Structure–activity relationship studies using peptide arrays: the example of HIV-1 Rev–integrase interaction†
Ronen
Gabizon
a,
Ofrah
Faust
a,
Hadar
Benyamini
a,
Sivan
Nir
a,
Abraham
Loyter
b and
Assaf
Friedler
*a
aInstitute of Chemistry, The Hebrew University of Jerusalem, Safra Campus, Givat Ram, Jerusalem, 91904, Israel. E-mail: assaf.friedler@mail.huji.ac.il
bDepartment of Biological Chemistry, The Alexander Silberman Institute of Life Sciences, The Hebrew University of Jerusalem, Safra Campus, Givat Ram, Jerusalem, 91904, Israel
First published on 22nd October 2012
Abstract
Peptide arrays are a powerful tool for characterizing protein interactions and for identifying and optimizing peptide ligands. Here we demonstrate the use of peptide arrays for performing detailed SAR studies of lead peptide inhibitors of the interaction between the HIV integrase and Rev proteins. The integration of viral DNA into the genome of the host cell, mediated by the viral integrase (IN) enzyme, is a crucial step in the HIV-1 replication cycle. We have recently found that IN activity is regulated by interactions with the HIV-1 Rev protein and identified three lead peptides derived from the Rev-binding interface in IN. Due to their ability to promote dissociation of the Rev–IN complex in HIV infected cells, these peptides caused IN activation leading to multi-integration, genomic instability and specific eradication of such infected cells. Here we explored the mechanism of action of these three IN-derived peptides as the basis for developing improved anti HIV-1 leads. Using peptide array screening, we found that the IN derived peptides bound IN and Rev in a similar pattern. The Rev-binding sites in IN also mediate IN oligomerization while the IN-binding sites in Rev are also involved in Rev oligomerization. A structural homology was found between the oligomerization domains of Rev and IN residues 171–188 and 211–220. We performed SAR studies of the lead inhibitory peptides using a peptide array containing truncated peptides, alanine scan, D-amino acid scan and N-methylated amino acid scan. We screened IN and Rev for binding this array of modified IN-derived peptides. The screening results showed that C-terminal positively charged residues were essential for the interaction of the IN 118–128 and IN 174–188 peptides with both Rev and IN. The peptides could be shortened and modified without loss of binding to IN and Rev. This provides a basis for the future development of shorter peptides with better pharmacological properties that inhibit the Rev–IN interaction. We conclude that peptide arrays are excellent tools to perform detailed SAR binding studies in one short efficient experiment. The SAR study by the peptide array method is a powerful tool for developing improved inhibitors based on a lead peptide sequence.
Introduction
Peptide arrays are powerful tools for characterizing protein interactions and identifying specific domains involved in mediating these interactions.1 Peptide arrays are used for specific detection of individual proteins in complex mixtures,2 for identifying binding sites between proteins,3–5 for determining the contribution of individual amino acid residues to the binding6,7 and for high throughput measurements of enzymatic activites.8,9 Here we demonstrate the use of peptide arrays for performing detailed SAR studies of lead peptide inhibitors of the interaction between HIV integrase and HIV Rev.The integrase protein (IN) of the Human Immunodeficiency Virus type 1 (HIV-1) catalyzes the integration of the viral cDNA into the host genome, a crucial step in the HIV-1 replication cycle.10 IN is a 288 residue protein, divided into three distinct structural domains (Fig. 1): (i) the N-terminal domain (residues 1–49), which is involved in IN oligomerization and may also be required for specific DNA binding;11,12 (ii) the catalytic core domain (residues 50–212) containing the three conserved acidic residues D64, D116 and E152, which are essential for all IN catalytic activities;13–16 and (iii) the C-terminal domain (residues 220–288), which is involved in DNA binding and IN oligomerization.17–19 IN is in equilibrium between dimeric and tetrameric states.20–22 Dimeric IN binds at each LTR end of the viral DNA during the 3′-end processing step in the cytoplasm.23 After nuclear import, the two LTR DNA-bound dimers approach each other in the presence of the cellular protein lens epithelium-derived growth factor (LEDGF/p75), forming a tetramer, and the integration proceeds to the strand-transfer step.24 LEDGF/p75 activates IN by tethering it to the host chromosomes.25–28 The structures of the isolated domains of IN, as well as structures of IN fragments containing two domains, have been solved.16,29–31 In all the structures IN crystallized as a dimer. The structures of full-length tetramers of integrase proteins from other viruses have been characterized.32,33 The importance of the integration step within the HIV replication cycle, together with the lack of mammalian homologs for IN, makes IN an attractive therapeutic target. However, only one IN inhibitor (Raltegravir) is currently used in the clinic as an anti-HIV drug.34 IN binds both viral proteins such as the reverse transcriptase35,36 and cellular proteins such as LEDGF/p75 and ini137–39 and these interactions play an important role in regulating IN activity. Small molecules and peptides that mimic or inhibit the protein–protein interactions of IN are emerging as promising drug leads.36,40–44
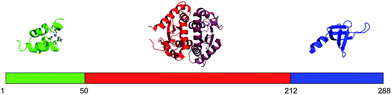 | ||
| Fig. 1 Structures of the IN domains. The N-terminal domain is shown in green (pdb 1eoe), the catalytic core domain in red (shown as a dimer, pdb 1ex4), and the C-terminal domain in blue (pdb 1ex4). | ||
Rev is a 116 residue protein required for promoting nuclear export of partially spliced and un-spliced viral RNA at the late phase of the viral life cycle.45,46 Rev binds to a specific sequence termed the Rev Response Element (RRE) in the viral RNA and the binding is accompanied by Rev oligomerization which is mediated primarily by Rev–Rev interactions.47–50 Each Rev monomer is comprised of a structured N-terminal domain containing two coplanar α-helices connected by a hairpin motif and a disordered C-terminal domain.51 Rev can also be expressed in early stages of infection from unintegrated viral cDNA.52–54 We have recently found that the early expressed HIV-1 Rev protein is one of the major regulators of IN activity. Early expressed Rev interacts with IN and inhibits IN activity in vitro and in vivo.55,56 Rev also interacts with LEDGF/p75 and promotes dissociation of the IN–LEDGF complex.57 The IN-binding sites in Rev have been identified, and peptides derived from these binding sites inhibited IN activity.55,56,58 We have identified the Rev-binding sites in IN and synthesized three peptides derived from these sites. The peptides were derived from residues IN 66–80, IN 118–128 and IN 174–188 and were termed by us INr1, INr2 and INS, respectively. All three peptides abrogated the IN–Rev interaction, resulting in activation of IN and stimulation of viral DNA integration in HIV-1 infected cells.59 INS also had a direct stimulatory effect on IN activity.60 Stimulation of integration by the combination of INr1, INr2 and INS resulted in genome instability leading to cell death and specific eradication of HIV-infected cells.61 However, these lead peptides are active at micromolar concentrations and their activity and stability need to be improved.
Here we explored the mechanism of action of these three IN-derived peptides as the basis for developing improved leads. Using peptide array screening, we found that the lead peptides bound Rev and IN in a similar pattern. We performed SAR studies of the lead inhibitory peptides using a peptide array containing truncated peptides as well as alanine scan, D-amino acid scan and N-methylated amino acid scan. We screened IN and Rev for binding this array of modified IN-derived peptides. The screening results showed that for two of the IN derived peptides (IN 118–128 and IN 174–188), C-terminal positively charged residues were essential for the interaction of the peptides with both Rev and IN. The peptides could be shortened and modified without loss of binding to IN and Rev. This provides a basis for the future development of shorter peptides with better pharmacological properties that inhibit the Rev–IN interaction. Our results show that peptide arrays are excellent tools to perform detailed SAR binding studies in one short efficient experiment.
Materials and methods
Protein expression and purification
The histidine-tagged Pet15b expression vector for HIV-1 integrase was a generous gift from Dr A. Engelman (Department of Cancer Immunology and AIDS, Dana Farber Cancer Institute, Division of AIDS, Harvard Medical School, Boston, MA, USA). The mutations F185K and C280S were introduced into the plasmid via a commercial site-directed mutagenesis kit (Stratagene). For expressing the protein, the plasmid was transformed into E. coli (Rosetta strain), and the culture was grown on LB-agar plates containing 100 μg ml−1 ampicillin and 34 μg ml−1 chloramphenicol. A single colony was picked to inoculate in 350 ml 2× YT medium containing ampicillin, chloramphenicol, 1% glucose and buffering salts (25 mM (NH4)2SO4, 50 mM KH2PO4, and 50 mM Na2HPO4). The culture was grown at 37 °C until the OD (600 nm) was ∼0.6, transferred to 22 °C, and 0.4 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) was added. The culture was further grown for 16 hours at 22 °C and the cells were harvested by centrifugation and frozen in liquid nitrogen.For purification, the frozen cells were thawed and dispersed in lysis buffer (HEPES 50 mM pH = 7.5, 1 M NaCl, 10% glycerol, 5 mM imidazole, 2 mM β-mercaptoethanol, 1 M urea) to which 0.2 mg ml−1 lyzozyme was added. The cells were lysed by microfluidization and the soluble fraction of the lysate was incubated with Ni-NTA beads (Invitrogen). The beads were washed with lysis buffer, followed by washing with lysis buffer + 100 mM imidazole. Elution of the bound protein was achieved with lysis buffer + 500 mM imidazole. 5 mM EDTA were added to the eluted protein, which was then loaded on a 500 ml Sephacryl S200 gel filtration column preequilibrated with HEPES 20 mM pH = 7.5, 1 M NaCl, 10% glycerol, 1 mM EDTA and 1 mM DTT. Fractions containing integrase were pooled, glycerol was added to 25%, and the protein was stored at −80 °C. The purity of the protein was estimated to be ∼95% from SDS-PAGE. Expression and purification of recombinant Rev was performed as described.62 Expression and purification of (His6LipoTEV)-Rev (HLT-Rev) was performed as described.62
Peptide array screening
Peptide arrays containing IN, Rev and LEDGF derived peptides and modified peptides (see ESI, Table S1† for sequences) were purchased from Intavis, Germany. The peptides were synthesized by standard Fmoc solid phase synthesis on a modified cellulose membrane. The array was blocked using 5% BSA in TBS-T (Tris 25 mM pH = 7.5, 150 mM NaCl, 0.05% Tween 20) for two hours at room temperature. Following a single wash with 2% BSA/TBS-T, the protein was diluted to 2 μM (HLT-Rev) or 1 μM (integrase) in 2% BSA/TBS-T + 10% glycerol + 1 mM EDTA + 1 mM DTT and incubated overnight with the array at 4 °C. The array was then washed once with 2% BSA/TBS-T and thrice with TBS-T, and incubated for 1 hour at room temperature with His-probe HRP (Santa Cruz) diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1500 in 2% BSA/TBS-T. The array was then washed once with 2% BSA/TBS-T and thrice with TBS-T, and the bound protein was detected using electrochemiluminescence. We used the program ImageJ to estimate the intensity of the spots in a semi-quantitative manner.
:1500 in 2% BSA/TBS-T. The array was then washed once with 2% BSA/TBS-T and thrice with TBS-T, and the bound protein was detected using electrochemiluminescence. We used the program ImageJ to estimate the intensity of the spots in a semi-quantitative manner.
For experiments performed with isolated Rev, skimmed milk was used instead of BSA. Detection of the bound protein was achieved with rat anti-Rev antibody, which was a generous gift from Ruth Brack Werner (German Research Center for Environmental Health, Munich, Germany). Screening the array with Rev and HLT-Rev gave identical results. However, HLT-Rev gave better signals and was therefore used for further analysis.
Sequence alignment between Rev and IN
The sequences of IN and Rev were aligned using Blast,63 MAFFT64 and TCOFFEE.65 For IN, we used the sequence of UniProt: P12497. For Rev, we used the sequence of UniProt: P04618. Proteins bearing these sequences were also used in the binding experiments presented in this study. The structures that were used to label secondary structures in the sequence alignment and to illustrate the sequentially similar α-helices are PDB: 1ex4 for IN and PDB: 2x7l for Rev. The C-terminal domain of Rev (residues 66–116) is intrinsically disordered51 and was not analyzed.Results
Interaction between IN derived peptides and Rev: design of a peptide array with modified IN-derived peptides
To explore how the IN-derived peptides bind Rev in a simple efficient experiment and to develop shorter peptides with improved pharmacological properties, we designed an array containing a library of modified peptides derived from the sequences of the two INrs and the INS peptides (IN 66–80, IN 118–128, and IN 174–188). The modifications included an alanine scan, truncations and systematic replacements of each residue in each peptide by the corresponding D-amino acid residue and N-methylated residue. The array was screened for binding both the recombinant wild type HIV-1 Rev protein and the HIV-1 integrase (Fig. 2A–C). Due to the low solubility of wild type HIV-1 integrase, we used the double mutant F185K/C280S which has higher solubility but similar activity to that of the wild type.66 We estimated the intensity of the spots for all peptides and compared them to the non-modified peptides (Table S1†).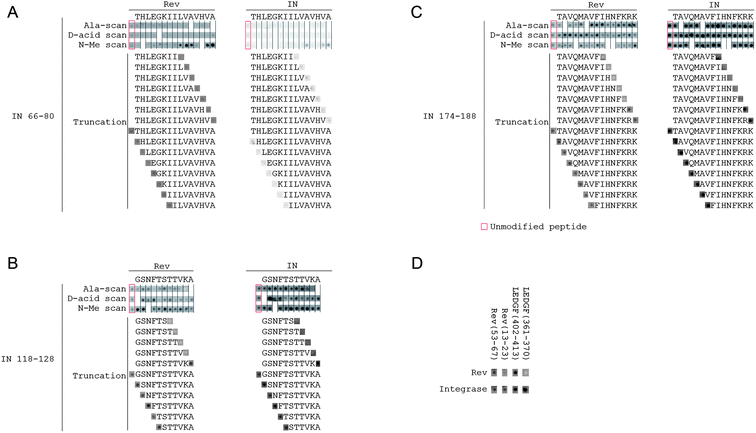 | ||
| Fig. 2 Binding of Rev and IN to an array of IN, Rev and LEDGF derived peptides. (A–C) INr1 (IN 66–80), INr2 (IN 118–128) and INS (IN 174–188). For each peptide, in the first three rows, each spot represents the binding of Rev or IN to a peptide with the indicated residue substituted by alanine, N-methylated or substituted by the corresponding D-amino acid residue. For truncations, the sequences of the truncated peptides tested are shown explicitly. See Materials and methods for details. The peptide sequences are detailed in Table S1.† (D) Binding of peptides derived from the IN-binding sites of LEDGF/p75 and Rev to IN and Rev. All experimental conditions are as described in Materials and methods. | ||
The Rev-binding sites in IN and the IN-binding sites in Rev may mediate protein oligomerization
Both Rev and IN bound the IN derived peptide array with very similar patterns (Fig. 2A–C), indicating that the Rev-binding sites in IN may also mediate IN–IN interactions. Only small differences were observed between the peptide binding patterns of Rev and IN. For example, the alanine scan shows that the binding of IN (174–188) to IN is significantly weakened by N184A substitution, an effect which is not observed for binding of Rev.The IN-binding sites in Rev (residues 13–23 and 53–67)56,58 are also involved in Rev oligomerization.51 Our screening is in agreement with this (Fig. 2D): peptides derived from both Rev 13–23 and Rev 53–67 bound both IN and Rev in the array screening.
Overlapping sites in LEDGF/p75 mediate its binding to both IN and Rev
Rev binds both LEDGF/p75 and IN and dissociates the IN–LEDGF/p75 complex.57 While it is believed that Rev dissociates the complex by directly masking the LEDGF/p75 binding site in IN,67,68 it is not clear what role a possible Rev–LEDGF/p75 interaction plays in this process. We therefore tested whether Rev can interact with the IN-binding peptides from LEDGF/p75 (Fig. 2D). Rev bound a peptide derived from LEDGF/p75 residues 402–413, indicating that the LEDGF/p75 binding sites for Rev and IN partly overlap. IN bound both LEDGF/p75 peptides as expected.Structural homology between the oligomerization domains of Rev and the IN residues 171–188 and 211–220
Based on the similar binding pattern of IN and Rev to the peptide array, we tested whether both proteins share homologous motifs that may explain their similar binding properties. A standard pairwise BLAST sequence comparison63 did not detect similarity between the sequences of IN and Rev. Using more sensitive sequence homology detection methods, we found that both MAFTT64 and TCOFFEE65 aligned Rev to the C-terminal part of IN in the same manner. This reveals structural similarities between the regions corresponding to the Rev oligomerization domains (residues 9–26 and 51–65)51 and sequences in IN (Fig. 3). Rev 11–27 bears a high similarity to IN 171–188, from which the INS peptide is derived. Rev 46–55 is similar to IN 211–220, which is located in the C-terminal domain of IN.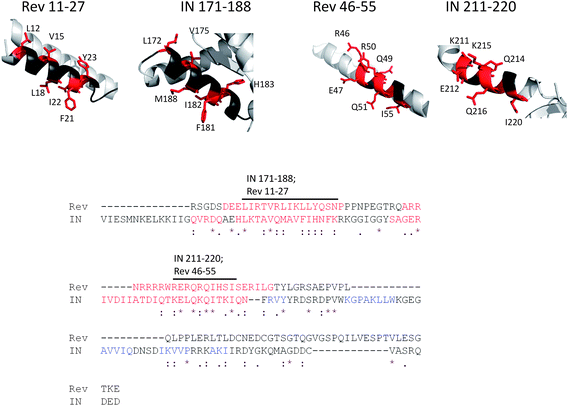 | ||
| Fig. 3 The similarity between the binding motifs of IN and Rev. (A) Sequence alignment (bottom) generated by both MAFFT64 and TCOFFEE65 aligned the sequence of Rev to the C-terminal part of IN. Letters representing residues of α-helices are colored red, and of β-sheets are colored blue. The structure of the C-terminal part of Rev from residue 66 was not determined since this part is intrinsically disordered. Two labeled stretches of residues in the alignment correspond to two sequentially similar α-helices of IN and Rev (top). One helix is IN 171–188/Rev 11–27 and the second helix is IN 211–220/Rev 46–55. Identical and highly similar residues that may contribute to binding are colored red and represented as sticks. | ||
SAR studies of the IN-derived peptides
The peptide array screening revealed which residues are important for the activities of INr1, INr2 and INS (Fig. 2A–C). N-Methylation of hydrophobic residues in the C-terminus of INr1 significantly improved the binding to Rev and IN. Both IN and Rev bound to the INr1 peptides on the array very weakly, thus preventing the identification of residues crucial for binding. Since INr1 is very hydrophobic, the low signals may be the result of low yields in the synthesis of these peptides or aggregation of the peptides due to their high local concentration in the array. Since not every spot is analyzed during the array synthesis, quantitative yields could not be obtained. For INr2 (IN 118–128), the alanine scan showed that residues V126 and K127 are essential for binding of both Rev and IN, and truncation of the peptide from the C-terminus down to V126 or further completely abolished the binding. The N-terminus of INr2 was not essential for binding IN or Rev. For the INS peptide the C-terminus is also the essential part for binding. Any truncation from the C-terminus down to K186 or further abolished the binding to both Rev and IN. However, no single C-terminal residue was essential for binding, since peptides with single substitutions of C-terminal residues to alanine retained their binding.Substitution of residues by D-amino acid or N-methylated residues did not abolish the binding to Rev or IN, even when important residues (as determined from the alanine scan) were substituted. The majority of the modified peptides bound IN and Rev with intensities similar to or higher than the non-modified peptide (Table S1†). This indicates that such stabilizing modifications may be introduced into the peptides without loss of activity.
Discussion
Peptide arrays have been previously used to characterize the sites which mediate interactions between proteins and specifically the contribution of single amino acid residues to the binding.3,6,7,69 This makes peptide arrays powerful tools for developing improved ligands for target proteins.70 Here, the use of peptide arrays enabled us to analyze the binding of hundreds of modified peptides, all based on the same three lead sequences, to IN and Rev in a high throughput manner. Therefore, we were able to quickly and efficiently identify the residues necessary for binding and to test the effects of modifications on the interaction. This may provide the basis for developing more potent and stable peptidomimetics that stimulate IN activity by inhibiting its binding to Rev.Structural homology between Rev and IN
The IN, Rev and LEDGF derived peptides in the array bound both Rev and IN with highly similar patterns. For Rev, these results show that its IN-binding sites (residues 13–23 and 53–67) are also involved in its oligomerization (Fig. 4A).51 As for the Rev-binding sites in IN, IN 66–80 and IN 118–128 were not reported to be involved in IN oligomerization. IN 66–80 is mostly buried within the folded monomer of the catalytic core domain (Fig. 4B). IN 118–128 and IN 174–188 both lie in exposed, positively charged regions on the protein surface in the crystal structures of the IN core domain (Fig. 4B and C). IN 174–188 is known to bind IN directly60 and since it is located at the edge of the dimerization interface of the IN catalytic core domain (Fig. 4B) it is likely to be involved in IN dimerization. These C-terminal residues (IN 174–188) were also reported to form crystal lattice contacts between IN dimers.31 IN 174–188 is also involved in the formation of high order oligomers of IN. Mutation in F185, which is located in this region, is commonly used to increase the solubility of recombinant IN.66 The results do not necessarily imply that the interaction between IN and Rev inhibits their oligomerization. The oligomerization domains of Rev and IN 174–188 contain many solvent exposed residues and therefore may be able to bind other proteins without interfering with the oligomerization process.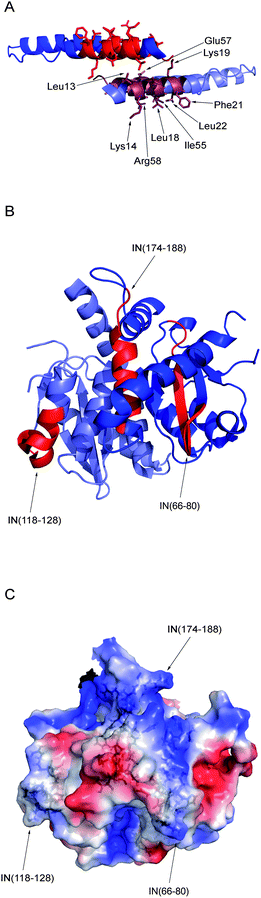 | ||
| Fig. 4 The structures of Rev–IN interacting domains. (A) Crystal structure of a Rev dimer, taken from pdb 2x7L.51 The exposed residues of the bottom monomer are indicated. (B) Crystal structure of the IN catalytic core domain dimer, taken from pdb 1ex4.67 For clarity, each domain is highlighted only in one of the monomers. (C) Surface charge density plot for the IN catalytic core domain. Blue – positive charge; red – negative charge. The peptides IN 66–80, IN 118–128 and IN 174–188 are shown as sticks and labeled. | ||
In light of the similarity between the binding patterns of Rev and IN to the peptide array, we identified a structural homology between the oligomerization domains of Rev and residues 171–188 and 211–220 of IN. This homology may indicate that domains involved in oligomerization of IN and Rev are also involved in their interactions with other proteins.
The effect of Rev binding on the IN–LEDGF interaction
The results of our peptide array screening may indicate that LEDGF/p75 binds Rev and IN via partly overlapping binding sites. The IN residues 118–128 and 174–188 are located in close proximity to the LEDGF/p75 binding pocket in IN.67 Protein docking indicates that Rev blocks the LEDGF/p75-binding site in IN upon binding IN residues 174–188.68 Therefore, the dissociation of the IN–LEDGF/p75 complex by Rev may be achieved by dual competition – Rev may bind to binding interfaces of both LEDGF/p75 and IN and thus block their interaction. Only future structural studies will reveal whether a ternary complex between IN, Rev and LEDGF/p75 is possible.Implications for development of inhibitors of the Rev–IN interaction
While the linear IN-derived peptides induce hyper-activity of IN and cause specific death of infected cells, their therapeutic value is limited due to their inherent low stability and bioavailability.71,72 For the peptide INr1, our results show that N-methylation of its C-terminal residues significantly improves its binding to both IN and Rev. However, the weak signals obtained from the array prevented the identification of residues essential for binding. For the peptides INr2 and INS, the positively charged C-terminus was important for the interaction with Rev or IN. The N-terminus of these peptides could also be truncated, resulting in a peptide bearing as little as 6 residues, without eliminating the binding to Rev and IN. Additionally, INr2 and INS could tolerate N-methylations and D-amino acid substitutions at almost any position, including the residues that are important for binding. These results may indicate that interactions of the backbone amide groups (especially hydrogen bonds) do not play a significant role in the binding of the peptides to Rev and IN, and that the protein-bound peptides are still relatively flexible. Therefore inversion of the configuration of a single amino acid, even an essential one, would not abolish the binding. This may provide the basis for developing more potent and stable peptidomimetics that stimulate IN activity by inhibiting its binding to Rev.In conclusion, in this study we used peptide arrays to perform a detailed characterization of the interactions of the INr1, INr2 and INS peptides with IN and Rev. The results provide important insights regarding the nature of the IN–Rev interaction and how the peptides modulate it, and are a promising starting point for developing these peptides into improved anti-HIV-1 lead compounds. The SAR study using peptide array is a powerful and efficient tool for developing improved inhibitors based on a lead peptide sequence.
Acknowledgements
AF is supported by a starting grant from the European Research Council under the European Community's Seventh Framework Programme (FP7/2007–2013)/ERC Grant agreement no. 203413. RG is supported by the Adams Fellowship Program of the Israel Academy of Sciences and Humanities. AF and AL are supported by a grant from Yissum, the Technology Transfer Company of the Hebrew University.References
- C. Katz, L. Levy-Beladev, S. Rotem-Bamberger, T. Rito, S. G. D. Rudiger and A. Friedler, Chem. Soc. Rev., 2011, 40, 2131–2145 RSC.
- M. Takahashi, K. Nokihara and H. Mihara, Chem. Biol., 2003, 10, 53–60 CrossRef CAS.
- G. S. Baillie, D. R. Adams, N. Bhari, T. M. Houslay, S. Vadrevu, D. Meng, X. Li, A. Dunlop, G. Milligan, G. B. Bolger, E. Klussmann and M. D. Houslay, Biochem. J., 2007, 404, 71–80 CrossRef CAS.
- C. Katz, H. Benyamini, S. Rotem, M. Lebendiker, T. Danieli, A. Iosub, H. Refaely, M. Dines, V. Bronner, T. Bravman, D. E. Shalev, S. Rüdiger and A. Friedler, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12277–12282 CrossRef CAS.
- S. Rudiger, L. Germeroth, J. Schneider-Mergener and B. Bukau, EMBO J., 1997, 16, 1501–1507 CrossRef CAS.
- R. A. Houghten, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 5131–5135 CrossRef CAS.
- H. M. Geysen, R. H. Meloen and S. J. Barteling, Proc. Natl. Acad. Sci. U. S. A., 1984, 81, 3998–4002 CrossRef CAS.
- X. Han, S. Shigaki, T. Yamaji, G. Yamanouchi, T. Mori, T. Niidome and Y. Katayama, Anal. Biochem., 2008, 372, 106–115 CrossRef CAS.
- J.-L. Reymond, Ann. N. Y. Acad. Sci., 2008, 1130, 12–20 CrossRef CAS.
- M. P. Sherman and W. C. Greene, Microbes Infect., 2002, 4, 67–73 CrossRef CAS.
- K. Carayon, H. Leh, E. Henry, F. Simon, J.-F. Mouscadet and E. Deprez, Nucleic Acids Res., 2010, 38, 3692–3708 CrossRef CAS.
- R. Zheng, T. M. Jenkins and R. Craigie, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13659–13664 CrossRef CAS.
- D. C. van Gent, A. A. Groeneger and R. H. Plasterk, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 9598–9602 CrossRef CAS.
- A. D. Leavitt, L. Shiue and H. E. Varmus, J. Biol. Chem., 1993, 268, 2113–2119 CAS.
- A. Engelman and R. Craigie, J. Virol., 1992, 66, 6361–6369 CAS.
- J. C. Chen, J. Krucinski, L. J. Miercke, J. S. Finer-Moore, A. H. Tang, A. D. Leavitt and R. M. Stroud, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8233–8238 CrossRef CAS.
- R. A. Puras Lutzke, C. Vink and R. H. A. Plasterk, Nucleic Acids Res., 1994, 22, 4125–4131 CrossRef.
- A. M. Woerner and C. J. Marcus-Sekura, Nucleic Acids Res., 1993, 21, 3507–3511 CrossRef CAS.
- A. Engelman, A. B. Hickman and R. Craigie, J. Virol., 1994, 68, 5911–5917 CAS.
- A. Faure, C. Calmels, C. Desjobert, M. Castroviejo, A. Caumont-Sarcos, L. Tarrago-Litvak, S. Litvak and V. Parissi, Nucleic Acids Res., 2005, 33, 977–986 CrossRef CAS.
- E. Deprez, P. Tauc, H. Leh, J.-F. Mouscadet, C. Auclair, M. E. Hawkins and J.-C. Brochon, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10090–10095 CrossRef CAS.
- E. Deprez, P. Tauc, H. Leh, J.-F. Mouscadet, C. Auclair and J.-C. Brochon, Biochemistry, 2000, 39, 9275–9284 CrossRef CAS.
- E. Guiot, K. Carayon, O. Delelis, F. Simon, P. Tauc, E. Zubin, M. Gottikh, J.-F. Mouscadet, J.-C. Brochon and E. Deprez, J. Biol. Chem., 2006, 281, 22707–22719 CrossRef CAS.
- A. Chen, I. T. Weber, R. W. Harrison and J. Leis, J. Biol. Chem., 2006, 281, 4173–4182 CrossRef CAS.
- G. Maertens, P. Cherepanov, W. Pluymers, K. Busschots, E. De Clercq, Z. Debyser and Y. Engelborghs, J. Biol. Chem., 2003, 278, 33528–33539 CrossRef CAS.
- S. Emiliani, A. Mousnier, K. Busschots, M. Maroun, B. Van Maele, D. Tempé, L. Vandekerckhove, F. Moisant, L. Ben-Slama, M. Witvrouw, F. Christ, J.-C. Rain, C. Dargemont, Z. Debyser and R. Benarous, J. Biol. Chem., 2005, 280, 25517–25523 CrossRef CAS.
- L. Vandekerckhove, F. Christ, B. Van Maele, J. De Rijck, R. Gijsbers, C. Van den Haute, M. Witvrouw and Z. Debyser, J. Virol., 2006, 80, 1886–1896 Search PubMed.
- M. Llano, D. T. Saenz, A. Meehan, P. Wongthida, M. Peretz, W. H. Walker, W. Teo and E. M. Poeschla, Science, 2006, 314, 461–464 CrossRef CAS.
- A. P. A. M. Eijkelenboom, R. Sprangers, K. Hård, R. A. Puras Lutzke, R. H. A. Plasterk, R. Boelens and R. Kaptein, Proteins: Struct., Funct., Bioinf., 1999, 36, 556–564 CrossRef CAS.
- A. P. A. M. Eijkelenboom, F. M. I. van den Ent, R. Wechselberger, R. H. A. Plasterk, R. Kaptein and R. Boelens, J. Biomol. NMR, 2000, 18, 119–128 CrossRef CAS.
- J.-Y. Wang, H. Ling, W. Yang and R. Craigie, EMBO J., 2001, 20, 7333–7343 CrossRef CAS.
- R. S. Bojja, M. D. Andrake, S. Weigand, G. Merkel, O. Yarychkivska, A. Henderson, M. Kummerling and A. M. Skalka, J. Biol. Chem., 2011, 286, 17047–17059 CrossRef CAS.
- G. N. Maertens, S. Hare and P. Cherepanov, Nature, 2010, 468, 326–329 CrossRef CAS.
- E. Serrao, S. Odde, K. Ramkumar and N. Neamati, Retrovirology, 2009, 6, 25 CrossRef.
- T. Tasara, G. Maga, M. O. Hottiger and U. Hübscher, FEBS Lett., 2001, 507, 39–44 CrossRef CAS.
- I. Oz Gleenberg, O. Avidan, Y. Goldgur, A. Herschhorn and A. Hizi, J. Biol. Chem., 2005, 280, 21987–21996 CrossRef CAS.
- P. Cherepanov, G. Maertens, P. Proost, B. Devreese, J. Van Beeumen, Y. Engelborghs, E. De Clercq and Z. Debyser, J. Biol. Chem., 2003, 278, 372–381 CrossRef CAS.
- M. Maroun, O. Delelis, G. Coadou, T. Bader, E. Ségéral, G. Mbemba, C. Petit, P. Sonigo, J.-C. Rain, J.-F. Mouscadet, R. Benarous and S. Emiliani, J. Biol. Chem., 2006, 281, 22736–22743 CrossRef CAS.
- E. Yung, M. Sorin, A. Pal, E. Craig, A. Morozov, O. Delattre, J. Kappes, D. Ott and G. V. Kalpana, Nat Med, 2001, 7, 920–926 CrossRef CAS.
- M. Maes, A. Loyter and A. Friedler, FEBS J., 2012, 279, 2795 CrossRef CAS.
- Z. Hayouka, J. Rosenbluh, A. Levin, S. Loya, M. Lebendiker, D. Veprintsev, M. Kotler, A. Hizi, A. Loyter and A. Friedler, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8316–8321 CrossRef CAS.
- S. Suzuki, E. Urano, C. Hashimoto, H. Tsutsumi, T. Nakahara, T. Tanaka, Y. Nakanishi, K. Maddali, Y. Han, M. Hamatake, K. Miyauchi, Y. Pommier, J. A. Beutler, W. Sugiura, H. Fuji, T. Hoshino, K. Itotani, W. Nomura, T. Narumi, N. Yamamoto, J. A. Komano and H. Tamamura, J. Med. Chem., 2010, 53, 5356–5360 CrossRef CAS.
- R. G. Maroun, S. Gayet, M. S. Benleulmi, H. Porumb, L. Zargarian, H. Merad, H. Leh, J.-F. Mouscadet, F. Troalen and S. Fermandjian, Biochemistry, 2001, 40, 13840–13848 CrossRef CAS.
- F. Christ, A. Voet, A. Marchand, S. Nicolet, B. A. Desimmie, D. Marchand, D. Bardiot, N. J. Van der Veken, B. Van Remoortel, S. V. Strelkov, M. De Maeyer, P. Chaltin and Z. Debyser, Nat. Chem. Biol., 2010, 6, 442–448 CrossRef CAS.
- L. Li, H. S. Li, C. D. Pauza, M. Bukrinsky and R. Y. Zhao, Cell Res., 2005, 15, 923–934 CrossRef CAS.
- V. W. Pollard and M. H. Malim, Annu. Rev. Microbiol., 1998, 52, 491–532 CrossRef CAS.
- N. R. Watts, M. Misra, P. T. Wingfield, S. J. Stahl, N. Cheng, B. L. Trus, A. C. Steven and R. W. Williams, J. Struct. Biol., 1998, 121, 41–52 CrossRef CAS.
- S. J. K. Pond, W. K. Ridgeway, R. Robertson, J. Wang and D. P. Millar, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1404–1408 CrossRef CAS.
- M. H. Malim and B. R. Cullen, Cell, 1991, 65, 241–248 CrossRef CAS.
- T. J. Daly, R. C. Doten, P. Rennert, M. Auer, H. Jaksche, A. Donner, G. Fisk and J. R. Rusche, Biochemistry, 1993, 32, 10497–10505 CrossRef CAS.
- M. A. DiMattia, N. R. Watts, S. J. Stahl, C. Rader, P. T. Wingfield, D. I. Stuart, A. C. Steven and J. M. Grimes, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5810–5814 CrossRef CAS.
- Y. Wu, Retrovirology, 2004, 1, 13 CrossRef.
- Y. Wu and J. W. Marsh, J. Virol., 2003, 77, 10376–10382 CrossRef CAS.
- S. R. Iyer, D. Yu, A. Biancotto, L. B. Margolis and Y. Wu, J. Virol., 2009, 83, 8662–8673 Search PubMed.
- A. Levin, Z. Hayouka, A. Friedler and A. Loyter, Nucleus, 2010, 1, 190–201 CrossRef.
- A. Levin, Z. Hayouka, R. Brack-Werner, D. J. Volsky, A. Friedler and A. Loyter, Protein Eng., Des. Sel., 2009, 22, 753–763 CrossRef CAS.
- A. Levin, J. Rosenbluh, Z. Hayouka, A. Friedler and A. Loyter, Mol. Med., 2010, 16, 34–44 CAS.
- Z. Hayouka, J. Rosenbluh, A. Levin, M. Maes, A. Loyter and A. Friedler, Biopolymers, 2008, 90, 481–487 CrossRef CAS.
- A. Levin, Z. Hayouka, M. Helfer, R. Brack-Werner, A. Friedler and A. Loyter, PLoS One, 2009, 4, e4155 Search PubMed.
- A. Levin, Z. Hayouka, M. Helfer, R. Brack-Werner, A. Friedler and A. Loyter, Biopolymers, 2010, 93, 740–751 CAS.
- A. Levin, Z. Hayouka, A. Friedler and A. Loyter, AIDS Res. Ther., 2010, 7, 31 CrossRef.
- P. Siman, O. Blatt, T. Moyal, T. Danieli, M. Lebendiker, H. A. Lashuel, A. Friedler and A. Brik, ChemBioChem, 2011, 12, 1097–1104 CrossRef CAS.
- S. F. Altschul, T. L. Madden, A. A. Schäffer, J. Zhang, Z. Zhang, W. Miller and D. J. Lipman, Nucleic Acids Res., 1997, 25, 3389–3402 CrossRef CAS.
- K. Katoh, K. Kuma, H. Toh and T. Miyata, Nucleic Acids Res., 2005, 33, 511–518 CrossRef CAS.
- C. Notredame, D. G. Higgins and J. Heringa, J. Mol. Biol., 2000, 302, 205–217 CrossRef CAS.
- T. M. Jenkins, A. Engelman, R. Ghirlando and R. Craigie, Analysis, 1996, 271, 7712–7718 CAS.
- P. Cherepanov, A. L. B. Ambrosio, S. Rahman, T. Ellenberger and A. Engelman, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17308–17313 CrossRef CAS.
- H. Benyamini, A. Loyter and A. Friedler, Biochem. Biophys. Res. Commun., 2011, 416, 252–257 CrossRef CAS.
- S. Kudithipudi, A. Dhayalan, A. F. Kebede and A. Jeltsch, Biochimie, 2012, 94, 2212 CrossRef CAS.
- U. Wiedemann, P. Boisguerin, R. Leben, D. Leitner, G. Krause, K. Moelling, R. Volkmer-Engert and H. Oschkinat, J. Mol. Biol., 2004, 343, 703–718 CrossRef CAS.
- Z. Hayouka, M. Hurevich, A. Levin, H. Benyamini, A. Iosub, M. Maes, D. E. Shalev, A. Loyter, C. Gilon and A. Friedler, Bioorg. Med. Chem., 2010, 18, 8388–8395 CrossRef CAS.
- M. Pernot, R. Vanderesse, C. Frochot, F. Guillemin and M. Barberi-Heyob, Expert Opin. Drug Metab. Toxicol., 2011, 7, 793–802 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2md20225e |
| This journal is © The Royal Society of Chemistry 2013 |