Exploring the structural determinants of selective phosphopeptide recognition using bivalent metal-coordination complexes†
Dziyana
Kraskouskaya‡
a,
Joel A.
Drewry‡
a,
Eugenia
Duodu‡
a,
Steven
Burger
b,
James
Eaton
a,
G. Andrés
Cisneros
b and
Patrick T.
Gunning
*a
aDepartment of Chemical and Physical Sciences, University of Toronto, 3359 Mississauga Road North, Mississauga, Ontario L5L 1C6, Canada. E-mail: patrick.gunning@utoronto.ca; Fax: +1-905-569-5425; Tel: +1-905-828-5354
bDepartment of Chemistry, Wayne State University, 5101 Cass Ave., Detroit, MI 48202, USA
First published on 12th December 2012
Abstract
We herein explore the structural determinants of selective phosphopeptide recognition by employing novel bivalent, bis-(Zn2+-dipicolylamine)-based coordination complexes against a select library of model phosphoserine-containing peptides of varying amino acid composition. The results demonstrated that Lewis acidic coordination complexes equipped with cationic binding groups might be best utilized as selective receptors for binding phosphopeptides with anionic side chain residues proximal to the phosphoserine residue.
Protein phosphorylation is one of the key post-translational mechanisms for mediating inter- and intra-cellular communication. Protein phosphorylation commonly occurs on serine (S),1 threonine (T)2 and tyrosine (Y)3 residues and can serve as both an on or off “switch” to a particular cellular signaling cascade. Access to synthetic agents capable of selective and potent binding to these phosphorylated sites might provide a means for the detection and inhibition of specific molecular pathways.4 Di-nuclear (Zn2+)2-bis-dipicolylamine ((Zn2+)2–BDPA) compounds have demonstrated significant potential as high affinity binders of phosphorylated peptide sequences.5–9 These receptors were shown to preferentially bind phosphate mono-esters within peptide sequences over other phosphate species, including inorganic phosphate.7 Much effort has been directed at fine-tuning these receptors' selectivity towards di-phosphorylated peptides by designing inhibitors that effectively bind two phosphorylated Y residues. Notably, it was demonstrated that such systems could effectively sense hyper-phosphorylated Tau peptides present in Alzheimer's samples10 and disrupt the association between a CTD and WW domain.11 However, only few studies12,13,14 have tried to address whether (Zn2+)2–BDPA receptors can selectively bind to clinically relevant mono-phosphorylated peptide sequences. We herein describe the development of novel bivalent receptors for delineating the structural facets required for selective recognition of mono-phosphoserine containing peptides and for more generally assessing the likelihood of identifying therapeutically viable phosphopeptide-selective binders.
In considering the design of a bivalent scaffold for binding pS and making additional complementary contacts with adjacent residues for purposes of deriving target peptide selectivity, we employed quantum mechanical calculations (Gaussian09) and molecular mechanical (MM) calculations (AMBER software) to aid our scaffold selection.15,16 First, the phosphate ester of pS is significantly closer to the peptide backbone than is found with pY, and by analogy, closer in proximity to the adjacent side chain residues. Thus, employing compact bivalent receptors, reflecting the relative distances between the two binding sites, may more effectively target pS-peptides. For this work, we sought to design and synthesize a novel coordination complex ligand which would serve as a stable, structurally rigid linker to append two Zn2+–DPA units, and to which a secondary binder could be readily coupled for recognizing adjacent residues of the peptide primary sequence (pS+X) for the purpose of deriving peptide specificity.
Briefly, we selected a rigid, planar and metabolically stable, 2′-substituted 1,4-dimethylbenzothiazole core from which two DPA metal-chelating groups could be readily appended from the 1,4-xylyl exogenous methyl groups of the heterocycle (Fig. 1A). The 1,4-xylyl BDPA configuration of DPA groups was selected due to the predicted positioning of the metal ions located below the plane of the benzothiazole. From the 2′-benzothiazole position, secondary binding groups (R) were appended to interact with proximal amino acid residues of the pS-peptide. Molecular Dynamic (MD) analysis of a benzothiazole modeled with a target peptide, Ac-pS-AAAA, showed that the core unit preferentially adopted a flat geometry atop the bound peptide (Fig. 1B) and the appended 2′ R group efficiently projected into space occupied by residues adjacent to pS, specifically, pS+2 and to a lesser extent, pS+3 (Fig. 1C). Thus, a family of (Zn2+)2–BDPA benzothiazole receptors were prepared possessing a variety of R groups at the 2′ position including hydrophobic, anionic and cationic species (Fig. 1D). Synthetic and computational protocols as well as compound characterization are provided in ESI.†
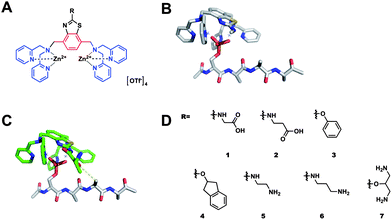 | ||
| Fig. 1 (A) Chemical structure of benzothiazole-based (Zn2+)2–BDPA ligands; (B) and (C) energy minimized structures (HF/6-31G*) of a 2′ substituted benzothiazole (Zn2+)2–BDPA complex bound to AcpSAAA; (D) library of benzothiazole based receptors. | ||
Since the 2′ pendant R groups were predicted to interact with residues proximal to pS we assessed receptor binding potency against model pS-containing glycine (G) hexa-peptide sequences. The G residues at positions pS+2 and pS+3 were iteratively substituted for an arginine (R), isoleucine (I), or glutamic acid (E), to represent a wide-scope of side chain functionality in the key interacting regions of the peptide. Modelling demonstrated that the size and rigidity of the benzothiazole core precluded R group interaction with the pS+1 residue. We reasoned that the variability of the model peptides would serve to determine the effect of the complementary interactions between the R group functionality and the chemical composition of the pS+2 and pS+3 peptide residue on the receptors' binding affinity and specificity. To assess the receptor affinity towards peptide sequences, a 5-carboxyfluorescein (FAM) fluorescent label was appended through a glycine linker at the N-terminus of the target peptide sequences and binding constants (Ka, Table S1, ESI†) were determined by a high throughput fluorescence intensity (FI) assay previously described.12 The binding affinities determined by FI assay are in close agreement with the values obtained by analogous isothermal titration calorimetry (ITC) binding experiments (Fig. 3, Table 1).
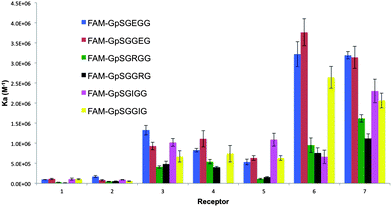 | ||
| Fig. 2 A column graph representing the binding affinities of the 7 bis-(Zn2+–DPA)-benzothiazole based receptors towards the range of pS-containing FAM-conjugated peptides. | ||
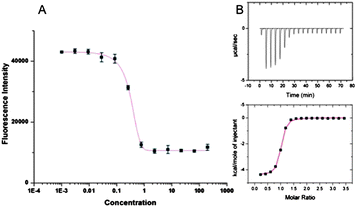 | ||
| Fig. 3 (A) Sample fluorescence intensity; (B) corresponding ITC trace obtained for binding of 7 to FAM-GpSGEGG. | ||
| Peptide | K a/M−1, (ITC) | K a/M−1, (FI) | ΔH/cal mol−1 | ΔS/cal mol−1 K−1 |
|---|---|---|---|---|
| FAM-GpSGEGG | 8.36 × 105 | 5.31 × 105 | −2973 | 17.1 |
| FAM-GpSGRGG | 1.59 × 104 | 1.09 × 105 | −1241 | 15.1 |
| FAM-GpSDLDL | 3.90 × 106 | 1.01 × 107 | −2193 | 22.8 |
| FAM-GpSDDDD | 1.25 × 107 | 1.18 × 107 | −2865 | 22.9 |
The results of the screen against the 6 labeled hexa-peptides are presented in Fig. 2. Several interesting trends were observed. First, regardless of the amino acid composition or position of the target amino acid (pS+2/pS+3) within the peptide, receptors containing cationic (5–7) and hydrophobic (3 and 4) pendant R groups displayed a greater affinity to all peptides as compared to receptors with anionic groups (1–2). This may be attributed to electronic repulsions between the anionic pS on the target peptide and the anionic carboxylate R group present on receptors, 1 and 2. Additionally, we reasoned that the carboxylate R group could form an intra-molecular bond with one of the Zn2+ metals, thus requiring an unfavourable ΔH component for dissociation to permit pS binding. Conversely, cationic receptors, 5–7, can experience intra-molecular electrostatic repulsion between the R group and the di-nuclear (Zn2+)2 core of the receptor, thereby constraining the receptor and reducing its entropic cost to binding.
Presuming the 2′ binding group of the receptor interacts with the pS+X residue(s), we anticipated preferential binding of anionic (1 and 2), hydrophobic (3 and 4) and cationic (5–7) receptors towards R, I and E containing peptides, respectively. Consistent with this prediction, the most potent cationic ligands, 6 and 7, preferentially bound the negatively charged peptides over both cationic and hydrophobic ones. The weakest affinity was observed for cationic sequences (∼5-fold weaker binding for pSGGRG as compared to pSGGEG). Surprisingly, the hydrophobic and anionic ligands (1–4) displayed the same trend and preferentially bound the I and E containing sequences. We suggest that the disfavored binding to cationic peptides may result from electrostatic repulsions between the receptor's intrinsic cationic core (Zn2+)2, and the positively charged R residue. Additionally, within the peptide itself, the guanidinium group of R may form an intra-molecular salt bridge interaction with the phosphate group. Although such a rigidified ionic complex may favor receptor binding entropically, there might be an enthalpic penalty associated with guanidinium–pS complex disruption to permit receptor–pS association. In contrast, we postulate that glutamate-containing peptides were more potently bound across the receptor family as no ionic intra-molecular interaction could be formed. Moreover, phosphate–glutamate repulsions may result in a relatively pre-organized extended peptide suitable for binding by this class of receptors. To test this hypothesis we conducted comparative ITC experiments. Encouragingly, ITC titrations showed that the binding of 5 (Table 1) and 7 (Table S3, ESI†) to the R-containing sequence was accompanied by an enthalpic penalty as compared to binding the anionic E-containing sequence. The observed difference in enthalpy (∼2 kcal mol−1), may be attributed to the unfavourable energy associated with disrupting the salt bridge. The preference of this library of compounds for binding negatively charged, and to a lesser extent, hydrophobic sequences, suggested that phosphopeptide binding selectivity of coordination complexes is primarily dictated by the Lewis acidic metal core of the receptor and by the characteristics of the primary peptide sequence. The secondary R group can subsequently confer moderate increases or decreases in the overall general affinity to phosphopeptides.
To gain further insight into the determinants of peptide binding selectivity for this receptor library and to test our general hypothesis, we screened against pS-containing peptides with greater anionic (pSDDDD, pSDLDL) and cationic character (pSRLRL). Consistent with the previous screen, cationic receptors 5, 6 and 7 displayed the highest affinity towards all peptides (Fig. 4). Among these complexes, only 7 displayed significant selectivity for the pSDDDD over the pSDLDL peptide. The di-cationic R group of 7 facilitates a larger number of hydrogen bonding and electrostatic interactions as compared to ligands 5 and 6. This experimental observation supported the in silico prediction that 2′ R groups can indeed reach and interact with the pS+2/3 residues proximal to pS within a peptide. The affinity of 5 and 6 towards di- and tetra-D sequences was enhanced by more than an order of magnitude as compared to the mono-glutamate sequence from the first screen (5:pSDLDL, Ka = 1.01 × 107 M−1cf.5:pSGEGG, Ka = 5.31 × 105 M−1). Since we presume the cationic R-group of 5 binds the anionic glutamate residue in pSGEGG, the enhanced affinity of 5 towards the pSDDDD should not arise from a significantly more favorable enthalpic component. Instead, we postulated that differences in Ka could be attributed to the differences in target peptide's structural and thermodynamic properties. As evidenced by ITC binding experiments, the enhanced affinity of 5 for pSDDDD over pSGEGG was entropically driven (5:pSGEGG, ΔS = 17 cal mol−1 K−1cf.5:pSDDDD, ΔS = 22.9 cal mol−1 K−1). This can be attributed to the increased rigidity of the pSDDDD secondary structure, which is constrained by electrostatic repulsions between both the pS and D residues. This phenomenon has been previously described and is observed in SH2 domain binding of more extended phosphorylated peptides.17
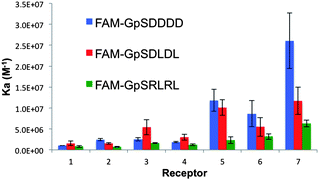 | ||
| Fig. 4 A column graph representing the binding affinities of the 7 bis-(Zn2+–DPA)-benzothiazole based receptors towards the three pS-containing FAM-conjugated peptides of the second screen. | ||
The pSRLRL sequence was the weakest bound peptide by all receptors. This is in agreement with the hypothesis that disrupting the intra-molecular pS–guanidinium electrostatic interaction was disfavored. However, the pSRLRL sequence was still relatively well accommodated by cationic receptors (Ka = 6.33 × 106 M−1 for 7). Analogous to the enhanced affinity of poly-anionic peptides, we suggest that the driving force for binding leucine–arginine peptides was the favourable preorganization of the peptide via hydrogen bonding (pS–guanidinium) as well as electrostatic repulsion between the two R side chain residues.
An encouraging result from the study was that overall, receptors 1–4 preferentially bound the pSDLDL sequence, while 5–7 preferentially associated with the pSDDDD sequence. Since the preorganization of both peptides is similar as approximated by the equal entropy terms (Table 1), the results indicate that receptors containing hydrogen-bond-donor R groups interact preferably with sequences containing more anionic pS+X residues and can derive selectively for such peptides. This work shows for the first time that coordination complexes might be effectively tuned to derive phosphopeptide selectivity by recognizing anionic character in the residues surrounding pS.
Conclusions
In conclusion, guided by computational modeling we have designed and synthesized a novel library of 2′ functionalized bis-(Zn2+–DPA)-benzothiazole compounds to explore the field of selective targeting of pS-containing peptides varying in proximal amino acid composition. The results of this study strongly suggest that efficient application of these Zn2+ complexes as high-affinity (Ka > 107 M−1) probes for intracellular phosphorylated peptides is limited to targeting the sequences rich in anionic residues in direct proximity to the pS. Moreover, the data suggests that coordination complexes equipped with a positively charged R group can derive phosphopeptide selectivity for poly-anionic peptide sequences. We are currently investigating whether the results can be applied to receptor design for recognition of both phosphotyrosine- and phosphothreonine-containing sequences.Notes and references
- A. M. Weaver and C. M. Silva, Biochem. Biophys. Res. Commun., 2007, 362, 1026–1030 CrossRef CAS.
- M. Frodin, EMBO J., 2002, 21, 5396–5407 CrossRef.
- Q. Su, S. Wang, D. Baltzis, L.-K. Qu, A. H.-T. Wong and A. E. Koromilas, Proc. Natl. Acad. Sci. U.S.A., 2006, 103, 63–68 CrossRef CAS.
- H. T. Ngo, X. Liu and K. A. Jolliffe, Chem. Soc. Rev., 2012, 41, 4928–4965 RSC.
- A. Ojida, Y. Mito-Oka, M.-A. Inoue and I. Hamachi, J. Am. Chem. Soc., 2002, 124, 6256–6258 CrossRef CAS.
- A. Ojida, Y. Mito-Oka, K. Sada and I. Hamachi, J. Am. Chem. Soc., 2004, 126, 2454–2463 CrossRef CAS.
- J. A. Drewry, S. Fletcher, H. Hassan and P. T. Gunning, Org. Biomol. Chem., 2009, 7, 5074 CAS.
- A. Ojida, M.-A. Inoue, Y. Mito-Oka and I. Hamachi, J. Am. Chem. Soc., 2003, 125, 10184–10185 CrossRef CAS.
- A. Ojida, M.-A. Inoue, Y. Mito-Oka, H. Tsutsumi, K. Sada and I. Hamachi, J. Am. Chem. Soc., 2006, 128, 2052–2058 CrossRef CAS.
- Y. Ishida, M.-A. Inoue, T. Inoue, A. Ojida and I. Hamachi, Chem. Commun., 2009, 2848–2850 RSC.
- A. Ojida, T. Sakamoto, M.-A. Inoue, S.-H. Fujishima, G. Lippens and I. Hamachi, J. Am. Chem. Soc., 2009, 131, 6543–6548 CrossRef CAS.
- J. A. Drewry, E. Duodu, A. Mazouchi, P. Spagnuolo, S. Burger, C. C. Gradinaru, P. Ayers, A. D. Schimmer and P. T. Gunning, Inorg. Chem., 2012, 51, 8284–8291 CrossRef CAS.
- J. A. Drewry, S. Fletcher, P. Yue, D. Marushchak, W. Zhao, S. Sharmeen, X. Zhang, A. D. Schimmer, C. Gradinaru, J. Turkson and P. T. Gunning, Chem. Commun., 2010, 46, 892 RSC.
- A. Ojida, Y. Mito-Oka, K. Sada and I. Hamachi, J. Am. Chem. Soc., 2004, 126, 2454–2463 CrossRef CAS.
- D. A. Case, et al., Amber 11, University of California, San Francisco Search PubMed.
- S. K. Burger, M. Lacasse, T. Verstraelen, J. Drewry, P. Gunning and P. W. Ayers, J. Chem. Theory Comput., 2012, 8, 554–562 CrossRef CAS.
- F. J. Dekker, N. J. de Mol, P. Bultinck, J. Kemmink, H. W. Hilbers and R. M. J. Liskamp, Bioorg. Med. Chem., 2003, 11, 941–949 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2md20342a |
| ‡ These authors equally contributed to the study. |
| This journal is © The Royal Society of Chemistry 2013 |