 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The Porphyromonas gingivalis HmuY haemophore binds gallium(III), zinc(II), cobalt(III), manganese(III), nickel(II), and copper(II) protoporphyrin IX but in a manner different to iron(III) protoporphyrin IX
Halina
Wójtowicz
a,
Marcin
Bielecki
a,
Jacek
Wojaczyński
b,
Mariusz
Olczak
a,
John W.
Smalley
c and
Teresa
Olczak
*a
aUniversity of Wrocław, Faculty of Biotechnology, Laboratory of Biochemistry, Tamka St. 2, 50-137 Wrocław, Poland. E-mail: Teresa.Olczak@biotech.uni.wroc.pl; Fax: +48 71 3752 608; Tel: +48 71 3752 612
bUniversity of Wrocław, Department of Chemistry, F. Joliot-Curie 14 St., 50-383 Wrocław, Poland
cUniversity of Liverpool, Department of Clinical Infection, Microbiology and Immunology, Institute of Infection and Global Health, The Ronald Ross Building, 8 West Derby Street, Liverpool L69 7BE, UK
First published on 18th January 2013
Abstract
Porphyromonas gingivalis, a major etiological agent of chronic periodontitis, acquires haem from host haemoproteins through a haem transporter HmuR and a haemophore HmuY. The aim of this study was to analyse the binding specificity of HmuY towards non-iron metalloporphyrins which may be employed as antimicrobials to treat periodontitis. HmuY binds gallium(III), zinc(II), cobalt(III), manganese(III), nickel(II), and copper(II) protoporphyrin IX but in a manner different to iron(III) protoporphyrin IX which uses His134 and His166 as axial ligands. The metal ions in Ga(III)PPIX and Zn(II)PPIX can accept only His166 as an axial ligand, whereas nickel(II) and copper(II) interact exclusively with His134. Two forms of pentacoordinate manganese(III) are present in the Mn(III)PPIX–HmuY complex since the metal accepts either His134 or His166 as a single axial ligand. The cobalt ion is hexacoordinate in the Co(III)PPIX–HmuY complex and binds His134 and His166 as axial ligands; however, some differences in their environments exist. Despite different coordination modes of the central metal ion, gallium(III), zinc(II), cobalt(III), and manganese(III) protoporphyrin IX bound to the HmuY haemophore cannot be displaced by excess haem. All of the metalloporphyrins examined bind to a P. gingivalis wild-type strain with higher ability compared to a mutant strain lacking a functional hmuY gene, thus corroborating binding of non-iron metalloporphyrins to purified HmuY protein. Our results further clarify the basis of metalloporphyrin acquisition by P. gingivalis and add to understanding of the interactions with porphyrin derivatives which exhibit antimicrobial activity against P. gingivalis.
Introduction
Porphyromonas gingivalis is a black-pigmented Gram-negative anaerobic bacterium which has been implicated as a major etiological agent in the development and progression of chronic periodontitis.1P. gingivalis produces several virulence factors which lead to direct connective tissue destruction and alveolar bone loss, as well as increased deleterious host immune responses.1,2 The bacterium can enter gingival epithelial cells where it can remain viable and is capable of spreading between host cells,3–5 which contribute to its survival in the oral cavity. Also, the ability of P. gingivalis to participate in biofilm formation on oral surfaces contributes to bacterial persistence and increases the potential for periodontal tissue destruction.6 Importantly, in a wider context, it has been demonstrated that periodontal diseases increase the risk of cardiovascular and respiratory diseases, diabetes mellitus, osteoporosis, rheumatoid arthritis, and preterm low-birth weight.7–9P. gingivalis is a haem† auxotroph and uptake of this compound is central for bacterial survival and the ability to establish infection.10 In P. gingivalis, several TonB-dependent outer-membrane haem receptors have been described, including HmuR, Tlr, IhtA, and HusB.10–13 In this work we focus on the HmuY haemophore that sequesters haem directly from haemoglobin to transfer it to the HmuR receptor.14–16 Analysis of the three-dimensional structure of the haem–HmuY complex has revealed a unique protein all-beta fold structure.17 Detailed characterization of the haem–HmuY complex has demonstrated that histidine residues at positions 134 and 166 serve as the iron ligands.17,18 Recently, we have shown that HmuY also binds iron(III) mesoporphyrin IX and iron(III) deuteroporphyrin IX.19
This work extends our recently published findings on the HmuY haem-binding protein by presenting data regarding its binding specificity towards selected non-iron metalloporphyrins, some of which have been shown to have antimicrobial activity against P. gingivalis.20 Our data demonstrate that HmuY binds gallium(III), cobalt(III), manganese(III), nickel(II), and copper(II) protoporphyrin IX but in a manner different to iron(III) protoporphyrin IX.
Experimental
Protein purification
P. gingivalis apo-HmuY lacking the first 25 amino-acid residues (NCBI accession no. CAM 31898) was over-expressed using the pHmuY11 plasmid and Escherichia coli ER2566 cells (New England Biolabs) and purified from a soluble fraction of the E. coli lysate as previously described.14 The recombinant form of HmuY does not possess the signal peptide, nor the following cysteine and four additional N-terminal amino-acid residues (CGKKK), which are not present in the mature soluble protein.15 In this study we also used HmuY variants where either His166 or His134 and Met136 residues were replaced with alanine residues giving the His166Ala or the His134Ala/Met136Ala HmuY variants, respectively.18 This allowed selective removal of each of the iron coordinating histidine residues along with the methionine residue that, due to its proximity to His134, plays an auxiliary role in iron coordination. HmuY variants were over-expressed and purified analogously to the wild-type HmuY as reported previously.18HmuY–metalloporphyrin titration experiments
Iron(III) protoporphyrin IX chloride (Fe(III)PPIX chloride; Fluka) and non-iron metalloporphyrins IX (Ga(III)PPIX chloride, Zn(II)PPIX, Mn(III)PPIX chloride, Ni(II)PPIX, Cu(II)PPIX, Co(III)PPIX chloride; Frontier Scientific) were prepared as described previously,21 and their complexes with HmuY were formed as reported earlier.17,19,20 For measurements of the metalloporphyrin concentration in 20 mM phosphate buffer, pH 7.4, containing 140 mM NaCl, the extinction coefficient of each non-iron metalloporphyrin was determined in this study by a combination of metal content determination using inductively coupled plasma-atomic emission spectroscopy (ICP-AES) analysis with a Liberty 220 spectrometer (Varian) equipped with a standard pneumatic nebulizer, and measurement of the absorbance spectra maxima of the respective metalloporphyrin solutions (ε417nm = 49.9 mM−1 cm−1 for Co(III)PPIX; ε407nm = 34.8 mM−1 cm−1 for Zn(II)PPIX; ε404nm = 90.6 mM−1 cm−1 for Ga(III)PPIX; ε370nm = 44.8 mM−1 cm−1 for Mn(III)PPIX; ε384nm = 55.5 mM−1 cm−1 for Ni(II)PPIX, and ε384nm = 53.5 mM−1 cm−1 for Cu(II)PPIX).Titration experiments were performed in 20 mM sodium phosphate buffer, pH 7.4, containing 140 mM NaCl and monitored by UV-visible absorption spectroscopy using an Agilent 8453E UV-Vis spectrophotometer (Agilent Technologies). The absorbance increase at the wavelength corresponding to the absorbance maximum of each complex was monitored for addition of each metalloporphyrin (1–20 μM) to HmuY solutions. Concentrations of HmuY used for titration experiments were optimised in preliminary experiments to give the best resolved titration curves depending on the affinity of each metalloporphyrin to HmuY (5 μM for Ga(III)PPIX and Zn(II)PPIX and 10 μM for the remaining metalloporphyrins). Titration curves were analysed using equations for a one-site binding model and Kd values determined as reported earlier19 using the OriginPro 8 software (OriginPro Corporation).
Concentrations of apo- and holo-HmuY were determined spectrophotometrically using the extinction coefficients (ε280nm = 36.86 mM−1 cm−1 and ε280nm = 59.26 mM−1 cm−1,18 and this study, respectively).
CD measurements
HmuY–metalloporphyrin complexes for the circular dichroism (CD) spectra measurements were prepared in 10 mM sodium phosphate buffer, pH 7.8. Solutions containing 200 μM wild-type HmuY and its His166Ala or His134Ala/Met136Ala variants were incubated with a 2-fold molar excess of metalloporphyrins for 24 h at 4 °C. Then, excess metalloporphyrin was removed from the solution by gel filtration and the protein concentration adjusted to 100 μM. CD spectra of the metalloporphyrin–HmuY complexes were recorded at 25 °C on a Jasco J-715 spectropolarimeter with a scan speed of 50 nm min−1, a response time of 2 s and a slit width of 1.0 nm. Measurements were made using a quartz cell with a 0.1 or 0.5 cm path length. Mean spectra were calculated from four independently recorded data sets.NMR measurements
The analysis of metalloporphyrin binding to HmuY was also carried out using 1H nuclear magnetic resonance (1H NMR) spectroscopy. For this purpose, metalloporphyrin–HmuY complexes were prepared as previously described.18,19 Final protein concentrations were in the range of 0.5–1.2 mM. 1H NMR measurements were performed using a Bruker Avance 600 spectrometer operating at a proton Larmor frequency of 600.13 MHz. The data were collected at 300 K with the chemical shift referenced to a residual water signal. The strong H2O/HDO resonance was suppressed by pre-saturation. For haem exchange experiments, 1 mg of Fe(III)PPIX chloride was dissolved in 100 μl of D2O containing two pellets of potassium carbonate. The resulting solution was added dropwise to the NMR tube containing the metalloporphyrin–HmuY complex dissolved in D2O.Bacterial strains and growth conditions
A P. gingivalis wild-type strain (A7436) and a hmuY mutant strain lacking a functional hmuY gene (TO4) constructed in strain A7436 were grown anaerobically on blood agar (ABA, Biocorp) plates and then in basal medium supplemented with 10 μM haem as previously described.14 Bacterial cells were depleted of iron and haem by passage of the cultures three times into BM without added haem and supplemented with dipyridyl (160 μM) as an iron chelator, until growth was significantly reduced, as measured by an OD at 600 nm, thus ensuring higher HmuY expression.14 The cells were collected by centrifugation (4000 × g for 10 min at 4 °C), washed twice in sterile 20 mM phosphate buffer, pH 7.4, containing 140 mM NaCl, suspended in fresh basal medium, and adjusted to OD600 = 1.0. The metalloporphyrins were added to 1 ml aliquots of bacterial cultures to a final concentration of 10 μM. Samples without added metalloporphyrins and culture medium without bacteria, but with metalloporphyrins, served as appropriate controls. After 1 h, cultures were centrifuged and the amounts of metalloporphyrins remaining in culture medium were determined by absorbance measurements at wavelengths corresponding to Soret band maxima of the respective metalloporphyrins (see extinction coefficient measurements above).Results and discussion
P. gingivalis HmuY binds non-iron metalloporphyrins
Non-iron metalloporphyrins are potent inhibitors of several proteins and therefore have potential for targeting bacterial infections. Amongst these, gallium(III) protoporphyrin IX is active against Yersinia enterocolitica, Neisseria gonorrhoeae, Haemophilus ducreyi and Staphylococcus aureus.22,23 Because it is similar in size and ionic radius to iron(III), gallium(III) is widely used as a non-redox iron substitute utilized by common bacterial transport systems.24 Recently we have shown that some non-iron metalloporphyrins reduce P. gingivalis growth and infection,20 and this study used UV-visible absorption, CD and 1H NMR spectroscopies to examine binding of selected non-iron metalloporphyrins to HmuY. All of the non-iron metalloporphyrins examined were bound to HmuY (Fig. 1 (left panel) and Table 1), although the coordination mode of each metalloporphyrin was different and depended on the central metal ion. The dissociation constants (Kd) of the metalloporphyrin–HmuY complexes were determined from titration experiments using UV-visible absorption spectroscopy (Fig. 1, right panel). For this purpose, metalloporphyrins at 1–20 μM and protein solutions at 10, 5, and 2 μM were examined. All experiments gave comparable values of Kd, suggesting general weak binding of respective metalloporphyrins to HmuY. In addition, to aid clarification of the binding modes of metal ions to HmuY, CD spectra of metalloporphyrins bound to the wild-type HmuY and to its variants (where either His134 and Met136 or His166 were replaced by Ala residues) were analysed (Fig. 2). We also investigated non-iron metalloporphyrin binding to HmuY by haem exchange experiments monitored by 1H NMR spectroscopy (Fig. 3 and data not shown). The potential contribution of HmuY to overall cellular binding of non-iron metalloporphyrins was assessed by analysing metalloporphyrin binding to the wild-type P. gingivalis cells and mutant cells lacking a functional hmuY gene (TO4).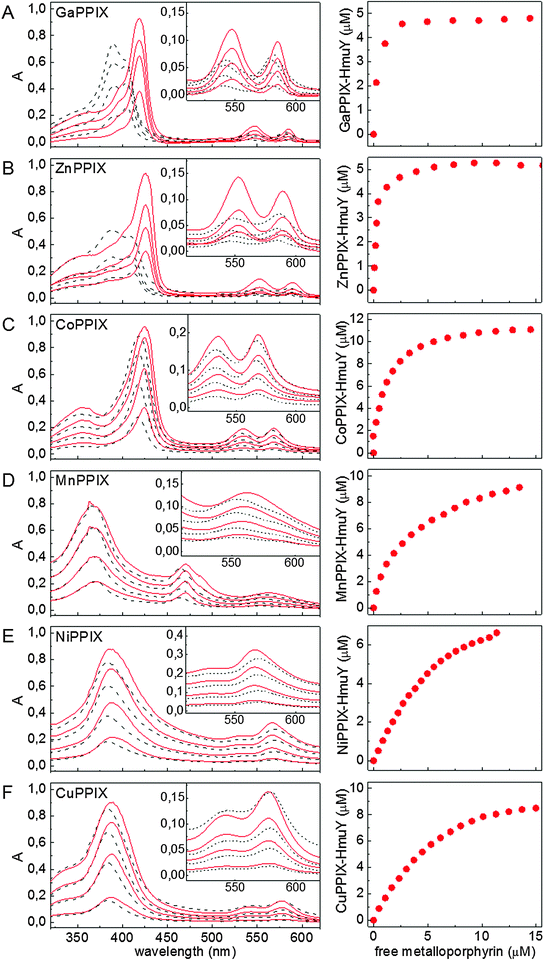 | ||
| Fig. 1 UV-visible absorption spectroscopic analysis of non-iron metalloporphyrin binding to the HmuY haemophore of P. gingivalis. The protein was titrated with increasing amounts of metalloporphyrins (1–20 μM) and the formation of metalloporphyrin–HmuY complexes was observed spectrophotometrically. HmuY was used at a concentration of 5 μM for Ga(III)PPIX and Zn(II)PPIX, and at 10 μM for remaining metalloporphyrins. Selected UV-visible absorbance spectra (left panel) and titration plots for Kd determination (right panel) are shown. Left panel: metalloporphyrins in 20 mM sodium phosphate buffer, pH 7.4, containing 140 mM NaCl (solid red line), HmuY–metalloporphyrin complex (dashed black line). Right panel: equilibrium concentration of protein–metalloporphyrin complexes plotted versus concentration of free metalloporphyrins in solution; presented data points were calculated from absorbance values at wavelengths corresponding to Soret band maxima (λmax) of the respective complexes and values of ελmax estimated using equations for a one-site binding model.19 (A) Ga(III)PPIX, (B) Zn(II)PPIX, (C) Co(III)PPIX, (D) Mn(III)PPIX, (E) Ni(II)PPIX, and (F) Cu(II)PPIX. | ||
| Metalloporphyrin/metalloporphyrin–HmuY complex | Soret region (nm) | β band (nm) | α band (nm) |
|---|---|---|---|
| a HmuY (10 μM) in 20 mM sodium phosphate buffer, pH 7.4, containing 140 mM NaCl or buffer alone was titrated with metalloporphyrins (1–20 μM) and absorbance spectra were recorded immediately after addition of 1 μl aliquots of metalloporphyrins. ND, not detected. | |||
| Fe(III)PPIX/HmuY–Fe(III)PPIX | 384, 363/411 | 487/529 | 611/559 |
| Ga(III)PPIX/HmuY–Ga(III)PPIX | 404, 398/422, 398 | 543/548 | 581/585 |
| Zn(II)PPIX/HmuY–Zn(II)PPIX | 407, 389/430, 402 | 548/553 | 586/590 |
| Co(III)PPIX/HmuY–Co(III)PPIX | 417, 388/427, 402 | 532/535 | 566/569 |
| Mn(III)PPIX/HmuY–Mn(III)PPIX | 465, 370/470, 370 | ND/ND | 553/559 |
| Cu(II)PPIX/HmuY–Cu(II)PPIX | 384/389 | 546/538 | 579/578 |
| Ni(II)PPIX/HmuY–Ni(II)PPIX | 384/389 | 536/527 | 569/566 |
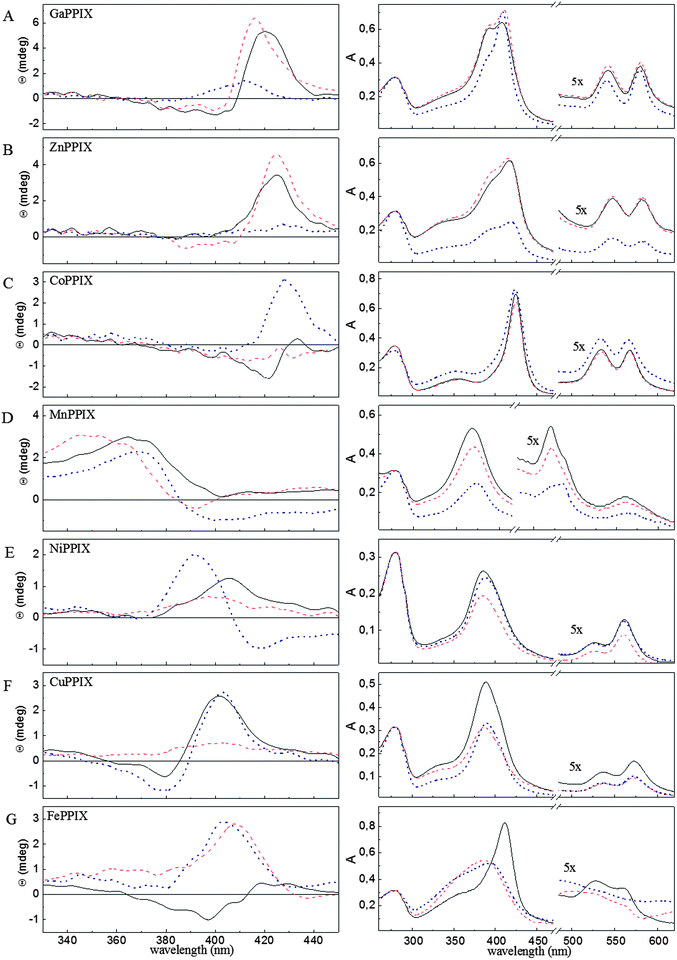 | ||
| Fig. 2 Circular dichroism (CD) analysis of non-iron metalloporphyrin binding to the HmuY haemophore of P. gingivalis. Soret region CD spectra (left panel) and absorbance spectra (right panel) of HmuY complexed with (A) Ga(III)PPIX, (B) Zn(II)PPIX, (C) Co(III)PPIX, (D) Mn(III)PPIX, (E) Ni(II)PPIX, (F) Cu(II)PPIX, and (G) Fe(III)PPIX. Spectra are shown for the wild-type HmuY (solid black line) and its variants: His166Ala (dotted dark blue line), His134Ala/Met136Ala (dashed red line). All samples were incubated in 10 mM phosphate buffer, pH 7.8, for 24 h at 4 °C and excess metalloporphyrin was removed from the solution by gel filtration. Spectra were recorded for 100 μM proteins in 10 mM sodium phosphate buffer, pH 7.8. | ||
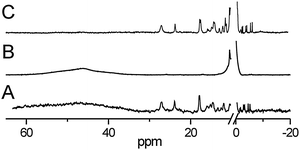 | ||
| Fig. 3 Binding of non-iron metalloporphyrins to the HmuY haemophore of P. gingivalis analysed by 1H NMR spectroscopy. (A) HmuY reconstituted with 5-fold molar excess of Fe(III)PPIX, (B) HmuY reconstituted with Zn(II)PPIX and treated with 1 equivalent of Fe(III)PPIX and (C) HmuY reconstituted with Ni(II)PPIX after addition of 1 equivalent of Fe(III)PPIX. | ||
Gallium and zinc accept His166 but not His134 as an axial ligand in Ga(III)PPIX and Zn(II)PPIX complexed with the HmuY protein
Ga(III)PPIX and Zn(II)PPIX binding to HmuY was confirmed on the basis of their UV-visible absorption spectra (Fig. 1A and B). The Soret red shift of Ga(III)PPIX–HmuY to 422 nm (Fig. 1A and Table 1) suggests attachment of an axial ligand.25–27 The absorption spectrum of the Ga(III)PPIX–HmuY complex (422, 548 and 585 nm for Soret, β and α bands, respectively) resembles pentacoordinate Ga(III)PPIX complexes with either histidine (as in myoglobin; 417, 545 and 584 nm) or tyrosine (as in Enterococcus faecalis catalase, KatA; 422, 548 and 588 nm).28,29 Ga(III)PPIX also binds to the HasA haemophore of Serratia marcescens30 and to the IsdH-NEAT3 haem transporter of S. aureus.31 Those complexes showed pentacoordinate ligation of Ga(III) with a tyrosine residue in IsdH-NEAT3, whilst hexacoordinate ligation via histidine-tyrosine in HasA.The Zn(II)PPIX–HmuY complex (Fig. 1B and Table 1) has distinctive Soret, α and β bands at 430, 553 and 590 nm which are red-shifted compared to other Zn(II)PPIX–protein complexes where the zinc atom is hexacoordinate, e.g., Zn(II)PPIX–myoglobin (416, 544 and 583 nm) or Zn(II)PPIX–cytochrome c (423, 549 and 584 nm).32,33 According to the general rule that the attachment of each axial ligand causes a red-shift of the Soret maximum, the above findings point to Zn(II) being hexacoordinate in the Zn(II)PPIX–HmuY complex.
After prolonged incubation and removal of excess of metalloporphyrins, the Ga(III)PPIX–HmuY and Zn(II)PPIX–HmuY complexes showed blue-shifted absorbance maxima, plus the appearance of additional Soret bands (at 398 and 394 nm, respectively) (Fig. 2A and B, and Table 2). Similar spectral characteristics were observed for Ga(III)PPIX and Zn(II)PPIX complexed with the His134Ala/Met136Ala and the His166Ala HmuY variants (Fig. 2A and B, and Table 2). Similar behaviour was also observed by others who have shown binding of Ga(III)PPIX and Zn(II)PPIX to myoglobin.29,34
| Metalloporphyrin | Metalloporphyrin–HmuY variant complex | Soret region (nm) | β band (nm) | α band (nm) |
|---|---|---|---|---|
| a All samples were incubated in 10 mM phosphate buffer, pH 7.8, for 24 h at 4 °C and excess of metalloporphyrins was removed from the solution by gel filtration. ND, not detected. | ||||
| Ga(III)PPIX | Wild-type | 412, 398 | 543 | 580 |
| His134Ala/M136Ala | 412, 398 | 543 | 580 | |
| His166Ala | 410 | 543 | 580 | |
| Zn(II)PPIX | Wild-type | 421, 394 | 546 | 583 |
| His134Ala/M136Ala | 421, 394 | 546 | 583 | |
| His166Ala | 423, 393 | 546 | 583 | |
| Co(III)PPIX | Wild-type | 425, 355 | 535 | 566 |
| His134Ala/M136Ala | 425, 355 | 535 | 566 | |
| His166Ala | 424, 354 | 533 | 568 | |
| Mn(III)PPIX | Wild-type | 370, 470, 489 | ND | 561 |
| His134Ala/M136Ala | 372, 470 | ND | 557 | |
| His166Ala | 376, 489 | ND | 559 | |
| Ni(II)PPIX | Wild-type | 389 | 527 | 562 |
| His134Ala/M136Ala | 386 | 526 | 560 | |
| His166Ala | 389 | 525 | 561 | |
| Cu(II)PPIX | Wild-type | 389 | 537 | 572 |
| His134Ala/M136Ala | 386 | 537 | 572 | |
| His166Ala | 389 | 537 | 572 | |
CD and absorbance spectra of the Ga(III)PPIX– and Zn(II)PPIX–HmuY complexes point to little or no change in the environment upon removal of His134 (Fig. 2A and B). However, replacement of His166 with an alanine residue strongly influences Ga(III)PPIX– and Zn(II)PPIX–HmuY formation. Changes in the Soret region of CD and absorbance spectra may result from different gallium coordination in Ga(III)PPIX complexed with the His166Ala variant (Fig. 2A), since removal of His166 diminishes the affinity of HmuY for Zn(II)PPIX resulting in a low A418nm/A280nm ratio and a flat CD spectrum in the Soret region (Fig. 2B).
1H NMR haem exchange experiments demonstrated that once bound to HmuY, neither Ga(III)PPIX nor Zn(II)PPIX could be displaced by haem, since no signals characteristic of Fe(III)PPIX coordinated by histidine ligands were found even after prolonged incubation (Fig. 3B and data not shown). Instead, a broad feature in the 40–60 ppm region was observed due to high-spin iron(III) protoporphyrin IX, probably attached outside the haem-binding pocket.
We demonstrated that both gallium(III) and zinc(II) metalloporphyrins are bound to HmuY with similar affinity to haem (Kd(Fe(III)PPIX) = 0.248; Kd(Ga(III)PPIX) = 0.341 μM; Kd(Zn(II)PPIX) = 0.249 μM), which further confirms the formation of stable complexes (Fig. 1A and B). However, it should be noted that the measurement of binding constants for haem in different solutions is complicated by the fact that it may exist as a dimer (μ-oxo dimer or π–π dimer).35,36 Similar effects have been shown for other metalloporphyrins. For example, Ga(III)PPIX forms a propionate-bridged dimer37 and Mn(III)PPIX forms dimers probably due to π–π stacking.38 In this study we have also noticed the presence of two distinct forms of Ga(III)PPIX and Zn(II)PPIX as two Soret maxima were observed for the solutions of each metalloporphyrin (Fig. 1). All these experiments were conducted in buffered aqueous solution, mimicking in vivo conditions, but allowing dimer formation. Therefore, extinction coefficients and Kd values estimated in this study under conditions applied should take into consideration the above-mentioned possibilities. In addition, the affinities of the complexes with Fe, Ga and Zn protoporphyrin IX should be reported as minimal values which exceed the limits of the assay at these concentrations of protein and sensitivities for detection of bound and free cofactors. However, although the binding of metalloporphyrins, described in this study, by HmuY is relatively weak, the haemophore will nevertheless efficiently bind heme from deoxyhaemoglobin, methaemoglobin and serum albumin16 and haemopexin (J. W. Smalley, unpublished findings). This may suggest that, for example, in contrast to the HasA hemophore, HmuY may bind and transport haem to the P. gingivalis HmuR receptor in a different manner, or that this weak binding may be necessary to store haem on the surface of P. gingivalis cells first and then transfer this molecule to other proteins.
Cobalt is hexacoordinate in the Co(III)PPIX–HmuY complex
The UV-visible spectrum of the Co(III)PPIX–HmuY complex (Fig. 1C and Table 1) with absorption maxima at 427, 535 and 569 nm matches that of low-spin hexacoordinate cobalt(III) in Co(III)PPIX–cytochrome c (426, 530 and 567 nm) and in Co(III)PPIX–methaemoglobin (433, 535 and 568 nm).39–42 Absorption spectra of Co(III)PPIX complexed with wild-type HmuY and with the His134Ala/Met136Ala variant are identical and their maxima are only slightly shifted (∼2 nm) with respect to those of the Co(III)PPIX–His166Ala complex (Table 2).The CD spectrum of the wild-type Co(III)PPIX–HmuY complex exhibits a weak negative Cotton effect, similar to that of the haem–HmuY complex, the latter being blue-shifted (Fig. 2C and G). Also, the intense positive band makes the CD spectrum of the Co(III)PPIX–His166Ala complex similar to that of the haem-His166Ala complex. In contrast, the CD spectrum of the Co(III)PPIX–His134Ala/Met136Ala complex is almost flat in the entire Soret region (Fig. 2C). These spectroscopic characteristics point to the Co(III)PPIX–HmuY complex being hexacoordinate, analogously to that of iron in haem–HmuY. However, based on the CD spectra, some differences in the environments of Co(III) and Fe(III) can be expected. This may be attributed to the fact that cobalt(III) binding to the sixth ligand may be rather weak due to anti-bonding electrons. Weak interaction with the sixth axial ligand might also explain the lower binding affinity of Co(III)PPIX to HmuY (Kd(Co(III)PPIX) = 1.05 μM) compared to that of haem. Even though unsymmetrical, interaction of Co(III)PPIX with the axial imidazole ligands is strong enough to prevent its displacement by haem in the haem-binding pocket of HmuY (data not shown).
Manganese is pentacoordinate in the Mn(III)PPIX–HmuY complex
The absorption spectral features of the Mn(III)PPIX–HmuY complex (Fig. 1D and Table 1) are similar to Mn(III)PPIX–substituted haemoglobin, myoglobin and cytochrome c, in having a 370 nm Soret band and an additional π–d transition band around 470 nm.43–45 As in all the above modified haemoproteins, the manganese ion in the Mn(III)PPIX-HmuY complex would be expected to be coordinated by a single histidine residue. Interestingly, the Mn(III)PPIX–HmuY absorbance spectrum shows a weak π–d transition band shoulder at 489 nm besides a distinctive maximum at 470 nm, whilst the spectra of Mn(III)PPIX–His134Ala/Met136Ala and Mn(III)PPIX–His166Ala have single π–d transition bands at 470 and 489 nm, respectively (Fig. 2D and Table 2). As a consequence, the absorbance spectra of the Mn(III)PPIX complexes with the HmuY mutants sum to give a spectrum similar to that of Mn(III)PPIX complexed with the wild-type protein. Also, the positive Cotton effect observed in the CD spectrum of the Mn(III)PPIX–HmuY seems to be a sum of spectra of the Mn(III)PPIX–His134Ala/Met136Ala and Mn(III)PPIX–His166Ala complexes (Fig. 2D). We conclude that Mn(III)PPIX–HmuY in solution is a mixture of two types of the complex where either His134 or His166 is accepted as a single axial ligand. Due to its relatively large ionic radius (0.82 Å), manganese(III) would be expected to protrude out of the porphyrin ring,40 replacing the axial symmetry (typical of hexacoordinate Fe(III)PPIX complexes) with a rhombic symmetry where only one imidazole ligand is accepted by the manganese ion, and which may offer a reason for the lower affinity of HmuY for Mn(III)PPIX (Kd = 3.90 μM) (Fig. 1D and 2D).In the 1H NMR spectrum obtained for the HmuY–Mn(III)PPIX system (data not shown), the positions of signals are typical for a high-spin (S = 2) manganese(III) complex and the general appearance of the spectrum is very similar to that observed for Mn(III)PPIX chloride itself.45,46 As expected, the possible coordination of histidine ligands does not change the spin state of manganese(III).47 Importantly, from a functional perspective, haem exchange experiments showed that formation of the pentacoordinate Mn(III)PPIX–HmuY complex with an axial imidazole ligand prevents its displacement by haem in the haem-binding pocket and may (in part) explain its antibacterial effects.
Weak binding allows Ni(II)PPIX and Cu(II)PPIX to be displaced by haem in the binding cavity of HmuY
UV-visible absorption spectra point towards similar modes of binding of Ni(II)PPIX and Cu(II)PPIX to HmuY (Fig. 1E and F, and Table 1). In both cases, complex formation causes only slight Soret band red shifts (5 nm) compared to free metalloporphyrins. The 389 nm Soret band for the Ni(II)PPIX–HmuY complex corresponds well with the fraction of the tetracoordinate nickel in Ni(II)PPIX-substituted haemoglobin (λmax 397 nm).26 The 389 nm Soret band for the Cu(II)PPIX–HmuY complex, which is close to that for Cu(II)PPIX in the vapour phase (387.5 nm), suggests that Cu(II) in this complex is tetracoordinate (Fig. 1F and Table 1).25The CD spectrum of Ni(II)PPIX–HmuY has a weak positive Cotton effect within the Soret region, and the Ni(II)PPIX–His166Ala complex shows a bisignate CD spectrum (Fig. 2E), whilst both Cu(II)PPIX–HmuY and Cu(II)PPIX–His166Ala show bisignate CD spectra (Fig. 2F). Nevertheless, both Ni(II)PPIX and Cu(II)PPIX complexed with His134Ala/Met136Ala show an almost completely flat Soret region CD spectrum and a blue-shifted Soret band (to 386 nm). We conclude that both Ni(II)PPIX and Cu(II)PPIX are bound specifically in the HmuY haem-binding cavity but each central metal ion interacts only weakly with His134. This is not an unexpected result since although both Ni(II)PPIX and Cu(II)PPIX share the peripheral substitution pattern of the native haem b, they exhibit a low affinity for axial ligands.26,27,48,49
In the case of Cu(II)PPIX binding, no metalloporphyrin-derived signals could be observed using 1H NMR, as they are broadened beyond detection due to the long electron relaxation times, which is typical for Cu(II)PPIX complexes.50 Addition of one molar equivalent of Fe(III)PPIX to a solution of either Ni(II)PPIX–HmuY (Fig. 3C) or Cu(II)PPIX–HmuY (data not shown) yields the 1H NMR pattern of paramagnetically shifted resonances characteristic of HmuY reconstituted with low-spin Fe(III)PPIX. This corresponds well with the fact that both Ni(II)PPIX and Cu(II)PPIX show low binding affinity to wild-type HmuY (Kd = 6.59 μM and Kd = 5.05 μM, respectively) (Fig. 1E and F). As a consequence, the majority of those metalloporphyrins are lost from the complex during gel filtration or dialysis against buffer (as indicated by the low A389nm–A280nm ratio) leaving some unoccupied haem binding sites that can be then filled with exogenous haem.
Biological implications
Recently we have shown that iron(III) mesoporphyrin IX and iron(III) deuteroporphyrin IX bind to HmuY and support planktonic and biofilm growth of P. gingivalis cells and infection of epithelial cells in a way analogous to iron(III) protoporphyrin IX.19,20 In contrast, however, Ga(III)PPIX and Co(III)PPIX exhibit antimicrobial activity against P. gingivalis.20 This effect has also been observed for the hmuY (TO4) and hmuY-hmuR (TO2) mutant strains,20 suggesting that the proteins encoded within the hmu operon may not constitute the only system used by P. gingivalis for uptake of metalloporphyrins. Here we demonstrated that all of the metalloporphyrins examined bind to the P. gingivalis wild-type strain with a greater ability compared to the TO4 mutant strain lacking a functional hmuY gene (Fig. 4). This suggests that HmuY may be used by P. gingivalis cells to bind free metalloporphyrins on the cell surface. The results herein further aid our understanding of the mechanisms of metalloporphyrin binding and utilization which might be exploited to reduce the virulence of P. gingivalis, e.g. photodynamic therapy.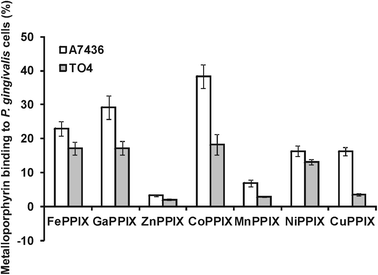 | ||
| Fig. 4 Binding of metalloporphyrins to P. gingivalis cells. P. gingivalis wild-type (A7436) and hmuY mutant (TO4) strains depleted of iron and haem were incubated in basal medium anaerobically at 37 °C for 1 h with metalloporphyrins (final concentration 10 μM). After centrifugation, the amount of metalloporphyrins remaining in the culture media was determined by absorbance measurements at wavelengths corresponding to their Soret λmax values. Results are shown as mean ± SD from two independent experiments performed in duplicate. | ||
Conclusions
This study showed that HmuY forms complexes with selected non-iron metalloporphyrins, though recognition of the ligand by HmuY depends on the central metal ion. Comparison of Ga(III)PPIX and Mn(III)PPIX binding to HmuY can serve here as a good example. The gallium ion appears to accept His166 but not His134 as an axial ligand, whilst the manganese ion accepts either of these histidines as a single axial ligand, resulting in two Mn(III)PPIX–HmuY complexes in solution, where either His134 or His166 serves as a manganese(III) axial ligand. Cu(II)PPIX and Ni(II)PPIX seem to interact only weakly with His134. Co(III)PPIX is the only one of the non-iron metalloporphyrins examined for which the central metal ion forms hexacoordinate complexes with both axial imidazole ligands of HmuY. Importantly, once bound to HmuY, neither Ga(III)PPIX, Zn(II)PPIX, Co(III)PPIX nor Mn(III)PPIX can be displaced by haem, pointing to their antimicrobial potential for subverting the normal haem acquisition process. In contrast, the low affinity displayed for Cu(II)PPIX and Ni(II)PPIX enables their easy displacement by haem. The observation that His134 and His166 are not equivalent in binding behaviour may indicate conformational changes and a complex mechanism of metalloporphyrin binding to HmuY. We hypothesize that once bound the porphyrins in which central metal ions are coordinated by His166 cannot be displaced by the haem. This may be an effect of a conformational change of the protein induced by protein–ligand interaction with His166 which can be simply a stronger ligand. The conformational change due to ligand binding was already suggested by different unfolding pathways of apo- and holo-HmuY17 and SPR experiments (our unpublished data). In the case of the porphyrins in which central metal ions are coordinated by His134 they can be displaced by haem in the haem-binding pocket. This picture may be complicated by the fact that HmuY may form dimers and tetramers. In monomeric proteins the type of coordination is determined by the protein conformation in the haem-binding pocket, whereas in oligomeric proteins subunit interactions can produce a mixture of different types of coordination.26,51,52 Therefore further studies are required to shed more light on detailed properties of metalloporphyrin binding to the HmuY hemophore, as well as to P. gingivalis cells.Acknowledgements
This work was supported in part by grant no. N401 029 32/0742 from the Department of Scientific Research, Ministry of Science and Higher Education (to TO), by Wroclaw Research Center EIT + under the project “Biotechnologies and advanced medical technologies – BioMed” (POIG 01.01.02-02-003/08/00) financed by the European Regional Development Fund (Operational Program Innovative Economy, 1.1.2) (to TO), and by the Department of Chemistry research project 1012/S/WCH/10/6 (to JW).References
- D. T. Graves, Y. Jiang and C. A. Genco, Curr. Opin. Infect. Dis., 2000, 13, 227 CrossRef CAS.
- R. G. Craig, Y. K. Yip, M. K. So, R. J. Boylan, S. S. Socransky and A. D. Haffajee, J. Periodontol., 2003, 74, 1007 CrossRef.
- R. J. Lamont, A. Chan, C. M. Belton, K. T. Izutsu, D. Vasel and A. Weinberg, Infect. Immun., 1995, 63, 3878 CAS.
- L. Vitkov, W. D. Krautgartner and M. Hannig, Oral Microbiol. Immunol., 2005, 20, 317 CrossRef CAS.
- O. Yilmaz, P. Verbeke, R. J. Lamont and D. M. Ojcius, Infect. Immun., 2006, 74, 703 CrossRef CAS.
- P. D. Marsh, Caries Res., 2004, 38, 204 CrossRef CAS.
- R. Leon, N. Silva, A. Ovalle, A. Chaparro, A. Ahumada, M. Gajardo, M. Martinez and J. Gamonal, J. Periodontol., 2007, 78, 1249 CrossRef.
- J. H. Meurman, M. Sanz and S. J. Janket, Crit. Rev. Oral Biol. Med., 2004, 15, 403 Search PubMed.
- E. D. Rosenstein, R. A. Greenwald, L. J. Kushner and G. Weissmann, Inflammation, 2004, 28, 311 CrossRef.
- T. Olczak, W. Simpson, X. Liu and C. A. Genco, FEMS Microbiol. Rev., 2005, 29, 119 CrossRef CAS.
- J. L. Gao, K. A. Nguyen and N. Hunter, J. Biol. Chem., 2010, 285, 40028 CrossRef CAS.
- T. Olczak, D. W. Dixon and C. A. Genco, J. Bacteriol., 2001, 183, 5599 CrossRef CAS.
- X. Liu, T. Olczak, H. C. Guo, D. W. Dixon and C. A. Genco, Infect. Immun., 2006, 74, 1222 CrossRef CAS.
- T. Olczak, A. Sroka, J. Potempa and M. Olczak, Arch. Microbiol., 2008, 183, 197 CrossRef.
- T. Olczak, H. Wojtowicz, J. Ciuraszkiewicz and M. Olczak, BMC Microbiol., 2010, 10, 134 CrossRef.
- J. W. Smalley, D. P. Byrne, A. J. Birss, H. Wojtowicz, A. Sroka, J. Potempa and T. Olczak, PLoS One, 2011, 6, e17182 CAS.
- H. Wojtowicz, T. Guevara, C. Tallant, M. Olczak, A. Sroka, J. Potempa, M. Sola, T. Olczak and F. X. Gomis-Ruth, PLoS Pathog., 2009, 5, e100041 Search PubMed.
- H. Wojtowicz, J. Wojaczynski, M. Olczak, J. Kroliczewski, L. Latos-Grazynski and T. Olczak, Biochem. Biophys. Res. Commun., 2009, 383, 178 CrossRef CAS.
- J. Wojaczynski, H. Wojtowicz, M. Bielecki, M. Olczak, J. W. Smalley, L. Latos-Grazynski and T. Olczak, Biochem. Biophys. Res. Commun., 2011, 411, 299 CrossRef CAS.
- T. Olczak, D. Maszczak-Seneczko, J. W. Smalley and M. Olczak, Arch. Microbiol., 2012, 57, 314 Search PubMed.
- J. W. Smalley, J. Silver, A. J. Birss, R. Withnall and P. J. Titler, Microbiology, 2003, 149, 1711 CrossRef CAS.
- J. Bozja, K. Yi, W. M. Shafer and I. Stojiljkovic, Int. J. Antimicrob. Agents, 2004, 24, 578 CrossRef CAS.
- I. Stojiljkovic, V. Kumar and N. Srinivasan, Mol. Microbiol., 1999, 31, 429 CrossRef CAS.
- Y. Kaneko, M. Thoendel, O. Olakanmi, B. E. Britigan and P. K. Singh, J. Invest. Med., 2007, 117, 877 CrossRef CAS.
- L. Edwards and D. H. Dolphin, J. Mol. Spectrosc., 1970, 35, 90 CrossRef CAS.
- J. A. Shelnutt, K. Alston, J. Y. Ho, N. T. Yu, T. Yamamoto and J. M. Rifkind, Biochemistry, 1986, 25, 620 CrossRef CAS.
- C. Findlay, L. C. Dickinson and J. C. Chien, J. Am. Chem. Soc., 1977, 99, 5168 CrossRef.
- M. Brugna, L. Tasse and L. Hederstedt, FEBS J., 2010, 277, 2663 CrossRef CAS.
- T. B. Pinter, E. L. Dodd, D. S. Bohle and M. J. Stillman, Inorg. Chem., 2012, 51, 3743 CrossRef CAS.
- N. Wolff, C. Deniau, S. Letoffe, C. Simenel, V. Kumar, I. Stojiljkovic, C. Wandersman, M. Delepierre and A. Lecroisey, Protein Sci., 2002, 11, 757 CrossRef CAS.
- Y. Moriwaki, J. M. M. Caaveiro, Y. Tanaka, H. Tsutsumi, I. Hamachi and K. Tsumoto, Biochemistry, 2011, 50, 7311 CrossRef CAS.
- S. Papp, J. M. Vanderkooi, C. S. Owen, G. R. Holtom and C. M. Phillips, Biophys. J., 1990, 58, 177 CrossRef CAS.
- H. Anni, J. M. Vanderkooi and L. Mayne, Biochemistry, 1995, 34, 5744 CrossRef CAS.
- H. Takashima, E. Fujimoto, C. Hirai and K. Tsukahara, Chem. Biodiversity, 2008, 5, 2101 CAS.
- K. A. de Villiers, C. H. Kaschula, T. J. Egan and H. M. Marques, JBIC, J. Biol. Inorg. Chem., 2007, 12, 101 CrossRef.
- C. Asher, K. A. de Villiers and T. J. Egan, Inorg. Chem., 2009, 48, 7994 CrossRef CAS.
- D. S. Bohle and E. L. Dodd, Inorg. Chem., 2012, 51, 4411 CrossRef CAS.
- L. D. Gruenke, J. Sun, T. M. Loehr and L. Waskell, Biochemistry, 1997, 35, 7114 CrossRef.
- X. Guo, C. Jiang and T. S. Shi, Inorg. Chem., 2007, 46, 4766 CrossRef CAS.
- V. Campo Dall' Orto, R. Carballo, J. A. Hurst and I. Rezzano, Spectrochim. Acta, Part A, 2005, 61, 2089 CrossRef CAS.
- L. C. Dickinson and C. W. Chien, J. Biol. Chem., 1973, 248, 5005 CAS.
- L. C. Dickinson and J. C. Chien, Biochemistry, 1975, 14, 3526 CrossRef CAS.
- L. C. Dickinson and J. C. Chien, J. Biol. Chem., 1977, 252, 6156 CAS.
- J. L. Heinecke, J. Yi, J. C. Pereira, G. B. Richter-Addo and P. C. Ford, J. Inorg. Biochem., 2012, 107, 47 CrossRef CAS.
- T. R. Janson, L. J. Boucher and J. J. Katz, Inorg. Chem., 1973, 12, 940 CrossRef CAS.
- G. N. La Mar and F. A. Walker, J. Am. Chem. Soc., 1975, 97, 5103 CrossRef CAS.
- J. K. M. Sanders, N. Bampos, Z. Clyde-Watson, S. L. Darling, J. C. Hawley, H. J. Kim, C. C. Mak and S. J. Webb, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, NY, 2000, vol. 3, pp. 1–112 Search PubMed.
- R. G. Alden, M. R. Ondrias and J. A. Shelnutt, J. Am. Chem. Soc., 1990, 112, 691 CrossRef CAS.
- J. G. Ma, M. Laberge, X. Z. Song, W. Jentzen, S. L. Jia, J. Zhang, J. M. Vanderkooi and J. A. Shelnutt, Biochemistry, 1998, 37, 5118 CrossRef CAS.
- I. Bertini, C. Luchinat and P. Parigi, Solution NMR of paramagnetic molecules, Elsevier, Amsterdam, 2001, pp. 174–181 Search PubMed.
- P. T. Manoharan, K. Alston and J. M. Rifkind, J. Am. Chem. Soc., 1986, 108, 7095 CrossRef CAS.
- E. W. Findsen, K. Alston, J. A. Shelnutt and M. R. Ondrias, J. Am. Chem. Soc., 1986, 108, 4009 CrossRef CAS.
Footnote |
| † Haem (iron protoporphyrin IX) as a common convention, regardless of the iron oxidation state, will be applied throughout this paper. |
| This journal is © The Royal Society of Chemistry 2013 |