 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A platinum complex that binds non-covalently to DNA and induces cell death via a different mechanism than cisplatin†
Kogularamanan Suntharalingama, Oscar Mendozaa, Alexandra A. Duarteb, David J. Mann*b and Ramon Vilar*a
aDepartment of Chemistry, Imperial College London, London SW7 2AZ, UK. E-mail: r.vilar@imperial.ac.uk
bDivision of Molecular Biosciences, Imperial College London, London SW7 2AZ, UK. E-mail: d.mann@imperial.ac.uk
First published on 19th February 2013
Abstract
Cisplatin and some of its derivatives have been shown to be very successful anticancer agents. Their main mode of action has been proposed to be via covalent binding to DNA. However, one of the limitations of these drugs is their poor activity against some tumours due to intrinsic or acquired resistance. Therefore, there is interest in developing complexes with different binding modes and mode of action. Herein we present a novel platinum(II)–terpyridine complex (1) which interacts non-covalently with DNA and induces cell death via a different mechanism than cisplatin. The interaction of this complex with DNA was studied by UV/Vis spectroscopic titrations, fluorescent indicator displacement (FID) assays and circular dichroism (CD) titrations. In addition, computational docking studies were carried out with the aim of establishing the complex's binding mode. These experimental and computational studies showed the complex to have an affinity constant for DNA of ∼104 M−1, a theoretical free energy of binding of −10.83 kcal mol−1 and selectivity for the minor groove of DNA. Long-term studies indicated that 1 did not covalently bind (or nick) DNA. The cancer cell antiproliferative properties of this platinum(II) complex were probed in vitro against human and murine cell lines. Encouragingly the platinum(II) complex displayed selective toxicity for the cancerous (U2OS and SH-SY5Y) and proliferating NIH 3T3 cell lines. Further cell based studies were carried out to establish the mode of action. Cellular uptake studies demonstrated that the complex is able to penetrate the cell membrane and localize to the nucleus, implying that genomic DNA could be a cellular target. Detailed immunoblotting studies in combination with DNA-flow cytometry showed that the platinum(II) complex induced cell death in a manner consistent with necrosis.
1. Introduction
Platinum complexes have received great attention as anti-cancer agents,1–6 largely due to the clinical success of cisplatin and its derivatives (such as oxaliplatin, nedaplatin, lobaplatin and carboplatin).7 These clinically approved platinum based agents interact with DNA by forming intra- and inter-stranded crosslinks (covalent Pt–DNA bonds).8–13 These modifications structurally kink DNA and inhibit transcription (by stalling polymerase), resulting in controlled cell death.11,14,15 The main limitation of these drugs is their poor activity against some tumors due to intrinsic or acquired resistance.16,17 Therefore new platinum drugs are needed that build on the mechanisms associated to clinically approved platinum drugs but bypass their inherent resistance.As an alternative, many platinum complexes bearing π-conjugated heterocyclic ligands have been developed and their anti-cancer properties investigated.18–21 The planar aromatic ligand allows these complexes to intercalate between base pairs. The first structural evidence of this binding mode was reported almost 40 years ago by Lippard et al.22 In this seminal paper, a platinum(II)–terpyridine complex was shown to intercalate non-covalently between adjacent DNA base pairs, resulting in the unwinding of the DNA. Similar complexes with leaving groups were shown to bind covalently, favourably targeting the N7 position on guanosine bases (like cisplatin).23–28 In recent years, vast libraries of platinum(II)–terpyridine compounds have been prepared (with modifications to the terpyridine ligand and various groups attached to the platinum center) and tested as potential anti-cancer agents.29 Many of these complexes have shown great promise; for example a report by McFadyen and co-workers showed a series of platinum(II)–terpyridine complexes with electron rich/deficient thiol ligands (attached directly to platinum) to be active against the L1210 murine leukemia cell line.30 Interestingly, a follow up study found the parent [Pt(terpy)Cl]+ species, with a labile chloro substituent to be relatively ineffective (IC50 = 450 μM), implying that platination is not a determinant of cytotoxicity.31 Other variations to groups attached to platinum (using picoline, thioalkylcarboranes, glycosylated acetylides) have resulted in higher water solubility and potency against cisplatin- and doxorubicin-resistant cell lines (human ovarian carcinoma).31–37
The majority of platinum(II)–terpyridine systems that have been reported to date (and shown anti-cancer activity), bind to DNA via intercalation or coordination. However some platinum(II)–terpyridines can bind via other non-intercalative modes. Indeed platinum(II)–terpyridine thiol and acetylide compounds have been shown to bind via electrostatic and hydrophobic interactions.38 Furthermore, the addition of sterically demanding groups such as t-butyl on the terpyridine ligand has yielded highly active anti-cancer agents that interact with the DNA grooves.34 When designing potential anti-cancer agents, non-intercalative platinum(II)–terpyridine complexes are largely overlooked in favor of metallo-intercalators. Given the success of groove binders such as Tallimustine39,40 and Brostallicin41,42 (both have undergone clinical trials) in cancer therapy, it is surprising that very few groove binding platinum(II)–terpyridine complexes have been reported. With the aim of exploring the ability of new metal complexes as DNA groove binders, we have prepared a platinum(II)–terpyridine complex (1) bearing two piperidine substituents at positions 2 and 2′ (see Scheme 1). The substituents were placed in these positions with the aim of hindering direct coordination to the platinum(II) centre and to potentially favour groove and phosphate binding (aided by the piperidine groups which at physiological pH are protonated). Besides its synthesis and full characterization, herein we also report the DNA binding properties of 1 (including docking studies to rationalize the interactions) as well as its behaviour in cells. We have carried out a series of experiments aimed at dissecting its cytotoxic mechanism of action.
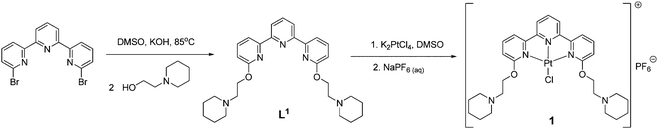 | ||
| Scheme 1 The synthetic procedure used to prepare the platinum(II)–terpyridine complex used in this study. | ||
2. Results and discussion
Synthesis and characterization
The new terpyridine ligand was prepared by reacting 6,6′′-dibromo-2,2′:6′2′′-terpyridine with two equivalents of 1-(2-hydroxyethyl)piperidine in DMSO under strong basic conditions (powdered KOH) at 85 °C for 24 h (see Scheme 1). The product was extracted into dichloromethane, washed several times with water and isolated after solvent removal. The resultant off-white solid was fully characterized by 1H and 13C NMR spectroscopy, mass spectrometry and elemental analysis. The attachment of the side arms was clearly illustrated by the downfield shift of the carbon signal corresponding to the site of substitution (from 157 ppm for 6,6′′-dibromo-2,2′:6′2′′-terpyridine to 163 ppm for L1). The ESI(+) mass spectrum showed a signal corresponding to the expected molecular ion, confirming the formation of the desired product.The new platinum(II)–terpyridine complex 1 was prepared by stirring L1 with K2PtCl4 in DMSO overnight. The product was isolated pure as the PF6− salt after anion exchange. The platinum(II)–terpyridine complex 1 was fully characterized by 1H NMR spectroscopy (which showed characteristic shifts of some of the aromatic signals upon coordination to platinum), UV/Vis spectroscopy, ESI(+) mass spectrometry and elemental analysis.
Before studying the interaction between 1 and DNA, the stability of the complex was investigated using UV-Vis and 1H NMR spectroscopies. The absorption spectrum for 1 (50 μM in Tris–HCl 10 mM/KCl 100 mM with and without cell lysate, 3.4 mg of protein) was recorded over 24 h (see Fig. S3 and S4, ESI†). The MLCT band was examined for any signs of platinum dissociation/reactivity. Over this period of time no significant changes were detected. Therefore the complex was deemed to be stable under the conditions used for the DNA binding and cellular assays. To probe the thermal stability of 1, its 1H NMR spectrum (80 mM in DMSO-d6) was recorded between 25–110 °C (see Fig. S2, ESI†). Over this temperature range the complex was found to be stable (displaying the expected chemical shifts and splitting patterns throughout).
DNA binding studies
UV-Vis spectroscopic titrations were carried out to determine the binding affinity and binding mode of 1 to DNA. Upon addition of aliquots of Calf Thymus DNA (ct-DNA), in the mM range, to a solution of 1 of known concentration (20 μM), the absorption band corresponding to the metal perturbed intra-ligand π → π* transition (300–320 nm) was observed to decrease (see Fig. S5, ESI†). From the absorbance, the concentration of bound and unbound metal complex was calculated and extrapolated to determine the binding constant. This was done by fitting the data to a reciprocal plot of D/Δεapversus D using the following equation: D/Δεap = D/Δε + 1/(Δε × K), details are provided in the Experimental section. The intrinsic binding affinity was found to be 6.10 ± 0.94 × 104 M−1. Notably this value is 1 to 2 orders of magnitude lower than those reported for platinum(II)–terpyridine intercalators, suggesting a non-intercalative binding interaction between 1 and ct-DNA.43–45The addition of ct-DNA to 1 resulted in hypochromicity (14%), no red shifts and sharp isosbestic points. These features are characteristic of compounds that interact with DNA via groove binding.46,47 The complex can be envisaged to engage bases in the grooves by π–π interactions (through the planar terpyridine moiety) and, at the same time, form hydrogen bonding and/or electrostatic interactions with the negatively charged phosphate backbone (via the piperidine side arms).
In order to investigate the binding mode further, fluorescent indicator displacement (FID) studies were carried out with two different dyes: a DNA intercalator, thiazole orange (TO) and a DNA minor groove binder, Hoechst 33258.48–51 Comparison of the DC50 values (the concentration required to displace 50% of the respective dyes from duplex DNA) obtained from the different dyes gives an insight into the binding mode (see Table S1 and Fig. S6, ESI†). The results clearly showed that Hoechst was preferentially displaced over TO (over 3-fold selectivity). Therefore 1 can be proposed to preferentially bind via the DNA grooves.
To gain further insight into the binding mode between this complex and DNA, circular dichroism studies were carried out. CD spectra of ct-DNA treated with 1 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DNA ratio of 0.5 or 1) showed no change in profile relative to the untreated control (only changes in the intensities of the bands related to helicity and base stacking; see Fig. S7, ESI†). These spectral changes are comparable to those reported for other metal containing groove binders.52–54
:DNA ratio of 0.5 or 1) showed no change in profile relative to the untreated control (only changes in the intensities of the bands related to helicity and base stacking; see Fig. S7, ESI†). These spectral changes are comparable to those reported for other metal containing groove binders.52–54
Computational docking studies were carried out to gain theoretical insight into the interaction between 1 and DNA. Thus the optimised structure of the metal complex (see ESI† for further details) was docked with a B-DNA structure (taken from the protein data bank; 1BNA) using Autodock.55 The docked model suggests that 1 interacts with the minor groove of DNA (see Fig. 1). The almost planar structure of the complex fits into the minor groove of the DNA in a parallel manner with respect to the DNA backbone. The two alkane chains of the complex remain out of the groove, pointing to DNA's backbone. The positively charged piperidines and the negatively charged phosphate groups from the backbone are linked by a hydrogen bond (2.10 Å). The estimated free energy of this binding was computed to be −10.83 kcal mol−1. Therefore the UV-Vis, FID, CD and computational docking data collectively suggest that 1 is a DNA groove binder.
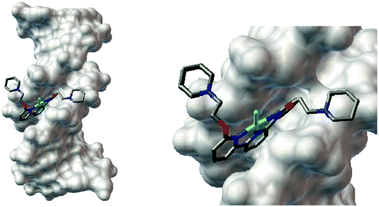 | ||
| Fig. 1 (a) Computational docking model (using the Autodock software) illustrating the interaction between 1 and B-DNA (PDB ID:1BNA). (b) Magnified view of the interaction between 1 and DNA. | ||
The bio-physical studies described thus far were all carried out within an incubation time of complex 1 and DNA of 2 h. Therefore further studies were carried out to determine whether the complex was able to coordinate to DNA over a longer period of time. Coordination was probed spectrophotometrically using the established selective precipitation of ct-DNA method which detected covalent binding of a given compound to DNA.56–58 A solution of ct-DNA was incubated with 1 at a bp:metal complex ratio of 5:1 at 37 °C; in a separate solution complex 2 (see Fig. 2)59 was incubated with DNA (bp:metal complex ratio of 5:1 and 37 °C) and used as a positive control (i.e.2 is able to platinate DNA).
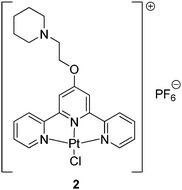 | ||
| Fig. 2 Platinum(II) complex used as positive control in the selective precipitation of ct-DNA method. This complex has been previously shown to interact with ct-DNA via direct coordination. | ||
At specific time intervals, aliquots were taken from these solutions and the DNA within the sample precipitated. The amount of unbound metal complex was determined using UV-Vis spectroscopy. The change in the absorbance of the supernatant was then plotted against time to determine the covalent binding ability. The amount of 1 in the supernatant remained constant throughout (while this was seen to decrease for the positive control, i.e. compound 2), suggesting that 1 was unable to bind covalently to DNA (see Fig. S8, ESI†). Agarose gel electrophoresis studies with supercoiled pUC19 plasmid DNA treated with different concentrations of 1 (at 37 °C for 24 h) also gave no detectable coordination (or nicking) to DNA (see Fig. S9, ESI†). The ability of complex 1 to coordinate to DNA bases was further probed by 1H NMR spectroscopic studies. Incubating 1 with guanosine over a 24 h produced no changes to the 1H NMR spectrum (see Fig. S10, ESI†), indicating that the platinum(II) does not coordinate to guanosine. Overall, the bio-physical and computational data are consistent with 1 binding to the minor groove of DNA in a non-covalent manner.
Cellular studies
The anti-proliferative properties of 1 were assessed using the MTS assay (using the CellTiter 96® AQueous Assay from Promega). Data for cisplatin under the same conditions are also reported for comparison. Four cell lines were initially tested: U2OS (human osteosarcoma), HEK 293T (human embryonic kidney), GM05757 (normal human fibroblast) and SH-SY5Y (neuroblastoma). The IC50 values were derived from dose–response curves and are summarised in Table 1. It should be noted that the IC50 values reported in Table 1 are for a 24 h incubation period – and therefore they might seem relatively high as compared to values previously reported for cisplatin for longer incubation times. Complex 1 was found to have a promising toxicity profile, with high potency toward the cancerous U2OS and SH-SY5Y cell lines (comparable to cisplatin in this 24 h incubation period) and reduced toxicity toward the normal GM05757 cell line (4-fold difference between the U2OS and GM05757 cell lines).| Compound | U2OS (μM) | HEK 293T (μM) | GM05757 (μM) | SH-SY5Y (μM) |
|---|---|---|---|---|
| 1 | 33 ± 4 | 68 ± 8 | 130 ± 12 | 52 ± 4 |
| Cisplatin | 48 ± 6 | 109 ± 12 | 74 ± 6 | 54 ± 4 |
Further toxicity studies with proliferating and non-proliferating NIH 3T3 mouse fibroblast cells (see Fig. S12 and S13, ESI,† for flow cytometry profile of the cells) showed that the complex was able to selectively induce cell death in proliferating but not quiescent cells (see Table 2). This is undoubtedly an attractive characteristic in terms of cancer therapy.
| Compound | Proliferating NIH 3T3 (μM) | Quiescent NIH 3T3 (μM) |
|---|---|---|
| 1 | 60 ± 12 | >500 |
| Cisplatin | 145 ± 13 | >500 |
In order to rationalise the varying degrees of toxicities across the different cell lines and to relate the results to genomic DNA interactions, further cell based studies were undertaken. Cellular uptake studies were performed to determine cell permeability and localization of 1. To do this, U2OS, HEK 293T, GM05757, SH-SY5Y and NIH 3T3 (for the latter, both proliferating and quiescent) cells were incubated for 24 h at 37 °C with a sub-lethal concentration (10 μM) of 1 and the platinum content determined for whole cell, nucleus and cytoplasm fractions using Inductively Coupled Plasma Mass Spectrometry (ICP-MS). Cellular platinum levels are expressed as ng platinum per mg protein and the results reported as a mean of four or five determinations for each data point (summarised in Fig. 3).
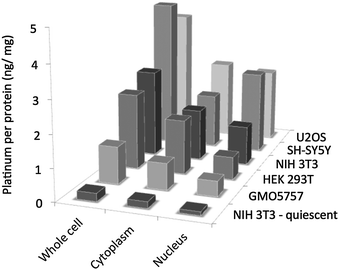 | ||
| Fig. 3 Cellular uptake data for 1. The amount of platinum (ng) per protein (mg) is expressed for the whole cell, cytoplasm and nucleus. The exact Pt/protein values and the corresponding errors are reported in Table S2 (ESI†). | ||
These studies showed that complex 1 was taken up well by all of the cell lines, except NIH 3T3 under non-proliferating conditions. The inability of 1 to penetrate the latter type of cell under starved conditions explains its lack of toxicity (IC50 >500 μM) under these conditions. The highest cellular uptake was observed for the cancerous U2OS and SH-SY5Y cell lines (15–18% of the administered complex at 10 μM enters the cell) and lowest for the normal human GM05757 cell line (in terms of the whole cell, the platinum content was found to be around 5-fold lower in this case). These variations in uptake may explain the differences in toxicities reported for the respective cell lines. Importantly, in all cases, 1 was found to enter the nucleus, giving access to genomic DNA. Therefore cellular toxicity could result from mechanisms related to genomic DNA damage. However it should be noted that a significant portion of the complex was also found in the cytoplasm, this could be due to off target interactions with traditional Pt-drug scavengers found in the cytoplasm61 like metallothionein or glutathione.
To establish whether 1 was able to induce DNA damage, immunoblotting analysis was carried out to monitor changes in the expression levels of biomarkers of the DNA damage response pathway (the phosphorylated forms of histone H2AX (γH2AX) and Chk2 protein kinase, both of which increase due to activation of the apical kinases ATM and ATR).62–66 Therefore U2OS cells were treated with 1 (10–60 μM) for 24 h at 37 °C and collected for immunoblotting analysis (see Fig. 4). After treatment with 60 μM of 1, a visible increase in γH2AX and phosphorylated Chk2 (with total Chk2 remaining constant) was observed (see Fig. 4). Therefore the increase in γH2AX and phosphorylated Chk2 expression indicates that 1 was able to induce DNA damage in cells. DNA damage can lead to a wide range of cellular responses; the p53 protein (which regulates cell cycle progression and induces apoptosis)67,68 plays a fundamental role in coordinating cellular response to DNA damage.69–71 Therefore it was of interest to monitor the expression of p53 and its downstream effector, p21, upon treatment with 1. Addition of 1 resulted in a dose dependent increase in p53 and p21 levels, suggesting that these downstream regulators play a part in the cellular response. In addition, treatment with 60 μM of 1 led to loss of cyclin D1, a pro-proliferative cyclin involved in G1 phase progression.72–74 Loss of cyclin D1 is another characteristic response to DNA damage.75–78
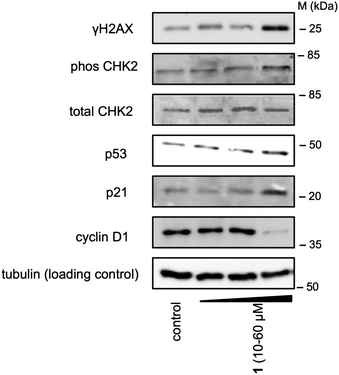 | ||
| Fig. 4 Analysis of protein expression in U2OS cells following treatment with 1 (10–60 μM) after 24 h incubation. Whole cell lysates were resolved by SDS-PAGE and analyzed by immunoblotting against γH2AX, phosCHK2, CHK2, p53, p21, cyclin D1, and α-tubulin (loading control). Results are representative of three independent experiments. MW = Protein Molecular Weight Marker. The relative ratios of the band intensities normalised against the loading control are reported in Fig. S15 (ESI†). | ||
To assess the effects of 1 on cell cycle distribution we performed DNA-flow cytometric studies. U2OS cells were treated with 1 (10 μM) for 24 h and then their cell cycle distribution compared to untreated cells (see Fig. 5). Upon addition of 1, cells were observed to undergo G1 phase arrest (+18.3 ± 1.5% increase in G1 relative to the untreated cells).
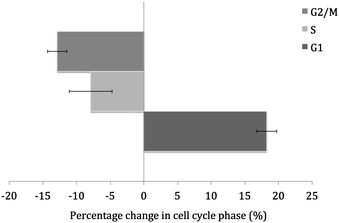 | ||
| Fig. 5 Graphical representation of the percentage change in phases of the cell cycle upon treatment of U2OS cells with 1 (10 μM). The values are an average of three independent measurements and the associated error for each value is represented in the form of capped error bars. | ||
Having established that 1 is able to damage DNA and induce G1 arrest, the mechanism of cell death was investigated. The most thoroughly studied and well-understood mechanism is apoptosis,79,80 and indeed many DNA damaging agents are known to exert their cytotoxic effects via this pathway.81–84 Cells that have undergone apoptosis express phosphatidylserine residues on the cell surface, which can be detected by annexin V. Thus, the annexin V–propidium iodide (PI) dual staining flow cytometric assay (see Section S11 in the ESI† for further details) was used to probe apoptosis. This assay found negligible levels of early or delayed apoptosis in U2OS cells treated with 1 (+3.0 ± 1.1% and +3.0 ± 1.7% increase in early and late apoptosis respectively, relative to the untreated cells). Further immunoblotting analysis found that cells treated with 1 displayed no change in BAX expression (a well-known pro-apoptotic protein) and non-proteolytic cleavage of pro-caspase-3 (see Fig. S16, ESI†). The addition of an apoptosis inducer, staurosporine, resulted in BAX up-regulation and pro-caspase-3 breakdown (see Fig. S17, ESI†). Therefore the data suggest that cell death invoked by 1 occurs in a non-apoptotic manner.
After ruling out apoptosis, other modes of cell death were investigated. To this aim cytotoxicity assays were conducted in the presence of necrosis and autophagocytosis inhibitors (necrostatin-1 and 3-aminobenzamide for necrosis and chloroquine for autophagocytosis). U2OS cells were treated with the inhibitors using conditions previously reported85 and the results are summarised in Fig. 6. Chloroquine had little effect on toxicity exhibited by 1, hence autophagocytosis is unlikely. 3-Aminobenzamide and necrostatin-1 had more profound effects on cell death induced by 1 (2-fold and 4-fold increase in IC50 value compared to the untreated control). Therefore cell death induced by 1 is likely to result from necrosis/necroptosis.
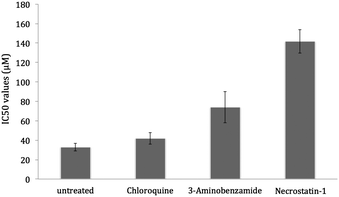 | ||
| Fig. 6 IC50 values obtained for 1 against the U2OS cell line in the absence and presence of necrosis and autophagocytosis inhibitors; necrostatin-1 (60 μM, 1 h), 3-aminobenzamide (2 mM, 1 h) and chloroquine (10 μM, 6 h). The values are an average of five independent measurements and the associated error for each value is represented in the form of capped error bars. | ||
Cells that have undergone necrosis are known to passively release HMGB186 (a chromatin component essential for cell survival87). Remarkably, in apoptotic cells, HMGB1 remains tightly bound to the chromatin, therefore the presence of extracellular HMGB1 serves as a marker for necrosis.88 U2OS cells were treated with toxic concentrations of 1 (50–200 μM) for 48 h and the extracellular media were probed for HMGB1 (see Methods section for detailed description). After treatment with 200 μM of 1, HMGB1 was clearly detected in the extracellular media, indicative of necrosis (see Fig. S17, ESI†). This was comparable to HMGB1 released from necrotic cells (induced by heating to 56 °C for 30 min). Therefore the cytotoxicity profiles in the presence of different inhibitors and the HMGB1 secretion data suggest that 1 induces necrotic cell death.
Necrosis has conventionally been overlooked as a mode of cell death for anti-cancer therapy. However it is now widely regarded as an “ancestral mode of programmed cell death” by many researchers in the cell death research community.89 The discovery of 432 genes90 and various proteins (in viruses)91 associated to the induction of necrosis has proved that it is in fact a highly regulated process.92–94 Necrosis results in the secretion of cytokines; therefore selective induction of necrosis in tumour cells can activate the immune system against tumours and improve the efficacy of anti-cancer agents.95 As DNA damage is known to trigger necrosis, we believe that 1 acts in a controlled manner, where it induces genomic DNA damage which ultimately leads to necrosis.
3. Conclusions
In summary, we report the DNA binding properties of a novel platinum(II) complex (1). The complex was found to interact with duplex DNA (with affinity in the order of 104 M−1) and bind to the minor groove of DNA (theoretical free energy of binding was −10.83 kcal mol−1). Longer term studies were used to demonstrate that DNA binding was not covalent. Cellular uptake studies demonstrated that the platinum complex was found to enter the nucleus, giving it access to genomic DNA. Complex 1 evoked a DNA damage response, giving rise to the accumulation of γH2AX, phosphorylated CHK2 and activation of p53, indicative of DNA damage. Compound 1 was found to have a promising cytotoxicity profile, displaying selective toxicity for cancerous cell lines (U2OS; IC50 = 33 μM and SH-SY5Y; IC50 = 52 μM) over the normal fibroblast cell line (GM05757; IC50 = 130 μM). The complex also showed selective potency for proliferating compared to quiescent NIH 3T3 cells. These characteristics are appealing in terms of anti-cancer therapy. Prolonged treatment with 1 led to cell death, which could be largely blocked by co-treatment with necrosis/necroptosis inhibitors. Taken together, our data indicate that groove binding platinum(II)–terpyridine complexes may be worth exploring as novel anti-cancer agents.4. Experimental details
Synthesis of L1
6,6′′-Dibromo-2,2′:6′,2′′-terpyridine (50 mg, 0.12 mmol) and 1-(2-hydroxyethyl)piperidine (73 mg, 0.28 mmol) were slowly added to a stirred suspension of powdered KOH (36 mg, 0.64 mmol) in DMSO (5 mL). The solution was stirred under a nitrogen atmosphere at 60 °C for 24 h. The reaction mixture was extracted with DCM (50 mL × 3), washed thoroughly with water (30 mL × 3) and dried over sodium sulphate. The solvent was removed under reduced pressure to yield the product as a brown solid (32 mg, 52%); 1H NMR (400 MHz, CDCl3): δH 8.41 (d, 2H, 3JHH = 8.0, tpy′ 3-H and 5-H), 8.45 (d, 2H, 3JHH = 8.0, tpy 5-H), 7.93 (t, 1H, 3JHH = 8.0, tpy′ 4-H), 7.76 (t, 2H, 3JHH = 8.0, tpy 4-H), 6.83 (d, 2H, 3JHH = 8.0, tpy 3-H), 4.69 (t, 4H, 3JHH = 6.0, ethyl 1-H), 2.95 (t, 4H, 3JHH = 6.0, ethyl 2-H), 2.67 (m, 8H, pip 1-H), 1.72 (m, 8H, pip 2-H), 1.53 (m, 4H, pip 3-H); 13C NMR (400 MHz, CDCl3): δC 163.1, 155.2, 153.6, 139.3, 137.5, 120.7, 113.8, 111.4, 63.3, 57.9, 55.1, 25.9, 24.2; ESI-MS calcd for C29H37N5O [M]+: 487.3 a.m.u.; found [M+H]+: 488.0 a.m.u.; anal. calcd for C29H37N5O1.5·HCl: C 64.91, H 7.57, N 12.74; found: C 64.23, H 7.13, N 12.97%.Synthesis of [Pt(L1)Cl](PF6), (1)
K2PtCl4 (0.23 mg, 0.055 mmol) was added to DMSO (3 mL) at 50 °C. To this, a solution of L1 (0.24 mg, 0.05 mmol) in DMSO (3 mL) was added dropwise and the resulting mixture was stirred for 15 h to yield a deep orange solution. The solution was then added dropwise to acetone (ca. 100 mL) and a yellow solid precipitated. The product was isolated by filtration and repeatedly washed with acetone. This solid was dissolved in the minimum amount of DMSO, to which an excess of NaPF6 (as an aqueous solution) was added dropwise. The resultant yellow precipitate was isolated by filtration and washed repeatedly with water, methanol and diethyl ether. The yellow solid was dried under reduced pressure to yield the platinum complex (23 mg, 55%); 1H NMR (400 MHz, DMSO-d6): δH 8.43 (d, 2H, 3JHH = 8.0, tpy′ 3-H and 5-H), 8.28 (d, 2H, 3JHH = 8.0, tpy 5-H), 8.14 (t, 1H, 3JHH = 8.0, tpy′ 4-H), 7.99 (t, 2H, 3JHH = 8.0, tpy 4-H), 7.02 (d, 2H, 3JHH = 8.0, tpy 3-H), 4.81 (t, 4H, 3JHH = 4.0, ethyl 1-H), 3.60 (t, 4H, 3JHH = 6.0, ethyl 2-H), 1.86 (m, 8H, pip 1-H), 1.71 (m, 8H, pip 2-H), 1.42 (m, 4H, pip 3-H); 31P NMR (400 MHz, DMSO-d6): δP −144.21 (sept, 1P, 1JPF = 1756, PF6); ESI-MS calcd for C29H37ClF6N5O2PPt [M]+: 862.2 a.m.u.; found [M-PF6+K+Na]+: 780.0 a.m.u.; anal. calcd for C29H37ClF6N6O2PPt·1.3DMSO: C 39.94, H 4.75, N 7.37; found: C 40.34, H 4.32, N 7.00%.Synthesis of 2
This compound was prepared as reported previously.22Determination of DNA affinity by UV-Vis titration
The UV-Vis spectra were recorded on a Perkin Elmer Lambda 25 spectrometer. In order to determine the binding constants of complex 1 with ct-DNA, the complex (20 μM) was titrated with a concentrated solution of ct-DNA (2.78 mM) in 50 mM Tris–HCl (pH 7.4)/100 mM KCl buffer. A 1 cm path-length quartz cuvette was used to carry out the measurements. The binding constants were obtained by fitting the data to a reciprocal plot of D/Δεapversus D using the following equation: D/Δεap = D/Δε + 1/(Δε × K), where the concentration of DNA is expressed in terms of base pairs (determined by measuring the absorption at 260 nm and the appropriate extinction coefficients), the apparent molar extinction coefficient εa = Aobserved/[complex], Δεap = [εa − εf] and Δε = [εb − εf]. εb is the extinction coefficient of the DNA bound complex and εf is the extinction coefficient of the free complex.Fluorescent intercalator displacement (FID) assay
A 26 base pair self-complementary strand (5′-CAA-TCG-GAT-CGA-ATT-CGA-TCC-GAT-TG-3′) was used (from Eurogentec S.A.). The oligonucleotide was dissolved in Milli Q water to yield 20 μM stock solution. This was then diluted using 10 mM potassium cacodylate (pH 7.4)/50 mM potassium chloride (60 mM K+) buffer to the appropriate concentrations. Prior to use in the FID assay, the DNA strand was annealed by heating to 95 °C for 5 min and then cooled down to room temperature overnight. Complex 1, thiazole orange (TO) dye and Hoechst dye were dissolved in DMSO to give 10 mM stock solutions of each. The corresponding solution was then diluted using 10 mM potassium cacodylate (pH 7.4)/50 mM potassium chloride (60 mM K+) buffer to the appropriate concentrations.Circular dichroism studies
Complex 1 was dissolved in DMSO to yield a 10 mM stock solution. This was then diluted using 50 mM Tris–HCl (pH 7.4) buffer to the appropriate concentrations. ct-DNA was dissolved directly in 50 mM Tris–HCl (pH 7.4) buffer. The CD spectra were recorded in a strain-free 10 mm × 2 mm rectangular cell path length cuvette. The data were obtained on an Applied Photophysics Ltd. Chirascan spectrometer. The CD spectra were measured in the wavelength region of 180–700 nm with the following parameters: bandwidth, 1 nm; spectral range, 230–360 nm; step-size, 0.5 nm; time-pep-point, 1.5 s. The CD spectra were collected and analysed using the Chirascan and Chirascan Viewer software respectively. CD studies were done for ct-DNA with and without varying amounts of 1 (1: base pair = 0.5 and 1) in Tris–HCl buffer.Agarose gel electrophoresis
Plasmid DNA (pUC19) was purchased from Sigma Aldrich. A DNA stock solution of 453 μg mL−1 in 10 mM Tris–HCl, pH 8.0, 1 mM EDTA storage buffer was prepared. The DNA binding of 1 was determined by monitoring the conversion of supercoiled plasmid DNA (form I) to nicked circular DNA (form II) and/or linear DNA (form III) using agarose gel electrophoresis. Solutions containing 256 ng (30 μM) of DNA and 0, 10, 20, 50, 100 and 200 μM of 1 with a total reaction volume of 15 μL were incubated at 37 °C for 24 h. After the time period stated, loading buffer (5 μL, containing 0.25% bromophenol blue, 0.25% xylene cyanol and 60% glycerol) was added and reaction mixtures were immediately loaded onto 1% agarose gels containing 1.0 μg mL−1 of ethidium bromide. The DNA fragments were separated by applying 40 V for 1 h in Tris–acetate EDTA (TAE) buffer. The DNA bands were analysed under UV light using the Fujifilm Image Reader LAS–3000.Cytotoxicity MTS assay
Cells (5 × 104 for U2OS, 5 × 104 for HEK 293T, 1 × 104 for GM05757, 5 × 104 for SH-SY5Y, 2 × 104 for NIH 3T3) were seeded in each well of a 96-well plate. After incubating the cells overnight, various concentrations of 1 (0.2–500 μM) were added and incubated for 24 h (total volume 100 μL). For the mode of death determination assays, the necrosis and autophagocytosis inhibitors, necrostatin-1 (60 μM), 3-aminobenzamide (2 mM) or chloroquine (10 μM) were added to the cells and incubated for 1 or 6 h prior to treatment with 1. The test compound was prepared as a 10 mM solution in DMSO and diluted further using DMSO. The final concentration of DMSO in each well was 4% (this concentration of DMSO was used at the untreated control). Cisplatin was prepared as a 10 mM solution in PBS:DMSO, 5:1 and diluted further using PBS. After the incubation period, 20 μL of the MTS solution (made up of 2 mg mL−1 MTS reagent + 100 μL of PMS 0.96 mg mL−1) was added to each well. The mixture was incubated for a further 1.5–4 h at 37 °C. After this time, an ELISA reader was used to record the absorption at 490 nm and the IC50 values were determined using the conventional method. IC50 values were calculated from the following equation: Abs IC50 = (Abs IC0 + Abs IC100)/2, where IC0 is the mean absorption at 490 nm of the media only and IC100 is the mean absorption at 490 nm of the cells only. Then the X-axis intercept of the dose–response curve at Abs IC50 (determined by the above equation) was considered as the IC50 value for each test compound. The dose–response curve is a plot of absorption at 490 nm (y-axis) and the concentration of test compound exposed to the cells (x-axis). Prior to plotting the curve the background absorption (due to the reaction of the test compound and MTS reagents) was subtracted from the absorption values obtained from the cells that had been incubated with the compounds.Cellular uptake studies
To measure the cellular uptake of 1, ca. 2 million U2OS, HEK 293T, GM05757, SH-SY5Y and NIH 3T3 (proliferating and non-proliferating conditions) cells were treated with 10 μM of the complex at 37 °C for 24 h. Then the media were removed, the cells were washed with PBS solution (×3), harvested and centrifuged. The cellular pellet was suspended in an appropriate volume of PBS to obtain a homogeneous cell suspension. The suspension was divided in two. One part was used to analyse the metal content in the whole cell and the other was used for the cytoplasmic and nucleus analysis. The Thermo Scientific NE-PER Nuclear and Cytoplasmic Extraction Kit was used to extract the separate cytoplasmic and nuclear fractions. From each of the fractions, aliquots were removed for protein determination using the Bradford assay (carried out according to the instructions of the manufacturer: Sigma Aldrich, micro-well plate protocol). The remaining cell suspension was mineralized with 65% HNO3 and then completely dried at 120 °C. The solid extracts were re-dissolved in 2% HNO3 and analysed using ICP-MS. Cellular metal levels were expressed as ng Pt per mg protein. Results are presented as the mean of 4 or 5 determinations for each data point. The fraction of the administered complexes that were taken up by the cells was calculated by dividing the amount of complex detected in the whole cell, cytoplasm and nucleus (i.e. the raw measurement in ng) by the total mass of metal corresponding to the concentration administered for each complex (10 μM converted to ng). The fractions were normalised so that all treated cells were accounted. The percentage of the administered complexes that were taken up by the cells was then determined by multiplying the normalised fractions by 100.Flow cytometry studies
In order to monitor the cell cycle, flow cytometry studies were carried out. U2OS cells were incubated with and without 10 μM 1 for 24 h at 37 °C. Studies on NIH 3T3 cells under normal and starved (proliferating and non-proliferating) conditions were also carried out. Cells were harvested from adherent cultures by trypsinization and combined with all detached cells from the incubation medium to assess the total cell viability. Following centrifugation at 1000 rpm for 5 min, cells were washed with PBS and then fixed with 70% ethanol in PBS. Fixed cells were collected by centrifugation at 2500 rpm for 3 min, washed with PBS and centrifuged as before. Cellular pellets were re-suspended in 50 μg mL−1 propidium iodide (Calbiochem) in PBS for nucleic acids staining and treated with 100 μg mL−1 RNaseA (Sigma). DNA content was measured on a FACSCalibur flow cytometer (BD Biosciences) using laser excitation at 488 nm and 10000 events per sample were acquired. Cell cycle profiles were analysed using the FlowJo software (Tree Star). For the annexin V experiments, the Invitrogen FITC Annexin V/Dead Cell Apoptosis Kit was used. The manufacturer's protocol was followed to carry out this experiment. Briefly, untreated and treated cells (1 × 105) were suspended in binding buffer (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4), then 5 μL FITC annexin V and 1 μL PI (100 μg mL−1) were added to each sample and incubated at room temperature for 15 min., after which more binding buffer (400 μL) was added while mixing gently. The samples were kept on ice prior to being read on the FACSCalibur flow cytometer (BD Biosciences) using laser excitation at 488 nm (10000 events per sample were acquired). Cell cycle profiles were analysed using the FlowJo software (Tree Star).Immunoblot analysis
For protein analysis by immunoblotting, U2OS cells (5 × 105 cells) were incubated with the indicated concentrations of 1 for 24 h at 37 °C. Cells were washed with PBS, scraped into SDS-PAGE loading buffer (64 mM Tris–HCl (pH 6.8)/9.6% glycerol/2% SDS/5% β-mercaptoethanol/0.01% bromophenol blue) and incubated at 95 °C for 10 min. Whole cell lysates were resolved by 12% sodium dodecylsulphate polyacrylamide gel electrophoresis (SDS-PAGE; 200 V for 1 h), followed by electron transfer to nitrocellulose membrane (400 mA for 1 h). Membranes were blocked in 5% (w/v) non-fat milk in PBST (PBS/0.1% Tween 20) and incubated with one of the following primary antibodies: anti-p53 (DO-1, Santa Cruz), anti-p21 (C-19, Santa Cruz), anti-Chk2 (2662, Cell Signalling Technology), anti-phospho-Chk2 (2661, Cell Signalling Technology), anti-BAX (P-19, Santa Cruz), anti-caspase-3 (E-8, Santa Cruz), anti-cyclin D1 (A-12, Santa Cruz), anti-phospho-Histone H2A.X (2577, Cell Signalling Technology) and anti-α-tubulin (TAT-1, Cancer Research UK). After incubation with horseradish peroxidase-conjugated secondary antibodies, immune complexes were detected with the ECL detection reagent (Millipore) and analysed using the Fujifilm Image Reader LAS-3000.Extraction of HMGB1 released into culture media
U2OS cells (1 × 106 cells) were incubated with 1 (50–200 μM) for 48 h at 37 °C to induce cell death. As a positive control, necrosis was induced by heating cells at 56 °C for 30 min, followed by incubation at 37 °C for an additional 1 h. The protein contained within the culture media was precipitated. Protein precipitation was carried out by adding methanol (4× volume), chloroform (1×) and ddH2O (3×), vortexing and then centrifugation at 5000 rpm for 2 min. The aqueous layer was removed and more methanol (4× volume) was added. Following mixing and centrifugation as before, the methanol was removed. The resultant protein precipitate was air dried, dissolved in SDS-PAGE loading buffer (64 mM Tris–HCl (pH 6.8)/9.6% glycerol/2% SDS/5% β-mercaptoethanol/0.01% bromophenol blue) and incubated at 95 °C for 10 min. The HMGB1 content was probed by immunoblotting analysis as described above. The anti-HMGB1 anti-body (3935, Cell Signalling Technology) was used in this experiment.Acknowledgements
The UK's Engineering and Physical Sciences Research Council (EPSRC) is thanked for funding (grant number: EP/H005285/1) and Johnsons Matthey for a loan of platinum.References
- Z. Guo and P. J. Sadler, Angew. Chem., Int. Ed., 1999, 38, 1512–1531 CrossRef CAS.
- T. W. Hambley, Science, 2007, 318, 1392–1393 CrossRef CAS.
- B. K. Keppler, Metal complexes in cancer chemotherapy, VCH, Weinheim, New York, 1993 Search PubMed.
- I. Kostova, Recent Pat. Anti-cancer Drug Discovery, 2006, 1, 1–22 CrossRef CAS.
- H. Sigel and A. Sigel, Metal complexes as anticancer agents, M. Dekker, New York, 1980 Search PubMed.
- C. X. Zhang and S. J. Lippard, Curr. Opin. Chem. Biol., 2003, 7, 481–489 CrossRef CAS.
- P. J. Dyson and G. Sava, Dalton Trans., 2006, 1929–1933 RSC.
- P. Carloni, M. Sprik and W. Andreoni, J. Phys. Chem. B, 2000, 104, 823–835 CrossRef CAS.
- H. C. Harder and B. Rosenberg, Int. J. Cancer, 1970, 6, 207–216 CrossRef CAS.
- J. A. Howle and G. R. Gale, Biochem. Pharmacol., 1970, 19, 2757–2762 CrossRef CAS.
- E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467–2498 CrossRef CAS.
- S. E. Sherman and S. J. Lippard, Chem. Rev., 1987, 87, 1153–1181 CrossRef CAS.
- D. Tullius Thomas, M. H. Ushay, M. M. Carolyn, P. C. John and J. L. Stephen, Platinum, Gold, and Other Metal Chemotherapeutic Agents, ACS, 1983, vol. 209, pp. 51–74 Search PubMed.
- S. M. Cohen and S. J. Lippard, Prog. Nucleic Acid Res. Mol. Biol., 2001, 67, 93–130 CrossRef CAS.
- Y. Jung and S. J. Lippard, J. Biol. Chem., 2006, 281, 1361–1370 CrossRef CAS.
- M. A. Fuertes, C. Alonso and J. M. Perez, Chem. Rev., 2003, 103, 645–662 CrossRef CAS.
- M. Galanski, M. A. Jakupec and B. K. Keppler, Curr. Med. Chem., 2005, 12, 2075–2094 CrossRef CAS.
- J. K. Barton and S. J. Lippard, Biochemistry, 1979, 18, 2661–2668 CrossRef CAS.
- A. Odani, R. Shimata, H. Masuda and O. Yamauchi, Inorg. Chem., 1991, 30, 2133–2138 CrossRef CAS.
- B. M. Zeglis, V. C. Pierre and J. K. Barton, Chem. Commun., 2007, 4565–4579 RSC.
- D. M. Fisher, R. R. Fenton and J. R. Aldrich-Wright, Chem. Commun., 2008, 5613–5615 RSC.
- P. J. Bond, R. Langridge, K. W. Jennette and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 4825–4829 CrossRef CAS.
- Z. D. Bugarcic, F. W. Heinemann and R. v. Eldik, Dalton Trans., 2004, 279–286 RSC.
- S. J. Lippard, Acc. Chem. Res., 1978, 11, 211–217 CrossRef CAS.
- G. Lowe, J. A. McCloskey, J. Ni and T. Vilaivan, Bioorg. Med. Chem., 1996, 4, 1007–1013 CrossRef CAS.
- G. Lowe and T. Vilaivan, J. Chem. Soc., Perkin Trans. 1, 1996, 1499–1503 RSC.
- D. Petrović, B. Stojimirović, B. Petrović, Z. M. Bugarčić and Ž. D. Bugarčić, Bioorg. Med. Chem., 2007, 15, 4203–4211 CrossRef.
- K. G. Strothkamp and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 2536–2540 CrossRef CAS.
- S. D. Cummings, Coord. Chem. Rev., 2009, 253, 1495–1516 CrossRef CAS.
- W. D. McFadyen, L. P. Wakelin, I. A. Roos and V. A. Leopold, J. Med. Chem., 1985, 28, 1113–1116 CrossRef CAS.
- W. D. McFadyen, L. P. Wakelin, I. A. Roos and B. L. Hillcoat, Biochem. J., 1986, 238, 757–763 CAS.
- E. L. Crossley, D. Caiazza and L. M. Rendina, Dalton Trans., 2005, 2825–2826 RSC.
- G. Lowe, A. S. Droz, T. Vilaivan, G. W. Weaver, J. J. Park, J. M. Pratt, L. Tweedale and L. R. Kelland, J. Med. Chem., 1999, 42, 3167–3174 CrossRef CAS.
- D.-L. Ma, T. Y.-T. Shum, F. Zhang, C.-M. Che and M. Yang, Chem. Commun., 2005, 4675–4677 RSC.
- J. A. Todd and L. M. Rendina, Inorg. Chem., 2002, 41, 3331–3333 CrossRef CAS.
- J. A. Todd, P. Turner, E. J. Ziolkowski and L. M. Rendina, Inorg. Chem., 2005, 44, 6401–6408 CrossRef CAS.
- S. L. Woodhouse, E. J. Ziolkowski and L. M. Rendina, Dalton Trans., 2005, 2827–2829 RSC.
- G. Arena, L. M. Scolaro, R. F. Pasternack and R. Romeo, Inorg. Chem., 1995, 34, 2994–3002 CrossRef CAS.
- C. Sessa, O. Pagani, M. G. Zurlo, J. de Jong, C. Hofmann, M. Lassus, P. Marrari, M. S. Benedetti and F. Cavalli, Ann. Oncol., 1994, 5, 901–907 CAS.
- J. Viallet, D. Stewart, F. Shepherd, J. Ayoub, Y. Cormier, N. DiPietro and W. Steward, J. Lung Cancer, 1996, 15, 367–373 CrossRef CAS.
- F. Caponigro, D. Lorusso, G. Fornari, C. Barone, M. Merlano, M. Airoldi, M. Schena, R. MacArthur, S. Weitman, M. G. Jannuzzo, S. Crippa, F. Fiorentini, A. Petroccione and S. Comis, Cancer Chemother. Pharmacol., 2010, 66, 389–394 CrossRef CAS.
- A. J. ten Tije, J. Verweij, A. Sparreboom, A. van der Gaast, C. Fowst, F. Fiorentini, J. Tursi, A. Antonellini, M. Mantel, C. M. Hartman, G. Stoter, A. S. T. Planting and M. J. A. de Jonge, Clin. Cancer Res., 2003, 9, 2957–2964 CAS.
- M. Howe-Grant, K. C. Wu, W. R. Bauer and S. J. Lippard, Biochemistry, 1976, 15, 4339–4346 CrossRef CAS.
- W. D. McFadyen, L. P. Wakelin, I. A. Roos and B. L. Hillcoat, Biochem. J., 1987, 242, 177–183 CAS.
- L. Messori, G. Marcon, A. Innocenti, E. Gallori, M. Franchi and P. Orioli, Bioinorg. Chem. Appl., 2005, 239–253 CrossRef CAS.
- R. Kieltyka, J. Fakhoury, N. Moitessier and H. F. Sleiman, Chem.–Eur. J., 2008, 14, 1145–1154 CrossRef CAS.
- C. Rajput, R. Rutkaite, L. Swanson, I. Haq and J. A. Thomas, Chem.–Eur. J., 2006, 12, 4611–4619 CrossRef CAS.
- E. Largy, F. Hamon and M. P. Teulade-Fichou, Anal. Bioanal. Chem., 2011, 400, 3419–3427 CrossRef CAS.
- D. Monchaud, C. Allain, H. Bertrand, N. Smargiasso, F. Rosu, V. Gabelica, A. De Cian, J. L. Mergny and M. P. Teulade-Fichou, Biochimie, 2008, 90, 1207–1223 CrossRef CAS.
- D. Monchaud, C. Allain and M. P. Teulade-Fichou, Nucleosides, Nucleotides Nucleic Acids, 2007, 26, 1585–1588 CAS.
- D. Monchaud and M. P. Teulade-Fichou, Methods Mol. Cell. Biol., 2010, 608, 257–271 CAS.
- P. U. Maheswari, S. Roy, H. den Dulk, S. Barends, G. van Wezel, B. Kozlevcǎr, P. Gamez and J. Reedijk, J. Am. Chem. Soc., 2005, 128, 710–711 CrossRef.
- B. Selvakumar, V. Rajendiran, P. Uma Maheswari, H. Stoeckli-Evans and M. Palaniandavar, J. Inorg. Biochem., 2006, 100, 316–330 CrossRef CAS.
- P. X. Xi, Z. H. Xu, X. H. Liu, F. J. Chen, Z. Z. Zeng, X. W. Zhang and Y. Liu, J. Fluoresc., 2009, 19, 63–72 CrossRef CAS.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Olson, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- J. K. Barton and E. Lolis, J. Am. Chem. Soc., 1985, 107, 708–709 CrossRef CAS.
- S. L. H. Higgins, A. J. Tucker, B. S. J. Winkel and K. J. Brewer, Chem. Commun., 2012, 48, 67–69 RSC.
- R. L. Williams, H. N. Toft, B. Winkel and K. J. Brewer, Inorg. Chem., 2003, 42, 4394–4400 CrossRef CAS.
- K. Suntharalingam, A. J. P. White and R. Vilar, Inorg. Chem., 2009, 48, 9427–9435 CrossRef CAS.
- K. Suntharalingam, D. J. Hunt, A. A. Duarte, A. J. P. White, D. J. Mann and R. Vilar, Chem.–Eur. J., 2012, 18, 15133–15141 CrossRef CAS.
- T. Boulikas and M. Vougiouka, Oncol. Rep., 2003, 10, 1663–1682 CAS.
- J. Y. Ahn, J. K. Schwarz, H. Piwnica-Worms and C. E. Canman, Cancer Res., 2000, 60, 5934–5936 CAS.
- S. Burma, B. P. Chen, M. Murphy, A. Kurimasa and D. J. Chen, J. Biol. Chem., 2001, 276, 42462–42467 CrossRef CAS.
- S. Matsuoka, G. Rotman, A. Ogawa, Y. Shiloh, K. Tamai and S. J. Elledge, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 10389–10394 CrossRef CAS.
- R. Melchionna, X.-B. Chen, A. Blasina and C. H. McGowan, Nat. Cell Biol., 2000, 2, 762–765 CrossRef CAS.
- E. P. Rogakou, D. R. Pilch, A. H. Orr, V. S. Ivanova and W. M. Bonner, J. Biol. Chem., 1998, 273, 5858–5868 CrossRef CAS.
- T. M. Gottlieb and M. Oren, Semin. Cancer Biol., 1998, 8, 359–368 CrossRef CAS.
- L. H. Hartwell and M. B. Kastan, Science, 1994, 266, 1821–1828 CAS.
- L. Gatti, R. Supino, P. Perego, R. Pavesi, C. Caserini, N. Carenini, S. C. Righetti, V. Zuco and F. Zunino, Cell Death Differ., 2002, 9, 1352–1359 CrossRef CAS.
- D. P. Lane, Nature, 1992, 358, 15–16 CrossRef CAS.
- B. Vogelstein, D. Lane and A. J. Levine, Nature, 2000, 408, 307–310 CrossRef CAS.
- H. Matsushime, M. E. Ewen, D. K. Strom, J. Y. Kato, S. K. Hanks, M. F. Roussel and C. J. Sherr, Cell, 1992, 71, 323–334 CrossRef CAS.
- H. Matsushime, D. E. Quelle, S. A. Shurtleff, M. Shibuya, C. J. Sherr and J. Y. Kato, Mol. Cell. Biol., 1994, 14, 2066–2076 CAS.
- S. W. Tam, A. M. Theodoras, J. W. Shay, G. F. Draetta and M. Pagano, Oncogene, 1994, 9, 2663–2674 CAS.
- Z. D. Lan, Z. Sever-Chroneos, M. W. Strobeck, C. H. Park, R. Baskaran, W. Edelmann, G. Leone and E. S. Knudsen, J. Biol. Chem., 2002, 277, 8372–8381 CrossRef CAS.
- L. L. Pontano and J. A. Diehl, Cell Cycle, 2009, 8, 824–827 CrossRef CAS.
- M. K. Santra, N. Wajapeyee and M. R. Green, Nature, 2009, 459, 722–725 CrossRef CAS.
- N. Suwaki, E. S. Child, L. M. Elphick and D. J. Mann, Arch. Biochem. Biophys., 2010, 497, 55–61 CrossRef CAS.
- J. F. Kerr, A. H. Wyllie and A. R. Currie, Br. J. Cancer, 1972, 26, 239–257 CrossRef CAS.
- J. F. R. Kerr, J. Pathol. Bacteriol., 1965, 90, 419–435 CrossRef CAS.
- S. Kasibhatla, T. Brunner, L. Genestier, F. Echeverri, A. Mahboubi and D. R. Green, Mol. Cell, 1998, 1, 543–551 CrossRef CAS.
- M. MacFarlane, N. A. Jones, C. Dive and G. M. Cohen, Mol. Pharmacol., 1996, 50, 900–911 CAS.
- Y. Sedletska, M. J. Giraud-Panis and J. M. Malinge, Curr. Med. Chem.: Anti-cancer Agents, 2005, 5, 251–265 CrossRef CAS.
- E. P. Simoes Magluta, F. C. Vasconcelos, R. C. Maia and C. E. Klumb, Cancer Invest., 2009, 27, 830–835 CrossRef.
- J. Pan, C. Cheng, S. Verstovsek, Q. Chen, Y. Jin and Q. Cao, Cancer Lett., 2010, 293, 167–174 CrossRef CAS.
- P. Scaffidi, Nature, 2002, 418, 191 CrossRef CAS.
- A. Agresti, Curr. Opin. Genet. Dev., 2003, 13, 170 CrossRef CAS.
- A. Raucci, R. Palumbo and M. E. Bianchi, Autoimmunity, 2007, 40, 285–289 CrossRef CAS.
- R. J. Ernst, A. C. Komor and J. K. Barton, Biochemistry, 2011, 50, 10919–10928 CrossRef CAS.
- J. Hitomi, D. E. Christofferson, A. Ng, J. Yao, A. Degterev, R. J. Xavier and J. Yuan, Cell, 2008, 135, 1311–1323 CrossRef CAS.
- F. K. Chan, J. Shisler, J. G. Bixby, M. Felices, L. Zheng, M. Appel, J. Orenstein, B. Moss and M. J. Lenardo, J. Biol. Chem., 2003, 278, 51613–51621 CrossRef CAS.
- A. L. Edinger and C. B. Thompson, Curr. Opin. Cell Biol., 2004, 16, 663–669 CrossRef CAS.
- L. Galluzzi and G. Kroemer, Cell, 2008, 135, 1161–1163 CrossRef CAS.
- G. Kroemer, W. S. El-Deiry, P. Golstein, M. E. Peter, D. Vaux, P. Vandenabeele, B. Zhivotovsky, M. V. Blagosklonny, W. Malorni, R. A. Knight, M. Piacentini, S. Nagata and G. Melino, Cell Death Differ., 2005, 12(suppl 2), 1463–1467 CrossRef CAS.
- M. S. Ricci and W. X. Zong, Oncologist, 2006, 11, 342–357 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3mt20252f |
| This journal is © The Royal Society of Chemistry 2013 |