Ring opening polymerization of mannosyl tricyclic orthoesters: rationalising the stereo and regioselectivity of glycosidic bond formation using quantum chemical calculations†
Siwarutt
Boonyarattanakalin
a,
Somsak
Ruchirawat
bc and
M. Paul
Gleeson
*d
aSchool of Bio-Chemical Engineering and Technology, Sirindhorn International Institute of Technology, Thammasat University, Pathum Thani 12121, Thailand
bLaboratory of Medicinal Chemistry, Chulabhorn Research Institute, 54 Moo 4, Vibhavadee-Rangsit Highway, Laksi, Bangkok 10210, Thailand
cChemical Biology Program, Chulabhorn Graduate Institute, and the Center for Environmental Health, Toxicology and Management of Chemicals, 54 Moo 4, Vibhavadee-Rangsit Highway, Laksi, Bangkok 10210, Thailand
dDepartment of Chemistry, Faculty of Science, Kasetsart University, 50 Phaholyothin Rd, Chatuchak, Bangkok 10900, Thailand. E-mail: paul.gleeson@ku.ac.th; Fax: +66-2-5793955; Tel: +66-2-562-5555 ext. 2210
First published on 22nd October 2012
Abstract
Quantum chemical calculations have been used to assess the physico-chemical origin of the stereo and regio-selectivity of polymerisation reactions of glycosyl tricyclic orthoesters. From the theoretical reaction pathway we find that subtle modulation of steric and electronic effects at the initiation event can dramatically influence the nature of the polymer products.
Infectious diseases such as tuberculosis (TB) remain a globally life-threatening health problem.1 TB is a particular problem in developing countries as the long term treatment of the disease using antibiotics is financially unviable.2 Further research is undoubtedly needed therefore, to allow the development of more, rapid and cost effective treatments.
Lipomannan (LM) is one of the key glycolipids that comprise the unique cell envelope of Mycobacterium tuberculosis (Mtb). Consisting of a α(1–6) mannopyranan backbone,3 LMs have been implicated during the infectious, virulent, and survival events in host mammalian cells.4,5 Thus, improved understanding of LM interactions with the host immune system should help in the development of improved treatments for TB.6 Unfortunately, many experimental studies on this glycolipid are impeded by the limited amount of naturally occurring oligosaccharides.
Yongyat et al.7 have previously reported a synthetic approach utilizing mannosyl tricyclic orthoesters as monomers for regio- and stereocontrolled polymerizations to generate α(1–6) mannopyranan. The Lewis acids, trimethylsilyl trifluoromethanesulfonate (TMSOTf) and boron trifluoride etherate (BF3·Et2O) were used as catalysts to promote the polymerizations of two of the monomers including 3,4-O-benzyl-β-D-mannopyranose 1,2,6-orthobenzoate (1) and 3,4-O-benzyl-β-D-mannopyranose 1,2,6-orthopivalate (2) (Fig. 1). From a single chemical transformation step, polymers of different lengths and differing degrees of regio- and stereo-selectivity were obtained. It was found that under the same conditions; (a) TMSOTf leads to longer and more selective α(1–6) mannan chains when compared to BF3 and (b) that the monomer 3,4-O-benzyl-β-D-mannopyranose 1,2,6-orthobenzoate (1) gives rise to longer, and more selective α(1–6) chains when compared to 3,4-O-benzyl-β-D-mannopyranose 1,2,6-orthopivalate (2).
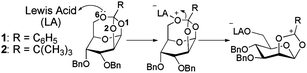 | ||
| Fig. 1 The initiation step of monomer 1 and 2, prior to the formation of α(1–6) mannopyranan. | ||
To facilitate the design of alternate methods to control regio- and stereo-selectivity, and to help improve reaction yields, quantum chemical (QC) calculations have been undertaken, the goal is to try and understand the physico-chemical origin of the control in polymerisation results described above. To this end, we explore the structures and energies associated with the critical activation step (Fig. 2).
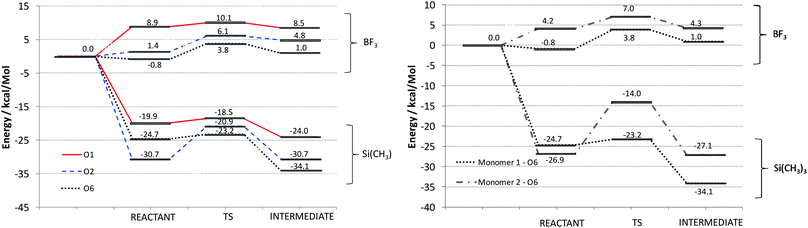 | ||
| Fig. 2 Reaction pathways obtained for monomer 1, at the O1, O2 and O6 positions, with two different initiators (left) and pathways obtained for monomers 1 and 2 using two different initiators, at the O6 position (right). | ||
Simulations were performed using complete molecular models of monomers 1 and 2, the active form of TMSOTf (i.e. Si(CH3)3+), and BF3. The initiation pathway (Fig. 1) was characterised using density functional theory (DFT) calculations at the M062X/6-31+G** level with a polarizable continuum model (PCM) consisting of dichloromethane. M062X is an increasingly popular DFT method that has performed better in recent benchmarking studies compared to the more common B3LYP method.8 Frequency analyses were used to fully characterise all minima and transition states and to compute thermochemical information. The potential energy surface was corrected for vibrational, rotational and translational motion.9 All minima and transition state energies are reported relative to the isolated, solvated reactants. All calculations were performed in Gaussian 09.10 Basis set superposition errors are known to be less significant for DFT based methods so were not evaluated.11 The energetic pathways associated with the initiation step of the 2 different glycosyl tricyclic orthoesters (GTO) are reported in Fig. 2. We have obtained the reactant (the non-bonded complex between monomer and initiator), the initiated intermediate, and the transition state that connects them. We begin our discussion with the preferred position of activation, followed by the initiator and finally the monomer.
GTOs contain three suitable Lewis bases on the three oxygen atoms (labelled as O1, O2, and O6 in Fig. 1) that are connected to the orthoester carbon (Corth). Upon monomer activation by a Lewis acid, the bond between the Corth and the activated oxygen atom is broken. The resulting carbocation ion intermediate is then capable of propagating the polymerisation process (Fig. 1). The first step in the process involves the formation of the initial non-bonded complex, or reactant as termed here.
The non-bonded reactant structures obtained with BF3 display B–O interaction distances of ∼1.6 Å while the larger (CH3)3Si+ based complexes display Si–O interaction distance of ∼1.85 Å. These distances drop to ∼1.5 Å and ∼1.7 Å respectively on reaction with Lewis acid to form the intermediate. We find that irrespective of the position of attachment, the (CH3)3Si+ based complexes formed with monomer 1 are considerably lower in energy than the corresponding BF3 based structures. Indeed, it should be noted that the large difference in binding energy observed (∼30 kcal mol−1) is considerably larger than the expected BSSE errors obtained for a system of comparable size using similar DFT methods (<5 kcal mol−1).12
Of the three possible oxygen atoms that can be activated (O1, O2, and O6, Fig. 1), activation at the O6 position is preferred thermodynamically irrespective of the Lewis acid (Fig. 2). This is consistent with the fact that the breaking of the O6–Corth bond leads to the least sterically hindered intermediate. The intermediate formed by the breaking of the O2–Corth bond is found to be lower in energy than that formed by the cleavage of the O1–C bond. Both the O1 and O2 intermediates form fused eight and six membered ring systems. However the O1 intermediate is the least stable as the resultant cyclic ring must span a longer distance between the C2 and C6 atoms. In contrast, activation at the O6 position results in a more stable bicyclic intermediate composed of a fused five and six membered ring system which spans the adjacent C1 and C2 atoms. The relative energy of the intermediates are also found to correlate reasonably well with the cleaved C+–O distance. We find that the shorter this distance on average over all the intermediates observed, the lower the energy of the intermediate. For the O1 position, the C+–O1 distances are ∼2.65 and 2.65 Å for (CH3)3Si+ and BF3 respectively, 2.44 Å and 2.34 Å for the O2 position respectively, and 2.43 Å and 2.35 Å for the C+–O6 position respectively (Fig. 3). Energetically, we find that BF3 results in less favourable intermediates than (CH3)3Si+ due to the fact that the positively charged (CH3)3Si+ can interact more effectively with the resultant monomer on cleavage. In addition, while the order of the stability of the 3 possible intermediates is the same, the BF3 intermediates are considerably higher in energy since they are zwitterionic structures in the non-polar solvent, rather than cationic for (CH3)3Si+. The latter Lewis acid forms the most stable intermediate, this being a pre-requisite for further polymerization. This helps to explain why the experimental yields with this Lewis acid are greater.7
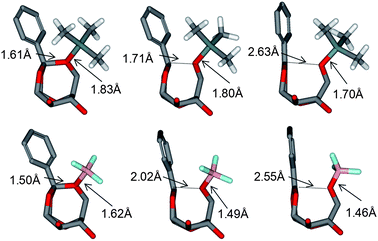 | ||
| Fig. 3 Optimized structures for monomer 1 pathway corresponding to the reactants (left), transitions states (middle) and intermediates (right) for (CH3)3Si+ (top) and BF3 (bottom). Benzyl groups and monomer H atoms have been removed for clarity. Key distances are given. The BF3 based initiator results in a later transition state, closer in structure to the intermediate. | ||
It is also necessary to explain why BF3 is experimentally observed to form less selective polymers compared to (CH3)3Si+. Based on the calculated data obtained here, this appears to be a function of both the kinetics and thermodynamics of the initiation step (Fig. 2). For the BF3 initiated process for monomer 1 the energies of the initial non-bonded complexes for positions 2 and 6 are similar and the corresponding barriers to reaction are rather close at 4.7 and 4.6 kcal mol−1, respectively. While the O1 barrier is the lowest of all, it is the least stable of all the BF3 based reactants, and also gives rise to the highest energy intermediate.
For (CH3)3Si+, we observe that the initial non-bonded complex formed at the O2 position is preferred over the O6 position due to the unfavourable steric interaction of the initiator with the C6 methylene. This effect is absent in case of the smaller BF3 Lewis acid. As is the case for BF3, the O1 position with (CH3)3Si+ also displays a low barrier to reaction, but the reactant and intermediates are also of high energy. It can therefore be concluded that the rate determining barrier associated with the O6 position is dramatically lower than that for the next preferable position (O2), being 1.5 and 9.8 kcal mol−1, respectively.
We postulate that the reaction to form the O6 intermediate is critical because it leads to the formation of the most preferable intermediate, which also displays a very high barrier to re-form the non-bonded complex (10.9 kcal mol−1). These results therefore suggest that (CH3)3Si+ will selectively form comparatively large quantities of the O6 intermediate, while BF3 can presumably activate at both the O2 and O6, and possibly O1. The subsequent nucleophilic attack of the carbocation intermediate by the sterically more accessible O6 nucleophile of an additional monomer is further complicated due to steric interference from the bridged ring system and the OBn substituents, as well as electronic effects from the ring oxygens. Polymerization with initiators at the O1 and O2 positions is expected to be much more challenging compared to the O6 position as the former two cases contain more sterically hindered points of attack. Attack at the C1 position of the O6-intermediate (Fig. 1) is preferential since it is both less sterically hindered and because the two adjacent oxygen atoms can better stabilize the resulting transition state with their lone pairs. Thus, it is clear that the monomer/initiator combination that give rise to the energetically most favourable and accessible intermediate, and which displays a large energy gap to the other possible intermediates, will result in the most selective, longer chained polymers.
From an analysis of the optimized transition state structures we indeed find evidence showing that the better the stabilizing effect on the C+ by the leaving group, the lower the energy of the transition state. We find that the C+–O distances in the transition state do indeed correlate with the relative energy. The longest interaction distance is observed for O1 position, being 1.86 and 2.19 Å for BF3 and (CH3)3Si+, respectively, compared to 1.73 and 2.08 Å, respectively, for O2, and 1.69 and 2.02 Å, respectively for O6. The results also show that the (CH3)3Si+ transitions states lie closer to the reactant state than BF3, consistent with the Hammonds postulate.
Finally, we investigated why monomer 2 is found to have experimentally lower reaction yields and poorer selectivity under the same experimental conditions as monomer 1. We therefore investigated how the replacement of the phenyl ring on the Corth of monomer 1 with tert-butyl affected the reaction profile at the preferred O6 position. We find that polymerization reactions involving monomer 2 with both BF3 and (CH3)3Si+ result in higher energy barriers to reaction, and higher intermediate energies (Fig. 2). We find that the O6-intermediate of monomer 2 obtained with BF3 is 3.3 kcal mol−1 higher in energy than the corresponding value for monomer 1, while that for (CH3)3Si+ is 7.1 kcal mol−1. This is due to the generally longer C+–O6 interaction distances, which is indicative of reduced electronic stabilization of the carbocation. This helps to explains why monomer 2 is found to have experimentally lower reactivity compared to monomer 1.7
Conclusions
The results presented here show that the initiation step is the critical step in the polymerisation process reported by Yongyat et al.7 Formation of α(1–6) bonds requires the selective activation of the first monomer at the O6 position, with favourable energies (increased quantities), leading to longer, more regio- and stereoselective linear chains. This can be achieved with the bulky initiator, TMSOTF and a stabilizing capping group (i.e. phenyl ring) that can effectively stabilise the resulting carbocation ion of the intermediate via resonance. It is also clear that the positively charged Lewis acid Si(CH3)3)+ results in lower energy intermediates than does neutral BF3 by providing more effective stabilization of the carbocation center. This is particularly important since the reaction is performed in non-polar solvent which cannot provide effective stabilization of the high energy species formed over the course of the reaction.The dimerization step of the O6-intermediate leads to the more selective formation of α(1–6) bond since the C1 position in this intermediate is the preferential point of attack from the most sterically unhindered O6 atom of another monomer. This is because the C1 position is less sterically hindered and the resulting transition state can be more effectively stabilized by the two adjacent oxygen atoms. With less sterically hindered initiators (i.e. BF3), or monomers with more poorly stabilizing, and bulky capping groups (i.e. tert-butyl), the relative preference for the α(1–6) glycosidic bond decreases. As a result, the polymerisation process proceeds in a disorderly fashion, via a sterically hindered and less energetically favourable pathway, resulting in shorter, more disorderly chains.
The information derived from such models could be used to guide the selection of the more optimal substituent at the orthoester position (R group in Fig. 1), and the initiators. Such calculations are currently in active use to design further experiments. QC methods are often seen as rather unapproachable, yet as seen here, they can provide a means to post-rationalise complex results and provide a method to quickly simulate alternative reagents in a matter of days, where a comparable synthetic approach may take days, weeks or even months.
Acknowledgements
Financial support for this work from the Chulabhorn Research Institute and the Thailand Research Fund (RSA5480016 for MPG and RSA5580059 for SB) are gratefully acknowledged.Notes and references
- M. W. Borgdorff, K. Floyd and J. F. Broekmans, Bull. W. H. O., 2002, 80, 217–227 Search PubMed.
- S. H. Kaufmann, Nat. Rev. Immunol., 2006, 6, 699–704 CrossRef CAS.
- P. Andersen, Nat. Rev. Microbiol., 2007, 5, 484–487 CrossRef CAS.
- V. Briken, S. A. Porcelli, G. S. Besra and L. Kremer, Mol. Microbiol., 2004, 53, 391–403 CrossRef CAS.
- N. N. Driessen, E. J. M. Stoop, R. Ummels, S. S. Gurcha, A. K. Mishra, G. Larrouy-Maumus, J. Nigou, M. Gilleron, G. Puzo, J. J. Maaskant, M. Sparrius, G. S. Besra, W. Bitter, C. M. J. E. Vandenbroucke-Grauls and B. J. Appelmelk, Microbiology, 2010, 156, 3492–3502 CrossRef CAS.
- P. Hutacharoen, S. Ruchirawat and S. Boonyarattanakalin, J. Carbohydr. Chem., 2011, 30, 415–437 CrossRef CAS.
- C. Yongyat, S. Ruchirawat and S. Boonyarattanakalin, Bioorg. Med. Chem., 2010, 18, 3726–3734 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- J. B. Foresman and A. Frisch, Exploring Chemistry With Electronic Structure Methods: A Guide to Using Gaussian, Gaussian Inc., Wallingford CT, ISBN0963676938, 1996 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Rev C01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- H. Valdés, V. Klusák, M. Pitoňák, O. Exner, I. Starý, P. Hobza and L. Rulíšek, J. Comput. Chem., 2008, 29, 861–870 CrossRef.
- D. Gleeson, B. Tehan, M. P. Gleeson and J. Limtrakul, Org. Biomol. Chem., 2012, 10, 7053–7061 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The 3D coordinates of the optimizated structures (mol2 format) and the tables of the enthalphies and free energies are provided. See DOI: 10.1039/c2md20178j |
| This journal is © The Royal Society of Chemistry 2013 |