small molecular anticancer drugs and therapeutic nucleic acids.5,10–16,28–32Here we present the development of photolabile plasmonic vesicles assembled from amphiphilic gold nanocrystals as a new type of optically traceable nanocarriers for synergistic ligand-directed cancer cell targeting and on-demand intracellular cargo release triggered by photo irradiation. Vesicular structures such as liposomes and polymersomes are promising nanocarriers under intense research because their unique structures allow for loading of a large number of therapeutic agents with diverse chemical characteristics.33–35 However, the majority of previously reported vesicles lack an intrinsic and adaptive imaging modality to follow their spatial and temporal distribution and the cargo release process, which are essential for the rapid screening of potential drug candidates.33–37
We previously reported that amphiphilic nanocrystals coated with mixed polymer brushes undergo amphiphilicity-driven self-assembly in aqueous media, forming vesicles with a monolayer of nanocrystals embedded in the shell of collapsed hydrophobic brushes, which is stabilized by the extended hydrophilic brushes.38 More recently, our results have shown that the use of amphiphilic gold nanoparticles with pH-sensitive polymers as the shell-forming brush led to pH-sensitive plasmonic vesicles that can release the encapsulated anticancer drugs in response to the acidic environment of intracellular compartments.39 In the present work, we have constructed plasmonic vesicles using gold nanoparticles coated with mixed polymer brushes of hydrophilic polyethylene glycol (PEG) and hydrophobic poly(2-nitrobenzyl acrylate) (PNBA) as building blocks, as illustrated in Fig. 1. This new design features a unique set of optical and structural properties to serve as multi-functional nanocarriers for cancer cell-targeted plasmonic imaging, photo-activated vesicle destruction, and optically detectible intracellular drug delivery. First, the PEG coating and the photo-reactive PNBA, which transforms into hydrophilic poly(acrylic acid) (PAA) upon photo-irradiation (Fig. 1), endow the plasmonic vesicles with excellent colloidal stability under physiological conditions and photo-regulated permeability for triggered cargo release. The PEG grafts also allow the facile conjugation of targeting ligands, i.e. folate, to direct the specific recognition of plasmonic vesicles to cancer cells. Second, plasmonic nanostructures exhibit localized surface plasmon resonance (LSPR) arising from the collective oscillation of free conduction electrons, and the ability to efficiently scatter light at the LSPR wavelength enables their use as bioimaging probes.40–44 Therefore, the gold nanoparticles can be employed as built-in optical probes for real-time intracellular imaging and tracing of the plasmonic vesicles. Third, the LSPR wavelength is highly sensitive to the proximity of other particles in an ensemble of plasmonic nanostructures, with decreasing interparticle distance leading to red-shifts of LSPR and increased scattering cross-sections.43,44 This distance-dependant plasmonic coupling imparts collective optical properties to the vesicle, which are distinctly different from those of discrete nanoparticles.45,46 As a result, disassembly of the vesicles into single nanoparticles, induced by the photo-irradiation-induced conversion of hydrophobic PNBA into hydrophilic PAA, gives rise to a dramatic change in optical properties, which provides a unique approach to optically monitoring the light-triggered cargo release inside live cells by plasmonic imaging. Scattering-based plasmonic imaging has emerged as a powerful imaging technique because the distance-dependent plasmonic coupling provides new possibilities to study dynamic biological processes.47–50 In comparison with commonly used fluorescent probes, the photobleaching-resistant scattering signal from plasmonic nanostructures offers a much longer imaging window, which is particularly interesting for the long-term tracking of intracellular events.
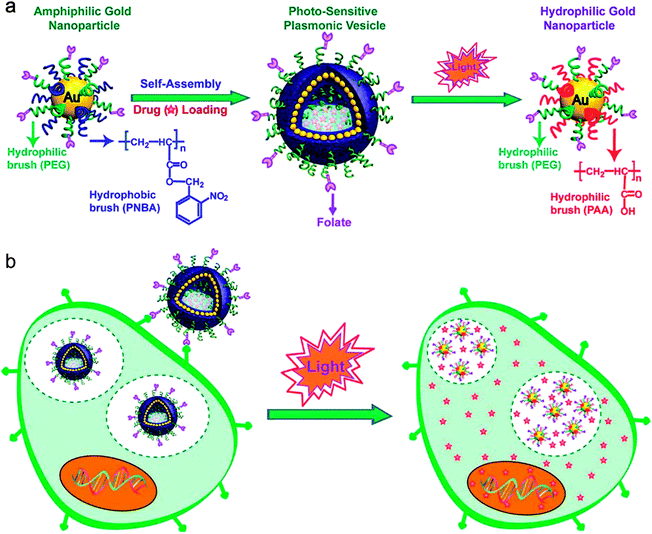 | ||
| Fig. 1 (a) Schematic illustration of self-assembly of amphiphilic gold nanoparticles with mixed polymer brush coatings into plasmonic vesicles and photo-responsive destruction of the vesicles. (b) Cellular binding and photo-regulated intracellular payload release of the plasmonic vesicles. | ||
2 Results and discussion
Amphiphilic gold nanoparticles with mixed polymer brushes of PEG and PNBA were synthesized through sequentially performed “grafting to” and “grafting from” reactions.38,51,52 In the “grafting to” step, lipoic acid-ended methoxy-PEG (MPEG-LA) and initiators of atom transfer radical polymerization (ATRP) were attached on 14 nm citrate-stabilized gold nanoparticles by forming Au–S bonds. In the following “grafting from” reaction, surface initiated ATRP of 2-nitrobenzyl acrylate (NBA) monomers were carried out to obtain the amphiphilic gold nanoparticles (Au@PEG/PNBA). This two-step synthetic approach offers flexible control over the molecular weight (MW) and relative ratios of the hydrophilic and hydrophobic brushes, which allows us to prepare a series of amphiphilic Au@PEG/PNBA nanoparticles with the PEG/PNBA ratio ranging from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:4 (Fig. S1†), while maintaining the same chain length of PEG (Mw = 5 kDa) and PNBA (Mw = 25 kDa) grafts and the graft density (0.4–0.5 chain per nm2) (Fig. S2 and S3†). The amphiphilic nanoparticles were assembled into vesicles following the film rehydration protocol we used previously.38 Electron microscopy observations (Fig. 2a and b) confirm the successful preparation of vesicles with a single layer of closely attached gold nanoparticles in the shell. The close packing of gold nanoparticles in the vesicle leads to strong interparticle plasmonic coupling, as manifested by the significant red-shift of the LSPR band relative to that (530 nm) of Au@PEG/PNBA in chloroform (Fig. 2c). We have found the spectral shift is highly dependent on the relative ratio of PEG and PNBA chains, with increasing PEG fraction leading to smaller red-shifts. For instance, the nanoparticles with PEG/PNBA ratios of 1:1, 1:2 and 1:4 gave rise to vesicles with LSPR centered at 550, 630, and 745 nm (Fig. S4†), respectively. Self-assembly of amphiphilic nanoparticles with homogeneously covered PEG and PNBA grafts into the vesicular shell necessitates conformational changes of the polymer grafts, in which PEG chains reorganize to face the aqueous environment on either side of the shell and the hydrophobic PNBA chains collapse to form the shell matrix. A larger fraction of PEG grafts is expected to cause stronger steric hindrance for the nanoparticles to approach each other, therefore leading to large interparticle distances and weaker plasmonic coupling. The plasmonic vesicles exhibited excellent colloidal stability in aqueous buffer solutions and storage at 4 °C for 3 months did not affect their morphology and hydrodynamic sizes (∼210 nm).
:1 to 1:4 (Fig. S1†), while maintaining the same chain length of PEG (Mw = 5 kDa) and PNBA (Mw = 25 kDa) grafts and the graft density (0.4–0.5 chain per nm2) (Fig. S2 and S3†). The amphiphilic nanoparticles were assembled into vesicles following the film rehydration protocol we used previously.38 Electron microscopy observations (Fig. 2a and b) confirm the successful preparation of vesicles with a single layer of closely attached gold nanoparticles in the shell. The close packing of gold nanoparticles in the vesicle leads to strong interparticle plasmonic coupling, as manifested by the significant red-shift of the LSPR band relative to that (530 nm) of Au@PEG/PNBA in chloroform (Fig. 2c). We have found the spectral shift is highly dependent on the relative ratio of PEG and PNBA chains, with increasing PEG fraction leading to smaller red-shifts. For instance, the nanoparticles with PEG/PNBA ratios of 1:1, 1:2 and 1:4 gave rise to vesicles with LSPR centered at 550, 630, and 745 nm (Fig. S4†), respectively. Self-assembly of amphiphilic nanoparticles with homogeneously covered PEG and PNBA grafts into the vesicular shell necessitates conformational changes of the polymer grafts, in which PEG chains reorganize to face the aqueous environment on either side of the shell and the hydrophobic PNBA chains collapse to form the shell matrix. A larger fraction of PEG grafts is expected to cause stronger steric hindrance for the nanoparticles to approach each other, therefore leading to large interparticle distances and weaker plasmonic coupling. The plasmonic vesicles exhibited excellent colloidal stability in aqueous buffer solutions and storage at 4 °C for 3 months did not affect their morphology and hydrodynamic sizes (∼210 nm).
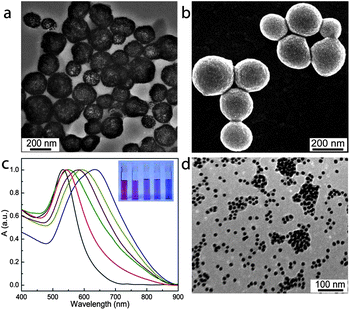 | ||
| Fig. 2 Transmission electron microscopy (TEM) (a) and scanning electron microscopy (SEM) (b) images of plasmonic vesicles of gold nanoparticles with a PEG/PNBA ratio of 1:2. (c) UV-vis spectra of the vesicles of gold nanoparticles with a PEG/PNBA ratio of 1:2 upon photo-irradiation for different periods (yellow line: 5 min, purple line: 8 min, green line: 10 min, and red line: 15 min) and corresponding photographs of the vesicle dispersions (inset). (d) TEM image of the disassembled vesicle after photo irradiated for 15 min. | ||
The shell-forming hydrophobic PNBA grafts carry a photolabile 2-nitrobenzyl ester moiety in their repeat units. Upon photo-irradiation (365 nm), this pedant group is cleaved from the polymer backbone, converting the hydrophobic PNBA chains into hydrophilic PAA.10,53 This well-established photochemical reaction has been used in a wide spectrum of applications in biological systems such as photo-triggered drug delivery and photodegradable hydrogels for tissue engineering and has shown excellent biocompatibility.5,10,13,54 We have examined the photo-irradiation induced destruction of plasmonic vesicles assembled from nanoparticles with a PEG/PNBA ratio of 1:2 (an average number of 93 PEG and 186 PNBA chains). Irradiation of the vesicle dispersion in PBS buffer (pH 7.4) using a hand-held UV lamp (2.3 mW cm−2) led to a rapid blue-shift of the LSPR peak from 633 nm to 547 nm within 15 min, accompanied by a gradual color change from deep blue to red (Fig. 2c). As discussed earlier, the LSPR of gold nanoparticles is extremely sensitive to the interparticle-spacing in the plasmonic vesicle. Carboxylic acid (pKa ∼ 4–4.5) groups in PAA are mostly deprotonated at pH 7.4, resulting in electrostatic repulsion between the neighboring particles to push the nanoparticles apart.10 When a large fraction of the PNBA is transformed into PAA, the gold nanoparticles become completely water-soluble. This process is accompanied with collapse of the vesicles (Fig. S5A†) and eventually disassembly of the vesicles into single nanoparticles or small clusters of nanoparticles (Fig. 2d and S5B†). Dynamic light scattering measurements show that the hydrodynamic size of the vesicles decreased from ∼210 nm to ∼30 nm after 15 min of photo-irradiation, consistent with the spectral blue-shift observed in UV-vis analysis. Complete disruption of the vesicles formed by gold nanoparticles with PEG/PNBA ratios of 1:1 and 1:4 required photo-irradiation of 14 min and 17 min, respectively.
The intense scattering light of plasmonic nanostructures at their LSPR wavelengths can be captured by a dark-field microscope.41,42,46,55,56 Although a 14 nm gold nanoparticle building block exhibits a negligible scattering signal, the strong interparticle plasmonic coupling in the vesicles give rise to both spectral red-shifts and an enhanced scattering cross-section, which enables imaging of the vesicles at the single-particle level.38,39 As shown in Fig. 3a, individual vesicles spread on a glass cover slip are red in color. After exposure to light irradiation in situ on the microscope (330–385 nm band-pass filter) for 15 min, most of the vesicles experienced evident color changes from red to orange and yellow-green (Fig. 3b and c). In line with this observation, scattering light of a representative vesicle exhibits a prominent spectral blue shift after exposure to light for 15 min, as shown in Fig. 3d.
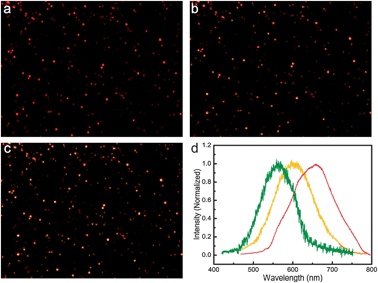 | ||
| Fig. 3 Dark-field images of the plasmonic vesicles before (a) and after photo irradiation for 10 min (b) and 15 min (c). (d) Representative scattering spectra of a single vesicle before (red line) and after photo irradiation for 10 min (yellow line) and 15 min (green line). | ||
We have used folate as the targeting ligand to promote the specific binding of the plasmonic vesicles to cancer cells. Folate is an intriguing small molecular targeting ligand, showing high affinity (Kd = 10−10 M) to folate receptors, which are highly overexpressed in varieties of human cancer types such as ovarian, breast and colon cancer.57 The MDA-MB-435 breast cancer cells used in this study have been demonstrated to overexpress folate receptor on their surfaces.58 Folate was introduced on the gold nanoparticle surfaces by replacing 30% of the mono-functional MPEG-LA with hetero-functional PEG carrying lipoic acid and folate at two ends during the “grafting to” reaction. We have found that the folate-conjugated plasmonic vesicles quickly bind to MDA-MB-435 cells after 40 min of incubation (Fig. 4a) and the scattering background from the cells is overwhelmed by the strong signal of the vesicles. In contrast, the vesicles without targeting ligands showed little binding to the cells (Fig. 4b). This is not surprising because the control vesicles are covered with a dense layer of PEG, which is known to effectively suppress non-specific binding to live cells.59 To further verify that the targeted vesicles are taken up by MDA-MB-435 cells through a receptor-mediated process, a competitive assay with free folic acid (1.0 mM) was conducted. Live cell imaging (Fig. 4c) showed that free folic acid effectively inhibited the binding of the vesicles to the cell surfaces, confirming the specificity of the targeted vesicles to folate receptors. To quantitatively determine the uptake of folate-targeted and non-targeted vesicles by the cells, we analyzed the Au element content in the cells by inductively coupled plasma mass spectroscopy (ICP-MS) analysis.60,61 As shown in Fig. 4e, the average number of gold nanoparticles in the cells was 4.8 × 105 and 1.8 × 104 for folate-targeted and non-targeted vesicles respectively after 40 min of incubation. The overall uptake of the targeted vesicle is about 27 times higher than the non-targeted one. Of equal importance is that the cellular uptake of the targeted vesicles decreased to the level of non-targeted vesicles when excessive folic acid (1.0 mM) was used to saturate the receptors on the cell surface. Taken together, our results have demonstrated that folate conjugated through the PEG space is indeed able to direct the specific recognition of the vesicles to targeted cancer cells.
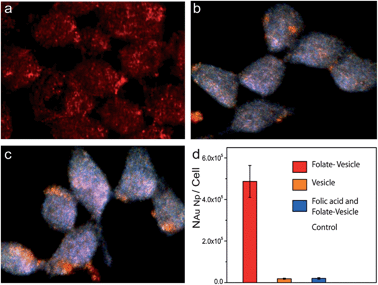 | ||
| Fig. 4 Dark-field imaging of live MDA-MB-435 cells incubated with folate-targeted vesicles (a), non-targeted vesicles (b), folate-targeted vesicles in the presence of free folic acid (c), and (d) the average number of gold nanoparticles uptake by cancer cell under different condition. | ||
Time-lapse dark-field imaging was performed to investigate photo-responsive properties of the vesicles inside live cells. Fig. 5 shows that the scattering color of vesicles inside cells under light-irradiation evolved from bright red to orange-yellow, accompanied by a concurrent scattering intensity drop, indicating the in situ photolysis of the vesicles, in contrast to the weak scattering background of the cells (Fig. S6†). We also noticed that the vesicle-labeled cells that are not exposed to light maintain the red color for 48 h throughout our experiment (Fig. S7†), confirming the possibility for localized processing of the vesicles by external photo-irradiation. Next, vesicles and organelle tracking dyes 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO) were co-delivered into live MDA-MB-435 cells. Dual-modality imaging of the scattering light of vesicles and the green fluorescence of DiO showed colocalization of the two signals (Fig. S8†), which confirmed the trapping of vesicles in organelles. After irradiation for 15 min, although the scattering color of the vesicles changed to orange, the distribution pattern remained unchanged, suggesting that the photolysis of the vesicles occurred inside intracellular compartments.
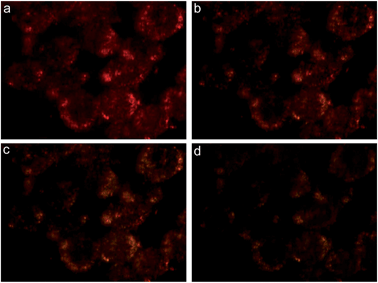 | ||
| Fig. 5 Dark-field images of live MDA-MB-435 cells incubated with folate-targeted vesicles exposed to light for 0 min (a), 5 min (b), 10 min (c) and 15 min (d). | ||
We further investigate the loading and photo-regulated release of a fluorescent model drug, i.e., doxorubicin (DOX) by the plasmonic vesicles. DOX, as a potent anticancer drug, suffers from side effects such as cardiac toxicity, and therefore DDS are actively explored for the targeted delivery of DOX.28,62 We previously demonstrated that DOX can be loaded in the plasmonic vesicles during the film rehydration preparation of the vesicles.39 Several groups have reported that preparing liposomes and polymersomes in acidic media followed by neutralization of the dispersion pH gives rise to a pH gradient across the vesicle membrane, which can drive the accumulation of DOX into the acidic vesicle cavity.63 Inspired by these results, we have investigated the possibility of loading DOX in the photosensitive plasmonic vesicles using the pH-gradient method. The plasmonic vesicle was firstly prepared by using pH 4.0 citric acid buffer for film rehydration. Afterwards, NaOH solution (0.1 M) was added to adjust the pH to 6.5, which gave rise to a pH gradient across the vesicle shell. We have found adding 10% DMSO in the solution is helpful for efficient accumulation of DOX in the aqueous cavity. Eisenberg and co-workers previously reported similar results in polystyrene-b-PAA (PS-b-PAA) polymersomes, which showed that dioxane can increase the permeability of the PS shell and lead to enhanced DOX loading in the vesicles.63,64 Our results (Fig. 6a) showed that the loading content of DOX rose in response to increasing DOX concentrations and leveled off at 30% when the weight ratio of DOX and vesicles reached 50%. Notably, the pH-gradient approach led to a higher DOX loading in comparison with the film rehydration method that we used previously,39 as displayed in Fig. 6b. Free unloaded DOX were removed by repeated centrifugation and redispersion three times. For the same concentration of DOX, the vesicle-loaded DOX consistently shows lower fluorescence than free drug molecules due to the fluorescence quenching of gold nanoparticles. Interestingly, the pH-gradient loading gave rise to a stronger fluorescence than the samples prepared using film rehydration (Fig. 6c), which is possibly due to the partial trapping of DOX in the vesicles shell during film rehydration. And consequently, the close contact of DOX with gold nanoparticles causes more prominent fluorescence quenching.46,65 We next investigated the photo-irradiation triggered DOX release from the vesicles. In the absence of external light, the encapsulated DOX has shown minimal premature release (<10%) over 72 h in PBS buffer (pH 7.4). In contrast, 15 min of irradiation led to the release of nearly 80% of the loaded drug (Fig. 6d). Even at 10 min, about 50% of the payload has been released from the plasmonic vesicles. This result is consistent with the spectroscopic data shown above, which suggest that introducing the hydrophilic moiety in the vesicle shell by the photochemical reaction enhances its permeability and the plasmonic vesicles completely disintegrated after 15 min of light exposure. In clear contrast, the plasmonic vesicles of gold nanoparticles grafted with PEG and poly(methyl methacrylate) (PMMA) lacked the photo-responsiveness and did not show any obvious drug release under photo irradiation.
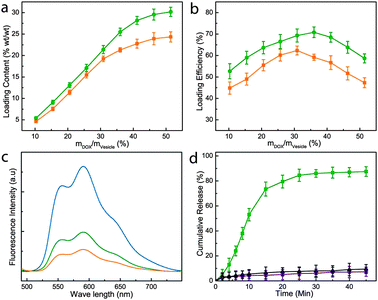 | ||
| Fig. 6 DOX loading content (a) and loading efficiency (b) in the vesicle based on the pH gradient method (green line) and film-rehydration method (orange line) as a function of the feeding weight ratio of DOX to the vesicles. (c) Fluorescence spectra of free DOX (blue line), DOX-loaded vesicles prepared based on the pH gradient method (green line) and film-rehydration method (orange line). (d) Release profiles of the DOX-loaded plasmonic vesicles in the presence (green line) and absence (purple line) of light irradiation, and DOX-loaded plasmonic vesicles of Au@PEG/PMMA in the presence of light irradiation (black line). | ||
The red fluorescence of DOX allows us to use combined dark-field and fluorescence imaging to demonstrate the potential of using the plasmonic vesicles for photo-triggered drug delivery. Fig. 7 shows the dual-modality imaging of the DOX-loaded plasmonic vesicles in live MDA-MB-435 cells. After 40 min incubation, the targeted plasmonic vesicles were taken up by the cells and exhibited patchy distribution inside the cell. The colocalized red scattering and fluorescence in dark-field and fluorescence imaging (Fig. 7a and c) suggests that drug molecules were retained in the vesicles. However, after exposure to light for 15 min, the dark-field imaging showed a color change from red to orange (Fig. 7e), and DOX became homogeneously distributed inside the cytosol (Fig. 7g) 20 min after the irradiation, indicating the photolysis of the plasmonic vesicles and the release of DOX. The red fluorescence signal from DOX in vesicles was mostly found in cell nuclei 60 min post-irradiation, and the dark-field imaging showed nearly no change (Fig. 7i–l). Importantly, the dual-modality imaging also shows that only the cell that underwent light irradiation exhibited the signal changes mentioned above, which is consistent with the high stability of vesicles. These results collectively confirm that the plasmonic vesicles in live cells maintain the photo-responsive property and are able to release the cargo efficiently. The other key finding is that the color change of plasmonic vesicles in dark-field imaging can serve as a built-in indicator for increased shell permeability, generating real-time feedback to signal the drug release.
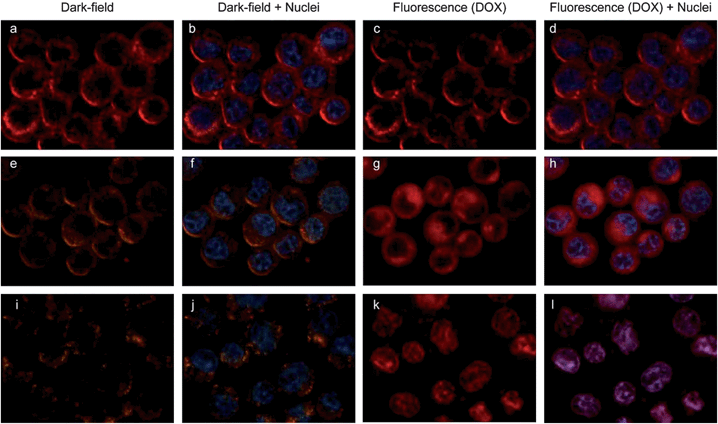 | ||
| Fig. 7 Uptake and dissociation of folate-conjugated vesicles in MDA-MB-435 breast cancer cells and the DOX release revealed by dual-modality imaging. Dark field (a, e and i), fluorescence (c, g and k), and the overlaid images (b, d, f, h, j and l) of live MDA-MB-435 cells labeled with DOX-loaded vesicles without light irradiation (a–d), exposed to light for 15 min and post-incubation for 20 min (e–h) and 60 min (i–l) after the photo-irradiation. DOX has a red fluorescence, and cell nuclei were stained with Hoechst 3342 exhibiting blue fluorescence. | ||
The current folate-targeted photo-labile plasmonic vesicles provide the opportunity to selectively deliver anticancer drugs to cancer cells by taking advantage of ligand-directed active targeting and the triggered release regulated by light irradiation. Cytotoxicity measurements (Fig. 8) showed that the plasmonic vesicles are highly biocompatible. Cells incubated with the drug-loaded vesicles maintained over 80% of cell viability, and targeted and non-targeted vesicles did not show significant difference, agreeing well the minimal premature drug release we observed. Clearly, the vesicles exposed to light irradiation showed greatly enhanced cytotoxicity due to the drug release, and the targeted vesicles are more efficient than the non-targeted across the concentration range we examined. The half-maximal inhibitory concentration (IC50) of folate-targeted vesicles against MDA-MB-435 cells is 0.44 μg mL−1, which is 5 fold lower than that of non-targeted vesicles, demonstrating a fairly high therapeutic effectiveness.
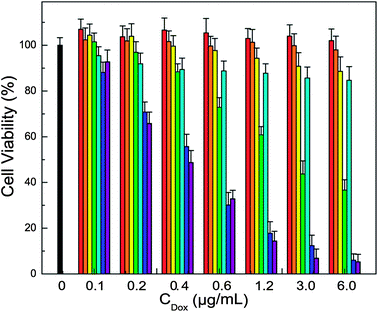 | ||
| Fig. 8 Cytotoxicity results of tested samples (black bar: control cells, red bar: folate targeted vesicles without light irradiation, orange bar: folate targeted vesicles with light irradiation, yellow bar: non-targeted DOX-loaded vesicles without light irradiation, green bar: non-targeted DOX-loaded vesicles with light irradiation, cyan bar: folate targeted DOX-loaded vesicles without light irradiation, blue bar: folate targeted DOX-loaded vesicles with light irradiation, and purple bar: free DOX at various concentrations of DOX). | ||
3 Experimental section
Materials
Doxorubicin (DOX), folic acid (FA), 2-nitrobenyl alcohol, acryloyl chloride, N,N,N′,N′,N′′-pentamethyl dieylenetriamine (PMDETA), copper(I) bromide, chloroauric acid (HAuCl4·3H2O), sodium citrate (99%), lipoic acid (LA), N-hydroxysuccinimide (NHS), dicyclohexylcarbodiimide (DCC), triethylamine (TEA) and trifluoroacetic acid (TFA) were purchased from Sigma-Aldrich. All solvents unless indicated were obtained from Sigma-Aldrich and used as received. Methoxy-poly(ethylene glycol)-amine (MeO-PEG-NH2) and t-butylcarbamoyl-PEG-NH2(BOC-PEG-NH2) with a molecular weight of 5 kDa was purchased from RAPP polymere. 2,2′-Dithiobis [1-(2-bromo-2-methyl-propionyloxy)]s ethane (DTBE) and the amphiphilic nanocrystals were synthesized according to our previous report.52 3,3-dioctadecyloxacarbocyanine perchlorate (DiO), and Hoechst 3342 were purchased from Life technology.Instruments and characterization
Transmission Electron Microscopy (TEM) observations were conducted on a Jeol JEM 2010 electron microscope at an acceleration voltage of 300 kV. Scanning electron microscopy images were obtained on a FESEM (JSM-6700F, Japan). UV-vis absorption spectra were recorded using a SHIMADZU UV-2501 spectrophotometer. Fluorescence spectra were collected on a Fluoromax-3 spectrometer (Horiba Scientific). Single-particle measurements were carried out in an Olympus71 inverted microscope with an oil-immersion dark field condenser, and colored images and scattering spectra were collected using a Photometrics CoolSNAP-cf cooled CCD camera and a PIXIS:100B spectroscopy CCD camera, respectively. To study the photoinduced dissociation of gold vesicles and the release of encapsulated DOX, UV irradiation was obtained from a Handheld UV lamp (UVGL-58, Upland CA. USA) at 365 nm. 1H NMR spectra were obtained with a Bruker AV300, using CDCl3 as the solvent. Gel permeation chromatography (GPC) was measured on a Shimadzu HPLC system using chloroform as the eluent, and the molecular weight was calibrated with polystyrene standards. Plasmonic vesicles or live cells were immobilized on a polylysine-modified glass cover slip for the imaging experiments. Inductively coupled plasma mass spectroscopy (ICP-MS) was measured on a Prodigy High Dispersion ICP-MS (Teledyne Instruments Leeman Labs Inc).Synthesis of amphiphilic folate conjugated FA-Au@PEG/PNBA
14 nm AuNPs were synthesized by reduction of HAuCl4 using trisodium citrate.52 The synthesis of amphiphilic 14 nm gold nanoparticles with hydrophilic FA-conjugated PEG (FA-PEG-LA) and hydrophobic poly(2-nitrobenzyl acrylate) (PNBA) grafts (FA-Au@PEG/PNBA) contains two steps: “grafting to” and “grafting from” reaction. In the “grafting to” reaction, a mixed solution of FA-PEG-LA and MPEG-LA with a molar ratio of 30:70 and DTBE in DMF was slowly added into the original 14 nm gold nanoparticle in water. The ligand exchange reaction was stopped after 12 h by repeatedly centrifuging (10000g, 20 min) the reaction mixer to remove free PEG and DTBE, leading to the PEG and initiator capped nanoparticles. In the “grafting from” reaction, ATRP of NBA monomer was carried out to synthesize Au@PEG/PNBA. Briefly, FA-Au@PEG/DTBE (60 nM) in 3 mL DMF and 2-nitrobenzyl acrylate monomer (0.6 g) were mixed in a 10 mL ampule, degassed, and filled with nitrogen. Then, CuBr (4 mg) and PMDETA (15 mg) dissolved in DMF (0.5 mL) was added through a syringe and the system was degassed again. After 30 min of stirring at room temperature, the ampule was placed in a preheated oil bath (65 °C) and the reaction was maintained for 10 h. Afterwards, the reaction mixture was centrifuged (11000g, 20 min) three times to remove the catalysts and unreacted monomer, and stored in chloroform at 4 °C for further use.
Preparation of vesicles and encapsulation of DOX
The Au@PEG/PNBA vesicle was prepared following the film rehydration method.38 Briefly, 0.2 mL solution (150 nM) of Au@PEG/PNBA in chloroform was first thoroughly dried on the wall of a glass vial under a steady flow of N2 gas. Afterwards, 1.0 mL citrate buffer (pH 4.0) was added to rehydrate the film under sonication at 30 °C. The mixture became clear in 3–5 min and was stirred for about 2 h, and then the pH of solution was adjusted to pH 6.5 using small volumes of NaOH aqueous solution. DOX was loaded into the plasmonic vesicles through a pH-gradient method. Typically, the vesicle dispersion was stored in a glass bottle and 10% (v/v) DMSO and DOX (as an aqueous solution) was added. After being maintained under stirring for 48 h, the drug-loaded vesicles were collected by centrifugation and dispersed in pH 7.4 buffer to remove unloaded DOX molecules. The loading content and loading efficiency were determined by measuring the absorbance of free DOX collected at 485 nm.ICP-MS sample preparation and measurements
For ICP-MS measurements, the folate-targeted vesicle, non-targeted vesicle and the mixture of folic acid (1 mM) and folate-targeted vesicle was incubated with preseeded MDA-MB-435 cells in 24 well plates (100000 cells per well) for 40 min. After incubation, the cells were washed three times with PBS buffer and then lysis buffer (300 μL) was added to the cells. The resulting cell lysate was digested using 3 mL of HNO3 and 1 mL of H2O2 for 24 h. 3 mL of aqua regia was then added, and the sample was allowed to react for another 3 h. This solution was diluted in 100 mL de-ionized water with 5% aqua regia, and centrifuged at 3000 rpm for 5 min to remove any cellular debris. All experiments were repeated 3 times, and each replicate was measured 3 times by ICP-MS.
The concentration of Au was then converted into the number of nanoparticles using the following eqn (1) and (2), where, r is the radius of the gold nanoparticle. The number of Au atoms (NAu atom) contained in each nanoparticle was determined by eqn (1), where VAu atom (0.017 nm3) refers to the size of a gold atom, leading to 84472 gold atoms per nanoparticle. The number of Au nanoparticles (N) can then be calculated by eqn (2), where MAu nanoparticle refers to the molar mass of the nanoparticle and M refers to the measured number of Au ions (atoms) from ICP-MS analysis.
| NAu atom = 4πr3/3VAu atom, | (1) |
| N = M/MAu nanoparticle, | (2) |
Photo-irradiation triggered release of DOX
To study the photo-induced dissociation of vesicles and the release of encapsulated DOX, the solution was exposed to a hand-held UV lamp (2.3 mW cm−2) for 50 min. Briefly, 3 mL of vesicle solution was placed in a standard cuvette, while the solution was exposed to a UV light source under stirring at a distance of 1 cm. The vesicles were separated through centrifugation and the amount of DOX released was measured by UV-vis spectrometer. All measurements were conducted in triplicate.Live cell imaging
MDA-MB-435 cells were incubated in folate-free RPMI 1640 medium with 10% fetal bovine serum and 1% streptomycin for 2 to 3 days (37 °C, 5% CO2). The live cells were transferred onto glass coverslips (treated with polylysine) in a 60 mm dish and allowed to grow for 2 days. Afterwards, the cover slip was rinsed with PBS and assembled into a flow-cell before the vesicle dispersion (0.5 nM) was injected to incubate with the cells for the live cell imaging. The dark-field image was captured using an Olympus71 inverted microscope with a Photometrics CoolSNAP-cf cooled CCD camera, illuminated by unpolarized white light through a dark-field condenser. To obtain the in situ photolysis of the plasmonic vesicles inside live cells, UV light was selected by a band-pass filter (330–385 nm). For colocalization studies of vesicles with DiO, the dye solution was mixed with the vesicle solution at 2 μg mL−1 and co-delivered into the cell. The fluorescence signal of DiO and dark-field image of the vesicles was detected before and after light irradiation separately. For the intracellular DOX release studies, MDA-MB-435 cells were treated with folate targeted DOX-loaded vesicles with nuclei counterstained with Hoechst 33342. The cellular fluorescence images were collected using wideband blue excitation (450–480 nm) provided by a mercury lamp, a long-pass dichroic filter (500 nm), and a band-pass emission filter (590–630 nm), and dark-field images were captured as described above.In vitro cytotoxicity
A total of 1.0 × 104 cells were seeded in each well of 96-well plates and incubated for 24 h to allow the cells to attach. The cells were exposed to folate targeted vesicles, non-targeted DOX-loaded vesicles, folate targeted DOX-loaded vesicles and DOX at 37 °C for 2 h. The concentrations of DOX were 0, 0.1, 0.2, 0.6, 1.2, 3, and 6 μg mL−1. After that, the medium was changed with fresh medium, and cultured in the incubator for 24 h. Cell survival efficiency was measured using the standard cell counting Kit-8 (CCK-8) assay according to the manufacturer’s suggested procedures. The data represent the average results of triplicate measurements. If necessary, the cells were exposed to UV light (2.3 mW cm−2) for 15 min.4 Conclusions
In summary, we have developed a new class of plasmonic nanocarriers for cancer-targeted imaging and photo-regulated drug delivery. One key finding is that the plasmonic vesicles can not only serve as nanocarriers for integrated plasmonic imaging and on-demand triggered drug release but also provide real-time feedback to sense the releasing process. The use of amphiphilic plasmonic nanostructures coated with environmental-responsive polymer brushes as self-assembly building blocks of the vesicles opens new avenues for the design of multi-functional DDS for combined ligand-directed active targeting and stimuli-triggered cargo release. Our ongoing efforts are directed to explore the concurrent delivery of hydrophobic anticancer drugs and water-soluble biomolecular therapeutics for combination therapy by taking advantage of the loading capacity of both hydrophobic shell and aqueous cavity of the plasmonic vesicles.Acknowledgements
H.D. thanks the Nanyang Assistant Professorship for financial support. This work is also supported by the INSIST program at Nanyang Technological University.Notes and references
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS.
- K. Riehemann, S. W. Schneider, T. A. Luger, B. Godin, M. Ferrari and H. Fuchs, Angew. Chem., Int. Ed., 2009, 48, 872–897 CrossRef CAS.
- G. D. Moon, S. W. Choi, X. Cai, W. Li, E. C. Cho, U. Jeong, L. V. Wang and Y. N. Xia, J. Am. Chem. Soc., 2011, 133, 4762–4765 CrossRef CAS.
- G. Han, C. C. You, B. J. Kim, R. S. Turingan, N. S. Forbes, C. T. Martin and V. M. Rotello, Angew. Chem., Int. Ed., 2006, 45, 3165–3169 CrossRef CAS.
- Y. Lee, S. Fukushima, Y. Bae, S. Hiki, T. Ishii and K. Kataoka, J. Am. Chem. Soc., 2007, 129, 5362–5363 CrossRef CAS.
- J. Z. Du, T. M. Sun, W. J. Song, J. Wu and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 3621–3626 CrossRef CAS.
- J. He, Y. J. Liu, T. Babu, Z. J. Wei and Z. H. Nie, J. Am. Chem. Soc., 2012, 134, 11342–11345 CrossRef CAS.
- T. Miyata, M. Jige, T. Nakaminami and T. Uragami, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1190–1193 CrossRef CAS.
- J. Q. Jiang, X. Tong, D. Morris and Y. Zhao, Macromolecules, 2006, 39, 4633–4640 CrossRef CAS.
- L. Polavarapu, M. Manna and Q. H. Xu, Nanoscale, 2011, 3, 429–434 RSC.
- V. Yesilyurt, R. Ramireddy and S. Thayumanavan, Angew. Chem., Int. Ed., 2011, 50, 3038–3042 CrossRef CAS.
- J. L. Vivero-Escoto, I. I. Slowing, C. W. Wu and V. S. Y. Lin, J. Am. Chem. Soc., 2009, 131, 3462–3463 CrossRef CAS.
- Q. N. Lin, Q. Huang, C. Y. Li, C. Bao, Z. Z. Liu, F. Y. Li and L. Y. Zhu, J. Am. Chem. Soc., 2010, 132, 10645–10647 CrossRef CAS.
- M. S. Yavuz, Y. Y. Cheng, J. Y. Chen, C. M. Cobley, Q. Zhang, M. Rycenga, J. W. Xie, C. Kim, K. H. Song, A. G. Schwartz, L. H. V. Wang and Y. N. Xia, Nat. Mater., 2009, 8, 935–939 CrossRef CAS.
- Y. C. Yeh, B. Creran and V. M. Rotello, Nanoscale, 2012, 4, 1871–1880 RSC.
- H. Masoud and A. Alexeev, ACS Nano, 2012, 6, 212–219 CrossRef CAS.
- K. Ock, W. I. Jeon, E. O. Ganbold, M. Kim, J. Park, J. H. Seo, K. Cho, S. W. Joo and S. Y. Lee, Anal. Chem., 2012, 84, 2172–2178 CrossRef CAS.
- M. H. Xiong, Y. Bao, X. Z. Yang, Y. C. Wang, B. Sun and J. Wang, J. Am. Chem. Soc., 2012, 134, 4355–4362 CrossRef CAS.
- A. Wijaya, S. B. Schaffer, I. G. Pallares and K. Hamad-Schifferli, ACS Nano, 2008, 3, 80–86 CrossRef.
- C. Y. Song, G. P. Zhao, P. J. Zhang and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 14033–14035 CrossRef CAS.
- S. H. Hu, S. Y. Chen and X. H. Gao, ACS Nano, 2012, 6, 2558–2565 CrossRef CAS.
- H. Kang, A. C. Trondoli, G. Zhu, Y. Chen, Y. J. Chang, H. Liu, Y. F. Huang, X. Zhang and W. Tan, ACS Nano, 2011, 5, 5094–5099 CrossRef CAS.
- W. Xiao, W. H. Chen, X. D. Xu, C. Li, J. Zhang, R. X. Zhuo and X. Z. Zhang, Adv. Mater., 2011, 23, 3526–3530 CrossRef CAS.
- R. Tong, H. D. Hemmati, R. Langer and D. S. Kohane, J. Am. Chem. Soc., 2012, 134, 8848–8855 CrossRef CAS.
- Y. L. Luo, Y. S. Shiao and Y. F. Huang, ACS Nano, 2011, 5, 7796–7804 CrossRef CAS.
- J. He, P. Zhang, T. Babu, Y. J. Liu, J. L. Gong and Z. H. Nie, Chem. Commun., 2013, 49, 576–578 RSC.
- K. Wang, D. S. Guo, X. Wang and Y. Liu, ACS Nano, 2011, 5, 2880–2894 CrossRef CAS.
- B. Yan, J. C. Boyer, D. Habault, N. R. Branda and Y. Zhao, J. Am. Chem. Soc., 2012, 134, 16558–16561 CrossRef CAS.
- N. Nishiyama, A. Iriyama, W. D. Jang, K. Miyata, K. Itaka, Y. Inoue, H. Takahashi, Y. Yanagi, Y. Tamaki, H. Koyama and K. Kataoka, Nat. Mater., 2005, 4, 934–941 CrossRef CAS.
- S. B. Lecommandoux, O. Sandre and F. Checot, Adv. Mater., 2005, 17, 712–718 CrossRef CAS.
- R. Huschka, O. Neumann, A. Barhoumi and N. J. Halas, Nano Lett., 2010, 10, 4117–4122 CrossRef CAS.
- J. Rodriguez-Hernandez, F. Checot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef CAS.
- D. E. Discher, V. Ortiz, G. Srinivas, M. L. Klein, Y. Kim, C. A. David, S. S. Cai, P. Photos and F. Ahmed, Prog. Polym. Sci., 2007, 32, 838–857 CrossRef CAS.
- P. Tanner, P. Baumann, R. Enea, O. Onaca, C. Palivan and W. Meier, Acc. Chem. Res., 2011, 44, 1039–1049 CrossRef CAS.
- C. Sanson, O. Diou, J. Thèvenot, E. Ibarboure, A. Soum, A. Brûlet, S. Miraux, E. Thiaudière, S. Tan, A. Brisson, V. Dupuis, O. Sandre and S. B. Lecommandoux, ACS Nano, 2011, 5, 1122–1140 CrossRef CAS.
- S. Egli, M. G. Nussbaumer, V. Balasubramanian, M. Chami, N. Bruns, C. Palivan and W. Meier, J. Am. Chem. Soc., 2011, 133, 4476–4483 CrossRef CAS.
- J. B. Song, L. Cheng, A. P. Liu, J. Yin, M. Kuang and H. W. Duan, J. Am. Chem. Soc., 2011, 133, 10760–10763 CrossRef CAS.
- J. B. Song, J. J. Zhou and H. W. Duan, J. Am. Chem. Soc., 2012, 134, 13458–13469 CrossRef CAS.
- L. M. Liz-Marzán and P. Mulvaney, J. Phys. Chem. B, 2003, 107, 7312–7326 CrossRef.
- P. K. Jain, X. H. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS.
- Y. W. Jun, S. Sheikholeslami, D. R. Hostetter, C. Tajon, C. S. Craik and A. P. Alivisatos, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17735–17740 CrossRef CAS.
- L. Xu, H. Kuang, C. Xu, W. Ma, L. B. Wang and N. A. Kotov, J. Am. Chem. Soc., 2011, 134, 1699–1709 CrossRef.
- B. M. Reinhard, S. Sheikholeslami, A. Mastroianni, A. P. Alivisatos and J. Liphardt, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2667–2672 CrossRef CAS.
- J. Wang, S. V. Boriskina, H. Y. Wang and B. M. Reinhard, ACS Nano, 2011, 5, 6619–6628 CrossRef CAS.
- F. Wang, Y. C. Wang, S. Dou, M. H. Xiong, T. M. Sun and J. Wang, ACS Nano, 2011, 5, 3679–3692 CrossRef CAS.
- C. Sonnichsen, B. M. Reinhard, J. Liphardt and A. P. Alivisatos, Nat. Biotechnol., 2005, 23, 741–745 CrossRef.
- M. J. Crow, K. Seekell, J. H. Ostrander and A. Wax, ACS Nano, 2011, 5, 8532–8540 CrossRef CAS.
- A. Pallaoro, G. B. Braun, N. O. Reich and M. Moskovits, Small, 2010, 6, 618–622 CrossRef CAS.
- K. L. Wustholz, A. I. Henry, J. M. McMahon, R. G. Freeman, N. Valley, M. E. Piotti, M. J. Natan, G. C. Schatz and R. P. V. Duyne, J. Am. Chem. Soc., 2010, 132, 10903–10910 CrossRef CAS.
- J. Yin, T. Wu, J. B. Song, Q. Zhang, S. Y. Liu, R. Xu and H. W. Duan, Chem. Mater., 2011, 23, 4756–4764 CrossRef CAS.
- L. Cheng, A. P. Liu, S. Peng and H. W. Duan, ACS Nano, 2010, 4, 6098–6104 CrossRef CAS.
- X. G. Jiang, C. A. Lavender, J. W. Woodcock and B. Zhao, Macromolecules, 2008, 41, 2632–2643 CrossRef CAS.
- A. M. Kloxin, A. M. Kasko, C. N. Salinas and K. S. Anseth, Science, 2009, 324, 59–63 CrossRef CAS.
- Z. Z. J. Lim, J. E. J. Li, C. T. Ng, L. Y. L. Yung and B. H. Bay, Acta Pharmacol. Sin., 2011, 32, 983–990 CrossRef CAS.
- L. Vigderman, P. Manna and E. R. Zubarev, Angew. Chem., 2012, 124, 660–665 CrossRef.
- P. S. Low, W. A. Henne and D. D. Doorneweerd, Acc. Chem. Res., 2007, 41, 120–129 CrossRef.
- R. Meier, T. D. Henning, S. Boddington, S. Tavri, S. Arora, G. Piontek, M. Rudelius, C. Corot and H. E. Daldrup-Link, Radiology, 2010, 255, 527–535 CrossRef.
- H. W. Duan and S. M. Nie, J. Am. Chem. Soc., 2007, 129, 3333–3338 CrossRef CAS.
- C. Kim, S. S. Agasti, Z. Zhu, L. Isaacs and V. M. Rotello, Nat. Chem., 2010, 2, 962–966 CrossRef CAS.
- L. Au, Q. Zhang, C. M. Cobley, M. Gidding, A. G. Schwartz, J. Chen and Y. N. Xia, ACS Nano, 2009, 4, 35–42 CrossRef.
- A. Agarwal, M. A. Mackey, M. A. El-Sayed and R. V. Bellamkonda, ACS Nano, 2011, 5, 4919–4926 CrossRef CAS.
- A. Choucair, P. Lim Soo and A. Eisenberg, Langmuir, 2005, 21, 9308–9313 CrossRef CAS.
- F. Ahmed, R. I. Pakunlu, A. Brannan, F. Bates, T. Minko and D. E. Discher, J. Controlled Release, 2006, 116, 150–158 CrossRef CAS.
- J. You, G. D. Zhang and C. Li, ACS Nano, 2010, 4, 1033–1041 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3nr01350b |
| This journal is © The Royal Society of Chemistry 2013 |