Stabilised G protein-coupled receptors in structure-based drug design: a case study with adenosine A2A receptor
Stephen P.
Andrews
* and
Benjamin
Tehan
Heptares Therapeutics Limited, BioPark, Broadwater Road, Welwyn Garden City, Hertfordshire AL7 3AX, UK. E-mail: steve.andrews@heptares.com
First published on 11th September 2012
Abstract
Significant progress has been made with stabilising G protein-coupled receptors (GPCRs) in recent years, and this has enabled the structures of several members of this important target class to be solved by X-ray crystallography. High resolution structural data is improving our understanding of GPCR activation and function, and is beginning to impact the drug discovery community. StaR® proteins are GPCRs which have been minimally engineered to impart thermostability. StaRs® are stable in detergent micelles and are suitable reagents for use with X-ray crystallography, biophysical screening techniques and fragment screening. This article reviews the role that StaRs® can play in the identification and optimisation of novel ligands for GPCRs by examining a specific case in which a preclinical candidate for the treatment of Parkinson's disease was developed. Compound 13 was identified following the virtual screening of experimentally enabled homology models of adenosine A2A receptor (A2AR) and was subsequently optimised using the structural insight provided by X-ray crystallography and Biophysical Mapping of closely related molecules. Compound 19 is an exemplar from this chemical series which displays low molecular weight and high oral bioavailability; it has a good pharmacokinetic profile and is highly efficacious in preclinical models of Parkinson's disease.
Introduction
The first structure of a G protein-coupled receptor (GPCR) to be solved by X-ray crystallography was that of rhodopsin, in 2000,1 but it was another seven years before the next GPCR structure was elucidated.2 However, exciting progress has been made since 2007, and during this productive period the X-ray structures of β1 (ref. 3) and β2 (ref. 4) adrenergic receptors, adenosine A2A receptor,5 chemokine CXCR4 receptor,6dopamine D3 receptor,7histamine H1 receptor,8 muscarinic acetylcholine M2 (ref. 9) and M3 (ref. 10) receptors, sphingosine 1-phosphate receptor 1,11 and the opioid receptors δ,12 κ,13 μ14 and nociceptin,15 have all been solved. Collectively, these 14 important receptors are activated by stimuli as diverse as alkaloids, hormones, light, neurotransmitters and cytokines; as drug targets they are implicated in conditions ranging from cardiovascular disease to cancer, and central nervous system disorders such as Alzheimer's disease; yet they constitute only a minor proportion of the GPCR superfamily. Indeed, there are some 800 known GPCRs, including olfactory receptors, and it has been estimated that they are the biological targets for >30% of all marketed drugs.16Initially, these high resolution structures were solved with the receptors in inactive states; however, several structures have now been solved with GPCRs in active-state conformations,17–19 and as co-complexes with agonist, inverse agonist and neutral antagonistligands, as well as with nanobodies,20 antibodies21 and G proteins.22 These recent advances in generating high-quality GPCR structural information have sparked much interest in the field and have greatly enhanced our understanding of receptor activation and function.16,23–30 Furthermore, this structural insight may now be used to drive the rational design of drug-like molecules with high degrees of potency and selectivity for their intended GPCR target by expediting the analysis of shape- and electrostatic-complementarity between the ligands and their binding sites. Such structure-based drug design (SBDD) approaches have been highly successful with soluble targets such as kinases and proteases, but the inherent challenges with stabilising, isolating and purifying GPCRs have precluded their use with these techniques until very recently.31–33
StaR® proteins are GPCRs which have been thermostabilised by the introduction of a small number of point mutations.34 These modified proteins are stable in detergent micelles and can be removed from their native cell membranes, purified, crystallised and used with a number of biophysical screening techniques.
This article reviews the applications of StaR® proteins in the discovery and development of new ligands for the adenosine A2A receptor (A2AR). These reagents have allowed, for the first time, the application of Biophysical Mapping techniques and the generation of X-ray structures of small molecules in co-complex with GPCRs during an on-going medicinal chemistry campaign. Furthermore, these techniques have enabled SBDD of a chemical series identified by virtual screening of A2AR which has led to the nomination of a preclinical candidate for the treatment of Parkinson's disease (PD).
Adenosine A2A receptor and Parkinson's disease
GPCRs can be sub-categorised into families according to their relative sequence similarities; of these, family A (also known as the rhodopsin family and comprising 701 receptors, including the olfactory receptor subclass), is by far the largest.24 Family A GPCRs are activated by a diverse range of stimuli, including low molecular weight amines, prostaglandins, peptides, glycoproteins and light, and it has been estimated that they are the therapeutic targets for 27% of all marketed small molecule drugs.35The four adenosine receptor sub-types (A1, A2A, A2B and A3) all belong to GPCR family A and are activated by extracellular adenosine. Selective agonist and antagonistligands are known for all of these receptors and have been reviewed elsewhere;36 a representative selection of A2AR ligands is shown in Fig. 1. The known A2AR agonists are almost exclusively derived from purine nucleosides and have been examined therapeutically as anti-inflammatory agents. Regadenoson is the only clinically approved A2AR agonist and is used as a coronary vasodilator in myocardial perfusion imaging.37
![A representative selection of A2AR ligands. A2AR agonists are typically closely related to adenosine, such as NECA and the coronary vasodilator, regadenoson.37 A2AR antagonists are generally derived from purine bases such as xanthine (e.g.caffeine, XAC and istradefylline95), adenine (e.g. ZM241385) or guanine (e.g. vipadenant97); some antagonists which are not derived from purines have recently shown efficacy in preclinical models of PD, such as Lu AA47070,98,99 SYN-115 (ref. 100) and 2-amino-8-(4-methylpiperazine-1-carbonyl)-4-phenyl-5H-indeno[1,2-d]pyrimidin-5-one.101 Preladenant is phase III clinical trials for the treatment of PD.45](/image/article/2013/MD/c2md20164j/c2md20164j-f1.gif) | ||
| Fig. 1 A representative selection of A2AR ligands. A2AR agonists are typically closely related to adenosine, such as NECA and the coronary vasodilator, regadenoson.37 A2AR antagonists are generally derived from purine bases such as xanthine (e.g.caffeine, XAC and istradefylline95), adenine (e.g. ZM241385) or guanine (e.g. vipadenant97); some antagonists which are not derived from purines have recently shown efficacy in preclinical models of PD, such as Lu AA47070,98,99 SYN-115 (ref. 100) and 2-amino-8-(4-methylpiperazine-1-carbonyl)-4-phenyl-5H-indeno[1,2-d]pyrimidin-5-one.101 Preladenant is phase III clinical trials for the treatment of PD.45 | ||
A2AR antagonists have generally been derived directly from xanthine, or contain heterocyclic scaffolds which are closely related to other purines such as adenine.36,38 Typical examples include the purine-like derivative ZM241385, a potent and selective A2AR inverse agonist which has been used widely as a research tool,39,40 and the xanthine derivative, caffeine, a weak, non-selective adenosine receptor antagonist.
Parkinson's disease (PD) is a neurological disease in which motor function is progressively impaired by the gradual loss of dopaminergic neurons in the striatum.41 Current treatments for PD rely upon restoring dopamine D2 receptor (D2R) signalling, most commonly by administering Levadopa (L-DOPA, a dopamine precursor).42 This can initially be effective at restoring motor function; however, efficacy often reduces as the disease progresses and the benefits of the treatment are generally accompanied by undesirable side effects such as dyskinesias.
A2AR antagonists have been evaluated as alternative therapeutic agents for the treatment of PD; they have shown efficacy in several preclinical models of PD and advantages in reducing the dyskinesias experienced by patients undergoing standard dopamine replacement therapies.38,43 A2AR is co-expressed with D2R in the striatum where the functional effects of the two receptors are in opposition: i.e. A2AR antagonism and D2R agonism invoke complementary mechanisms for the treatment of PD. A1R is widely distributed throughout the CNS and periphery; however, the low density of A2BR and A3R in the brain, as well as the relatively discrete localisation of A2AR in the striatum with respect to dopamine receptors, make A2AR an attractive CNS target for the treatment of PD.44
Preladenant (SCH420814; Merck) has shown efficacy in multiple preclinical models of PD, including the haloperidol-induced catalepsy and 6-hydroxydopamine-lesion turning models in rodents, as well as the MPTP-lesion model in cynomolgus monkeys.45 Preladenant is currently being evaluated in phase III clinical trials for the treatment of PD.
Part I: applications of stabilised GPCRs
This section describes the thermostabilisation process for generating StaR® proteins and considers the implications that this has on receptor conformation, using A2AR as a specific example. Applications of A2A StaRs® are then reviewed in the context of drug discovery, from enabling the refinement of homology models for virtual screening, to facilitating the application of crystallography and biophysical screening techniques, including Biophysical Mapping. Specific examples are delineated with standard ligands such as XAC, ZM241385 and caffeine. These techniques are then revisited in Part II, which reviews their applications in the discovery and optimisation of a novel, non-purine series of A2AR antagonists for the treatment of Parkinson's disease.GPCRs and their stabilisation
GPCRs (also known as seven transmembrane receptors or 7TMs) are located within cell membranes and are broadly characterised as folded proteins possessing an extracellular N-terminal domain, an intracellular C-terminal domain and a transmembrane domain comprising seven pseudo-parallel helices which are linked together by intracellular and extracellular loops (Fig. 2).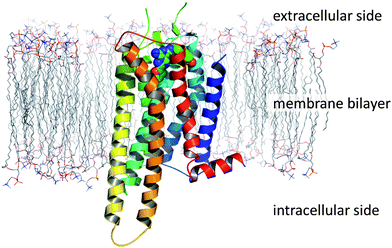 | ||
| Fig. 2 The topology of a GPCR, as exemplified by the crystal structure of A2A StaR2 in complex with ZM241385 (PDB code 3PWH; simulated lipid bilayer). | ||
These membrane-spanning proteins are inherently unstable when isolated from cells, whereupon they rapidly unfold. However, this stability can be modulated with point mutations introduced viaprotein engineering, or by preparing co-complexes of GPCRs with antibodies, both of which can facilitate crystallisation.46 Alternatively, chimeric fusion proteins have been prepared with T4 lysozyme47 (which has a high propensity to crystallise), and other fusion partners48 which have allowed the structures of several GPCRs to be solved.29,49
Thermostabilisation by site-directed mutagenesis
Site-directed mutagenesis (SDM) has been used to identify point mutations which dramatically increase the stability of GPCRs in detergent micelles and simultaneously lock the receptors into an antagonist, inverse agonist or an agonist conformation. The receptors generated during this process are known as StaRs®.34A2AR is soluble in detergents such as dodecylmaltoside (DDM), and the binding activity of the inverse agonist ZM241385 is maintained in this system at 4 °C.50 For wild-type (WT) A2AR solubilised in 2% DDM, the temperature at which 50% of binding is retained after 30 minutes of incubation (melting temperature, Tm) is 23 °C. However, the stability of the receptor was significantly increased following SDM experiments which identified point mutations to improve its apparent Tm. In some cases it is possible to predict these mutations based on the large in-house database of known stabilising mutations or from those in the public domain.46 Stabilising mutations are then combined to give a StaR®.
A2A StaR1 contained four thermostabilising mutations (A54L, T88A, K122A and V239A), and its Tm was found to be 12 °C higher than that of WT A2AR in decylmaltoside (DM; Table 1).34 A2A StaR1 was sufficiently stable for use with some biophysical screening technologies. A2A StaR2 contained a further four mutations (R107A, L202A, L235A and S277A) and was found to have a Tm that was 25 °C higher than that of WT A2AR in DM; it was stable enough for crystallisation.51
StaR® proteins can be prepared in different pharmacological conformations (i.e. active or inactive states of the receptor), and it is possible to control this by judicious choice of the ligand used in the thermostabilisation process. This has particular relevance when generating StaRs® for use as reagents in drug discovery as, for example, StaRs® in agonist (active) conformations generally bind to agonistligands with similar or increased affinities to the corresponding WT receptor, whereas they typically have lower affinity towards inverse agonistligands.18
StaR1 and StaR2 were both stabilised in inverse agonist (inactive) conformations (Table 1). StaR1 was found to bind to a number of neutral antagonists and inverse agonists with similar affinities to the native receptor (Fig. 3); however, the agonists CGS21680 and NECA showed a loss of affinity for StaR1 of approximately 100-fold. StaR2 was found to bind to antagonists and inverse agonists with higher affinity than the WT receptor whilst showing a lowered affinity towards agonists (Fig. 3).
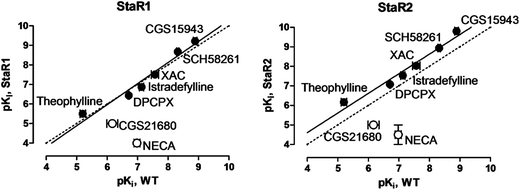 | ||
| Fig. 3 Binding affinities of A2AR ligands measured against StaR1 and StaR2, each plotted against affinities measured at WT A2AR. Filled circles denote neutral antagonist and inverse agonistligands; open circles denote agonistligands. | ||
Several A2A StaRs® have been generated in agonist (active) conformations, including GL26 and GL31.18,52 GL26 contained four thermostabilising mutations (L48A, Q89A, T65A and A54L) and was found to have a Tm 20 °C higher than that of WT A2AR in DM (Table 1). GL31 contained the same four thermostabilising mutations as well as a mutation to remove a potential glycosylation site (N154A). GL31 was sufficiently stable to be co-crystallised with the agonistligandsadenosine and NECA.52 As might be expected from the discussion above, GL26 and GL31 bind to agonistligands with similar affinity to the native receptor, whereas inverse agonistligands show a marked decrease in their affinity for these receptors in active-state conformations (e.g. ZM241385 shows ∼350-fold drop in affinity for GL26 vs. WT A2AR).18 However, neutral antagonistligands, such as istradefylline, bind to WT A2AR and GL26 with similar affinities.
The ability to evaluate the pharmacological profile of a ligand in binding assays with active and inactive StaRs® is a potentially powerful tool, particularly when used in combination with molecular modelling. This ‘reverse pharmacology’ approach contrasts with the conventional requirement to determine a ligand's pharmacology in a cellular or phenotypic assay, and may herald a complementary new approach to GPCR drug discovery. This technique enables the identification of compounds which have an ability to select either an active or inactive receptor conformation, independently of the cellular system. This topic will be the subject of future publications.
GPCR homology modelling
The recent success in generating crystal structures of GPCRs has subsequently led to an increase in the number and quality of GPCR homology models that have been constructed.29,53,54 However, the sheer volume and range of sequence identities found within this receptor superfamily, coupled with the conformational flexibility that they exhibit (particularly in the loop regions), mean that GPCR homology modelling remains a formidable task.53,55–57 For this reason, techniques such as molecular dynamics and knowledge-based constraints are often applied to refine models. Furthermore, SDM data can be used to highlight which residues within the receptor play a key role in the binding of ligands and can facilitate the generation of ‘experimentally enabled’ homology models.Experimentally enabled homology models of A2AR
Experimentally enabled homology models are homology models which are refined by comparing the sequence identity of the receptor in question with known GPCR crystal structures and taking into account specific motifs and structural features as well as mutagenesis data. Typically, each residue of the receptor is mutated to multiple side chains during the thermostabilisation process, resulting in the formation of >1000 mutants; the wealth of SDM data generated has utility both in designing StaR® constructs as well as in building and refining homology models. In particular, the data can be used to highlight which residues affect ligand binding and can enable a much better overall placement of the ligand in the receptor's binding site (Fig. 4). Where available, Biophysical Mapping data can also be used to refine models and optimise binding poses (vide infra).![The residues which were found to reduce the binding of [3H]-ZM241385 during the StaR® stabilisation process and are within 5 Å of residues known to affect the binding of triazolotriazineantagonists are shown in green (mapped onto the crystal structure 3PWH with ZM241385 shown in pink).62,63](/image/article/2013/MD/c2md20164j/c2md20164j-f4.gif) | ||
| Fig. 4 The residues which were found to reduce the binding of [3H]-ZM241385 during the StaR® stabilisation process and are within 5 Å of residues known to affect the binding of triazolotriazineantagonists are shown in green (mapped onto the crystal structure 3PWH with ZM241385 shown in pink).62,63 | ||
Experimentally enabled homology models of A2AR have been created using both MODELLER58 and MOE,59 with manual readjustment of the clustalW60 alignment onto the β1 adrenergic GPCR crystal where necessary.61 Special consideration was given to residues which were shown to affect ligand binding in both published SDM studies62,63 and during the thermostabilisation process, and the majority of these residues were found to line the anticipated ligand binding site. Further validation was obtained by docking a small set of known A2Aligands (including ZM241385) into each of the structures and comparing these models' enrichment rates against a decoy set of similarly sized molecules. Thus, two models were used for parallel virtual screening experiments with no added constraints (virtual screening results are discussed in Part II).
Virtual screening of A2AR
3D X-ray crystallographic data of A2AR has been available since 2008 and its availability has led to the publication of several articles detailing the virtual screening of ligands against this receptor.64,65 More recently, molecular modelling and virtual screening have also been performed with insights from active-state crystal structures of A2AR.66,67In some cases, hit rates greater than 35% have been achieved, highlighting the significant improvements that can be achieved with insights from crystal structures.68,69 However, significant user bias is typically used in the preparation of these models; for example, in one case, the dipole moment of a residue shown to be essential for all previously identified ligands was modified,68 and in another case, side chains were tuned and “structural waters” were retained to ensure high hit rates in test sets prior to engaging in final virtual screens.69
It is common practise to prepare models in this way to ensure that significant hit rates are obtained during virtual screening, and post processing of pharmacophores is often carried out in order to maximise the chance of success. However, as others have pointed out, care must be taken when preparing a model and post-processing the data generated so that ligands of interest are not excluded.70 These authors review a virtual screen based on the β2 receptor crystal structure which gave a hit rate of ∼30%, as well as numerous other virtual screens using homology models of GPCRs that achieved hit rates ranging from <1% to ∼45%. This variation is not surprising; as an earlier docking and scoring assessment study has highlighted, significant variation is often observed in virtual screening hit rates across a wide range of targets.71 This study showed that docking programs may be successful in generating poses similar to known binding modes; however, associated scoring functions were less successful at correctly identifying them. It was also noted that, while the application of knowledge about a target can improve hit rates, it is often to the detriment of the diversity of the leads identified.
During our own virtual screening against the experimentally enabled homology models of A2AR described above, no residue-specific constraints, modified weightings or structural waters were used, so as to maximise the novelty and diversity of the ligands identified (vide infra).
Screening GPCRs with biophysical techniques
Biophysical screening techniques can be highly sensitive and offer the ability to detect weak ligand–receptor interactions and perform fragment screening.72,73 These methods may be used to complement more conventional screening approaches and have been widely applied in the screening of soluble protein targets such as kinases and proteases. Membrane proteins have generally been much less amenable to biophysical screening; however, recent advances in generating stabilised GPCRs have facilitated the application of these techniques and offered new opportunities for the discovery of drugs for these receptors.Surface plasmon resonance
Surface plasmon resonance (SPR) can be used to measure protein–ligand binding affinities and kinetics in real time;74 however, the method requires the receptor to be captured onto a suitable biosensor surface, placing high demand on the protein's stability. As such, StaRs® are suitable candidates for these applications and A2A StaR1 has been captured onto nickel nitrolotriacetic acid-functionalised chips via C-terminal His-10 affinity tags and screened by SPR.75,76 Binding affinities of a set of known A2AR ligands were measured directly by SPR and the data for each ligand could be fitted to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 interaction model. The affinities of this set of ligands ranged from 5.7 μM to 0.68 nM and showed good agreement with the values measured on WT A2AR in cellular assays.
:1 interaction model. The affinities of this set of ligands ranged from 5.7 μM to 0.68 nM and showed good agreement with the values measured on WT A2AR in cellular assays.
Once validated, this SPR screening protocol was used to assess the binding of a library of fragment-sized molecules.75 Single-point screening was initially performed at 200 μM with injections of XAC as a positive control at regular intervals. XAC was found to perform consistently and the receptor remained active throughout the study. Fragment hits were re-analysed in a concentration–response format to determine their binding affinities, which ranged from 10–5000 μM.
Target-immobilised NMR screening
Target-immobilised NMR screening (TINS) is a sensitive technique which has been used to detect the binding of ligands to various classes of proteins, including stabilised GPCRs.75 TINS measures the binding of small molecules (MW typically <300 Da) to immobilised target and reference proteins in parallel, in a dual cell NMR sample holder. Binding to the target receptor is observed as a reduction in the 1H NMR signal of the ligand, and the 1H NMR spectrum recorded with the reference protein is subtracted from that of the target protein to mitigate effects caused by non-specific binding.A2A StaR2 has been immobilised on sepharose resin and screened by the TINS method using one third of the Zobio fragment collection (molecules selected at random). Thus 531 fragment-sized molecules were screened at 500 μM using OmpA (a stable membrane protein) as a reference protein.77 Using a target/reference ratio of ≤0.7 for the well-resolved 1H NMR signals, 94 fragments were found to bind specifically to StaR2 (conversely, only 6 fragments were found to bind specifically to OmpA which is known to exhibit minimal specific binding to small molecules).
Having established which molecules bound to A2AR via the TINS technique, the 94 hit molecules were screened against WT A2AR in radioligand binding assays with [3H]-ZM241385 and [3H]-NECA.77 Competitive binding and dissociation rates of the radioligands were examined with the fragments in order to determine whether they were acting as orthosteric or allosteric ligands.
Seven competitive orthosteric ligands with diverse chemical structures were identified, with IC50 values measuring between 70 and 1000 μM in concentration–response studies with [3H]-ZM241385. Several non-competitive ligands were also identified. Measurement of their effects on the dissociation rates of radioligands at both A2AR and A1R suggested that the TINS methodology had identified positive and negative allosteric modulators which were selective for A2AR.77
Capillary electrophoresis
Capillary electrophoresis (CE) is a high resolution, microscale technique which uses a capillary and electrodes with a high voltage power supply to separate charged and uncharged particles. CE has been used to detect ligand–protein interactions and has shown utility in microscale binding assays for the detection of weak ligands, including fragment-sized molecules.78,79 In a proof-of-concept study, a selection of known A2AR ligands with a wide range of potencies, including the fragment-sized molecule, caffeine, were found to bind to A2A StaR1, whereas negative control ligands did not bind to the receptor (Fig. 5).80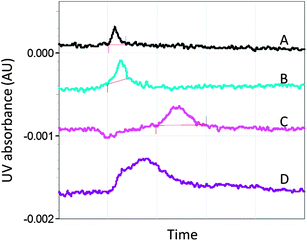 | ||
| Fig. 5 Electropherograms showing the relative migration time of the A2AR antagonist, 8-cyclopentyl-1,3-dipropylxanthine (DPCPX), under different conditions (corrected for differences in electro-osmotic flow).80,102 (A) DPCPX is injected into the capillary and gives a single peak. (B) DPCPX is injected with the competitive ligand, caffeine, present in both the injection buffer and running buffer. In this control run, caffeine does not produce an additional peak or cause a change in the DPCPX peak profile.102 (C) DPCPX is injected into the capillary containing A2A StaR1. The broadening and shift of the peak are indicative of an interaction between A2A StaR1 and DPCPX. (D) DPCPX and caffeine are injected into the capillary containing A2A StaR1 with caffeine in the running buffer. The DPCPX peak partly returned to its unbound peak profile, indicating that competitive binding had occurred between the two ligands and the receptor. Negative control ligands such as aspirin were found not to affect the profile of DPCPX in this way. | ||
Biophysical Mapping
Biophysical Mapping (BPM) can provide insight into the key structural interactions required for a ligand to bind to a GPCR and valuable structural information in the absence of a crystal structure. The use of a stabilised receptor is crucial to the success of this methodology which uses SPR screening techniques.Using the chosen StaR® as a template, it is possible to make an additional point mutation in the binding site of interest and compare the effect of this mutation on the binding of various ligands. By making a panel of such StaRs®, each with a different binding site mutation, it is possible to ‘map’ and rank the residues required for ligand binding. This can assist with prioritisation where a number of predicted binding modes exists, and can allow the comparison of binding interactions across different chemical series. The choice of mutants to be prepared can be influenced by SDM data and by residues thought to be of importance from molecular modelling.
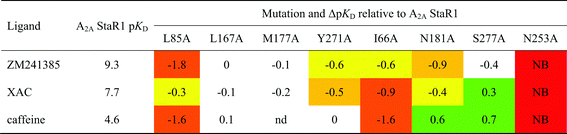
In the case of adenosine A2AR, a set of eight StaRs® was prepared, each with a different point mutation in the orthosteric binding site of the StaR1 backbone (Table 2). The receptors were then individually captured onto SPR chips to allow cross-screening against an array of ligands from different chemotypes and with a wide range of binding affinities.81
|
The study provided key structural information which facilitated lead optimisation of compounds identified by virtual screening before X-ray crystallography data was available for this receptor. The BPM process was also used to build knowledge and gain an understanding of the modes of binding of non-proprietary A2Aligands, such as ZM241385 and the xanthineligands XAC and caffeine (Fig. 6 and 7).
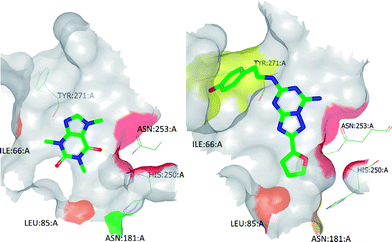 | ||
| Fig. 6 BPM heat maps of caffeine (left) and ZM241385 (right) to show the most significant ligand-binding site interactions. Heat map colouring is explained in Table 2. | ||
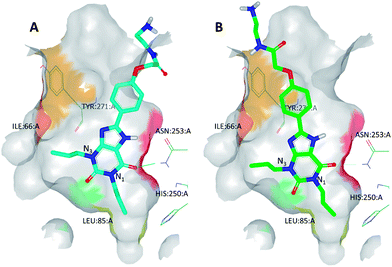 | ||
| Fig. 7 (A) The best-scoring binding mode of XAC observed during molecular modelling studies; (B) the refined binding mode of XAC which takes into account BPM data. This refined binding mode was later found to be broadly in agreement with the binding mode observed by X-ray crystallography (see Fig. 9). Heat map colouring is explained in Table 2. | ||
For example, docking XAC into the homology model of A2AR initially resulted in several plausible binding modes but these could be more accurately ranked by taking into account the BPM data generated for this ligand (Fig. 7). SPR screening of XAC against the panel of binding-site mutants revealed that, in particular, mutation to alanine of Ile66, Leu85, Tyr271 or Asn181 resulted in reduced binding, while the mutant S277A showed a higher affinity towards XAC than the parent receptor, StaR1. These data are in agreement with the binding mode shown in Fig. 7B, in which one of the hydrophobic propyl chains of XAC (N3-propyl) sits in a lipophilic pocket lined by Ile66 and the other (N1-propyl) sits in a pocket containing Ser277. Thus the mutant I66A (which has a reduced lipophilic contact with N3-propyl compared to StaR1) shows a reduction in binding, whereas S277A (which, after removal of the polar hydroxyl group, has a more favourable interaction with N1-propyl) shows an increase in binding relative to StaR1. The binding mode predicted during this process was later found to be broadly in agreement with the X-ray crystal structure of XAC in complex with A2AR (vide infra).
Crystallisation of A2AR
The pace of obtaining GPCR structures is rapidly increasing as crystallisation techniques are honed and matured but, nonetheless, generating and solving the X-ray structures of GPCRs in co-complex with their ligands remains a challenging and resource-intensive exercise. Advances in improving the relatively low expression levels of these receptors and increasing their stability in detergent micelles have contributed greatly to this field,82 as have developments with detergents and crystallisation techniques, such as lipidic cubic phase (LCP) crystallisation.16,49,83,84 To date, all GPCR crystal structures have been obtained using co-crystallisation techniques rather soaking experiments. Further developments in the field may allow apo structures to be solved, for example through further thermostabilisation the receptor; however, in this case, the requirements for additional thermostability must be balanced with the number of required mutations.The first A2AR structure to be reported was of an inactive receptor conformation, solved to 2.6 Å resolution with ZM241385 bound, using the T4 lysosyme technology (PDB code 3EML).5 Structures of A2AR have since been solved in both active and inactive conformations, including by using fusion protein (T4 lysosyme) techniques in combination with LCP crystallization,5,19 as well as with thermostabilisation51,52,85 and antibody co-complexation21 techniques.
Crystallisation of A2A StaR2 in complex with antagonistligands
The high thermostability of A2A StaR2 has enabled its crystallisation with several ligands of varying degrees of size and potency, including ZM241385 (PDB code 3PWH).51 The binding mode of this ligand in the A2A StaR2 structure showed some similarities to that seen in the T4 lysosyme structure, 3EML; for example, the triazolotriazine core of ZM241385 was located between Phe168 and Ile274, and the furan was seen to be H-bonding to Asn253 (Fig. 8; the phenyl ring of Phe168, not shown, sits in the plane of the page, π-stacking with the triazolotriazine).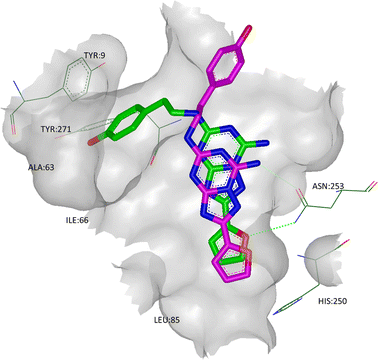 | ||
| Fig. 8 The X-ray crystal structure of ZM241385 in co-complex with A2A StaR2 (ligand shown in green; PDB code 3PWH). Overlaid in pink is the binding mode of ZM241385 observed in the T4 lysosyme structure (PDB code 3EML). | ||
In the StaR® structure, the phenolic ring of ZM241385 binds in a cleft between helices TM1, 2 and 7 (defined by Glu13, Ala63, Ile66, Ser67, Leu267, Met270, Ile274, His278, and Tyr271), with the hydroxyl group H-bonding to the backbone carbonyl oxygen of Ala63.51 This additional H-bond may, in some part, explain the relatively high affinity of ZM241385 when compared to its non-hydroxylated counterpart, as well as its high selectivity for A2AR vs. A1R.57 In the T4 lysosyme and antibody structures (which show close overall agreement in their ligand orientations), the phenolic tail of ZM241385 has been placed in solvent, suggesting that there may be multiple binding modes for this ligand.
A2A StaR2 has also been crystallised with two xanthine derivatives: the high affinity ligand, XAC (PDB code 3REY), and the low affinity fragment-sized ligand, caffeine (PDB code 3RFM).51 Both xanthine derivatives show similarities to ZM241385 in that their core heterocycles H-bond to the side chain carbonyl oxygen of Asn253, π-stack with Phe168 and make hydrophobic contacts with Ile274 (Fig. 9). Interestingly, however, the side chain amide of Asn253 in the XAC structure undergoes a rotation to optimise the H-bond formed. Furthermore, XAC extends much further out of the core of the binding site, where the propyl side chains can be seen to interact with Ile66 to one side, as well as Asn181 and Leu85 to the other. These interactions are consistent with BPM data (vide supra), as are the interactions between the phenyl group and polar tail with Tyr271. The polar tail of XAC binds in the cleft between TMs 1, 2 and 7 (in a similar way to ZM241385), although the residues in question are in different rotameric states (particularly Tyr9 and Tyr271), effectively creating a deeper and narrower cleft than is seen in the 3PWH structure. Interestingly, the cleft between TMs 1, 2 and 7 seen in the XAC structure is identical to that seen in the structure with caffeine, even though caffeine does not bind in this region, suggesting that this is a low energy orientation of these residues.
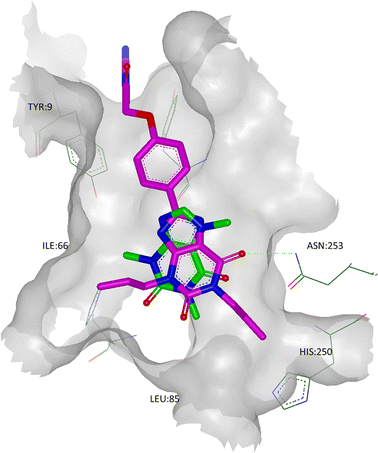 | ||
| Fig. 9 The crystal structure of A2A StaR2 in co-complex with caffeine (ligand shown in green; PDB code 3RFM). Overlaid in pink is the X-ray structure of XAC from its co-complex with A2A StaR2 (PDB code 3REY) to show the similarities in the placement of the xanthine cores. Note that in the 3REY structure, the amide head group of Asn253 had rotated 180°. | ||
These inactive A2A StaR2 co-complexes with the inverse agonists ZM241385 and XAC, and the neutral antagonist, caffeine, differ significantly from the active-state A2AR structures which have been produced either with T4 lysozyme technology (bound to UK-432097 (ref. 19)) or the A2A StaR®, GL31 (bound to NECA and adenosine52). These differences shed some light on the activation process of A2AR, as the binding of agonistligands results in a significant contraction of the binding site when compared to that seen with inverse agonist or neutral antagonistligands. This contraction results from an inward shift of TM7 toward the core of the receptor, an extracellular upward movement of TM3 by 2 Å, an inward movement of the proline bulge in TM5, and the rigid body rotation of TM6 around Phe242. At a molecular level, changes in the binding site from the ground state A2A StaR2 structure to the active state GL31 are driven by the agonists forming H-bonds from the adenine core to Asn253, and from the ribose group to Ser277 and His278.
By examining the binding of agonists, inverse agonists and neutral antagonists to both active and inactive StaRs® in this way, it is possible to understand the subtleties of the different interactions that these ligand classes make with the receptor. Indeed, by solving crystal structures of these different receptor conformations, it is possible to visualise the movements of the receptor and understand the driving forces of activation, which, in turn, can expedite the discovery and development of pharmaceutical agents.86
Part II: structure-based drug design with StaRs®
Part I revealed how stabilised GPCRs can enable the application of various screening techniques with these challenging receptors, thus providing a means to identify novel chemical starting points for a drug discovery programme, for example by facilitating the use of biophysical screening techniques and fragment screening. These approaches are complemented by the ability to generate experimentally enabled homology models from SDM data. Such models expedite the in silico screening of compound libraries, as well as the docking of hit and lead molecules, and they can be further refined by taking into account data generated during the BPM process. Furthermore, highly stabilised GPCRs can be crystallised from detergents and this has allowed the determination of the 3D structure of these receptors as co-complexes with pharmaceutically relevant ligands such as ZM241385, XAC and caffeine.The remainder of this article will show how the application of these techniques facilitated the rapid and efficient optimisation of a hit A2AR antagonist which was identified by virtual screening of experimentally enabled homology models. The techniques described in Part I were used in the development of a preclinical candidate for the treatment of PD.
Identification and optimisation of novel A2AR antagonists
Virtual screening of the experimentally enabled A2AR homology models was performed61 with 545000 compounds from a collection of commercial catalogues (CAP) and a subset of BioFocus' SoftFocus™ library, using Glide software.87,88 The CAP set had been pre-filtered to remove compounds with unwanted functional groups and retain those which had properties that were anticipated to be required of leads for a GPCR in the CNS. From the outset, a major objective was to identify novel chemotypes as A2AR antagonists, in particular, those which did not contain purine-like templates or furans (both of which were prevalent amongst A2AR antagonists).Following 3D visualisation and post-processing analyses, 230 compounds were screened against WT A2AR in a radioligand binding assay with [3H]-ZM241385; 20 compounds were active at concentrations below 55 μM, which corresponded to a 9% hit rate.61 Of particular note, the most potent compound showed pKi = 8.5, ligand efficiency (LE)89,90 = 0.52 and lipophilic ligand efficiency (LLE)91 = 5.4 (MW = 310.4, clogP = 3.1). The 10 most potent hits, which showed pKis of 5.53–8.46, LEs of 0.27–0.52 and LLEs between 2.1 and 5.4, are shown in Table 3 and Fig. 10.
| Compound | pKi | LE | LLE | clogP | PSA | MW |
|---|---|---|---|---|---|---|
| 1 | 8.46 | 0.52 | 5.4 | 3.1 | 84.9 | 310.4 |
| 2 | 5.15 | 0.47 | 4.5 | 0.7 | 72.2 | 222.3 |
| 3 | 5.75 | 0.44 | 3.9 | 1.9 | 61.7 | 264.3 |
| 4 | 6.15 | 0.36 | 3.2 | 3.0 | 66.6 | 327.4 |
| 5 | 5.65 | 0.33 | 3.7 | 1.9 | 85.7 | 331.3 |
| 6 | 5.62 | 0.31 | 2.6 | 3.0 | 76.7 | 367.9 |
| 7 | 5.91 | 0.30 | 3.2 | 2.7 | 78.4 | 367.4 |
| 8 | 5.33 | 0.29 | 3.4 | 1.9 | 79.8 | 340.4 |
| 9 | 5.70 | 0.29 | 3.9 | 1.8 | 80.1 | 363.4 |
| 10 | 5.53 | 0.27 | 2.1 | 3.4 | 95.9 | 382.4 |
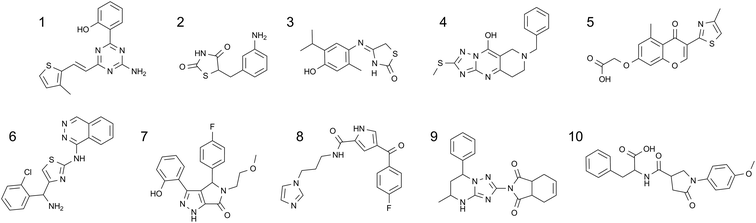 | ||
| Fig. 10 The chemical structures of the top 10 hits identified by virtual screening with experimentally enabled homology models of A2AR. | ||
These molecules represented a diverse range of chemotypes and showed very little resemblance to A2AR antagonists that were known at the time. Indeed, the structural similarity scores of these top 10 hits ranged from 0.31–0.19, when compared to the most similar A2AR ligands in the literature (Tanimoto similarities on FCFP4 fingerprints).61
Optimisation of 1,3,5-triazineantagonists
Compound 1 was the most potent and ligand efficient hit identified during this virtual screening campaign. It was found that LE could be further improved in this series by replacing the vinylthiophene functionality with an isopropyl group, affording fragment-sized molecule 11 (Fig. 11).61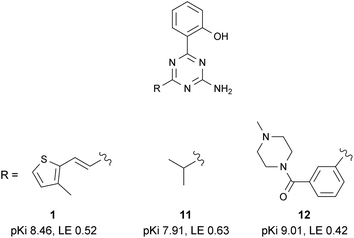 | ||
| Fig. 11 Selected SAR within the 2-amino-1,3,5-triazine series. | ||
BPM analysis of several members of this 1,3,5-triazine series highlighted that the molecules bound deeply within the binding site and revealed a general trend with key interactions between these ligands and binding site residues Leu85, Tyr271, Ile66, Ser277 and Asn181 (see compound series 2 in ref. 81).
Molecular modelling and BPM were used in synergy to further increase the potency and selectivity of this series. For example, it was found that one of the substituents of compound 12 occupies the same part of the binding site as the ribose ring of the natural ligandadenosine. This is a particularly druggable pocket which is known to contain a number of water molecules in energetically unfavourable positions.23
Structure-based drug design of 1,2,4-triazineantagonists
Analysis of the shape and binding mode of 12 led to the proposal that a 3-amino-1,2,4-triazine scaffold would maintain the key hydrogen bond to Asn253 and π-stacking interaction with Phe168, whilst more efficiently accessing the so-called ribose pocket. Indeed, 13 was subsequently identified as a potent ligand for A2AR and a candidate for further optimisation using structure-based methods (Table 4).85|
|
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|---|
| A2AR pKi | 6.93 | 7.29 | 8.40 | 7.67 | 8.11 | 8.85 | 8.46 |
| LE | 0.50 | 0.50 | 0.55 | 0.50 | 0.53 | 0.57 | 0.48 |
| A1R pKi | 6.56 | 7.25 | 7.36 | 6.71 | 7.07 | 9.79 | 7.50 |
| rPPB (%) | ND | 98 | 99 | 98 | 82 | 98 | 92 |
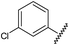
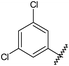
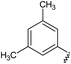
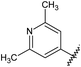
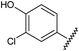
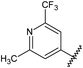
Consideration was given to designing molecules in this series which bind more favourably to A2AR than A1R and, to this end, GRID maps were calculated from homology models for each receptor.92,93 This systematic energetic analysis allowed the comparison of the overall shape, size and electrostatics of the binding sites of each of the receptor sub-types.23 In particular, there are two residues in the A2AR binding site which are correspondingly larger in the A1R binding site (A2AR: Ser277 and Ala59 vs. A1R: Thr and Val, respectively), giving an overall larger pocket for A2AR.
Furthermore, analysis of BPM data and GRID maps generated with H-bond acceptor and donor probes, as well as with water and lipophilic probes, facilitated the rational introduction of both lipophilic and polar substituents onto the template of compound 13 which could increase potency and selectivity. It was found that small substituents at position 5 of the phenyl group on triazine-C6 were well-placed to occupy a lipophilic hot spot within the A2AR/A1R binding sites and gave an overall increase in affinity, whereas substituents at position 3 of the same ring would project out of the tighter A1R pocket, giving selectivity against this off-target receptor (Fig. 12). This hypothesis was supported by SAR for a number of 1,2,4-triazine derivatives in which A2AR potency and selectivity could be increased, for example, as exemplified with chloride substituents in 13, 14 and 15 (Table 4).
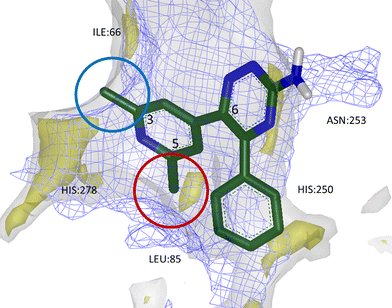 | ||
| Fig. 12 Calculated GRID maps to show the surfaces of A2AR (grey) and A1R (blue mesh) binding sites, as well as lipophilic hot spots within A2AR (yellow). 3,5-Disubstituted aryl groups at triazine-C6 were found to increase potency and selectivity for A2AR; small substituents at position 3 could break the surface of A1R binding site, imparting selectivity (blue circle), while lipophilic groups at position 5 were found to sit favourably within a lipophilic hot spot and increase affinity (red circle). | ||
Furthermore, it was hypothesised that the introduction of a hydrogen-bonding group at the 4-position of the same ring may introduce an additional interaction with His278 or binding-site water molecule. Indeed, pyridyl analogues such as 17 were found to be more potent than their phenyl counterparts and the introduction of this additional heteroatom favourably modified the physicochemical properties of the compounds which showed good solubility, long half-lives in rat liver microsomes and relatively low levels of binding to plasma proteins (Tables 4 and 5).85
| In vitro ADME profile | In vivo rat pharmacokinetic profile | ||||
|---|---|---|---|---|---|
| 1 mg kg−1 (IV) | 2 mg kg−1 (PO) | ||||
| RLM T½ | 86 min | Plasma clearance | 42 mL min−1 kg−1 | T max | 0.4 h |
| rPPB | 92% | V d (ss) | 4.6 L kg−1 | C max | 244 ng mL−1 |
| Kinetic solubility | 35 μM | Terminal T½ | 1.2 h | Terminal T½ | 1.1 h |
| AUCinf | 397 ng h mL−1 | AUCinf | 846 ng h mL−1 | ||
| Brain:plasma (0.5 h) | 3.2 | F po | 100% | ||
| CSF:brain (0.5 h) | 0.036 | ||||
The corresponding phenol derivatives were also found to have very high affinities for A2AR. Compound 18 was found to be a potent ligand in which the hydroxyl substituent was placed directly onto a GRID hotspot identified with the water and H-bond donor/acceptor probes (hotspots not shown; see WaterMap discussion below). This compound also displayed very slow receptor off-rates, as determined by SPR analysis against StaR1 (kd = 0.001 and >1.0 s−1 for compounds 18 and 13, respectively).81 The ability to examine receptor kinetics as part of the screening cascade during lead optimisation can be advantageous and, during this programme of work, facilitated the identification of compounds which later showed potent in vivo efficacy.85
Crystallisation of A2AR in complex with 1,2,4-triazine derivatives
As part of the process of optimising the 3-amino-1,2,4-triazine series, a number of these ligands were crystallised as complexes with A2A StaR2 (e.g.17: PDB code 3UZA and 18: PDB code 3UZC; Fig. 13).51 These two ligands showed similar binding modes to the heterocyclic core of ZM241385, in that the central aminotriazine core π-stacks with Phe168 whilst making hydrophobic contacts with Ile274. Two H-bonds are also observed from the aminotriazine motif: one from the amino group to the side chain carbonyl oxygen of Asn253, and the other from the N4 nitrogen of the triazine ring to the side chain NH2 of Asn253 (Fig. 13). The phenyl substituent at position 5 of the triazine core sits in a hydrophobic pocket defined by residues His250, Leu85, Met177, Asn181 and Trp246, of which Leu85, Asn181 and Met177 were previously examined via BPM.81 In the BPM study, mutation to alanine of either Leu85 or Asn181 was shown to significantly affect the binding of 17 and 18 (previously reported as ligands 3d and 3b, respectively, in ref. 81). The main differences in the binding of these two relatively similar ligands arise from the interactions their differing R-groups make with the protein (Table 4). For example, the hydroxyl group of 18 can be seen to H-bond to His278, deep within the ribose pocket of the receptor. This H-bond has the effect of pulling 18 closer to His278, resulting in a displacement of the atoms of the phenol ring by ∼1 Å when compared to those of the dimethylpyridine ring in 17. This pyridyl substituent of 17 is unable to make a similar H-bonding interaction with His278, thus doesn't move closer to the residue; however, it does occupy the ribose pocket of A2AR, as defined by residues His278, S277A (StaR® mutation), Val84, Aal63 and Ile66. Ser277 is located in the bottom of the ribose pocket and it has been found that mutation of this residue to alanine is highly stabilising for the inactive StaR® conformation. The mutation has been found to have little effect on the pharmacology of inverse agonists and antagonists;51 however, it cannot be ruled out that it has an effect on the binding modes of compounds 17 and 18 which have moderately increased affinities for StaR1 when it contains the additional mutation, S277A (increases in pKD of 0.9 and 0.3 log units, respectively).81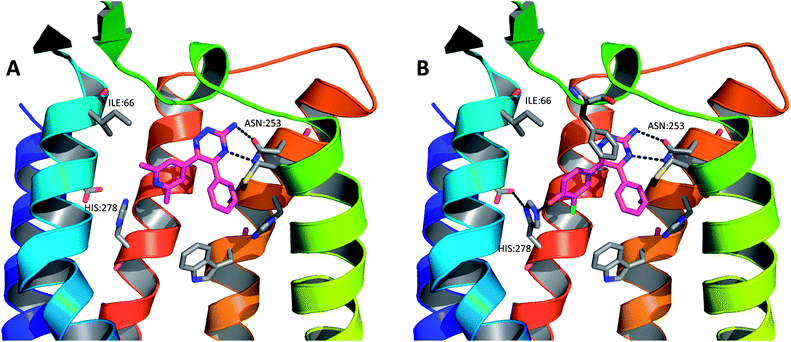 | ||
| Fig. 13 The X-ray crystal structures of (A) compound 17 in complex with A2A StaR2 (PDB code 3UZA) and (B) compound 18 in complex with A2A StaR2 (PDB code 3UZC). In both structures, the aminotriazine can be seen H-bonding to Asn253; in (B), the hydroxyl group of 18 can be seen to H-bond to His278. Helices 3 and 4 have been removed from the GPCRs for clarity. | ||
An analysis of the binding site of the 3PWH structure (after removal of the ligand), using WaterMap software from Schrödinger, showed that these highly ligand efficient molecules completely displace the two clusters of water molecules that are high in energy (relative to bulk solvent), located in the ribose binding pocket and in the hydrophobic pocket occupied by the phenyl substituent on triazine-C5.23,85 The WaterMap software was used to calculate the enthalpic and entropic energies of waters relative to bulk solvent using a molecular dynamics simulation on a full, explicit water network. It has also been calculated using SZMAP that pyridyl analogues such as 17 are able to stabilise high energy water molecules within this network via interactions with their pyridylnitrogen atom, whereas phenol derivatives such as 18 are able to completely displace these water molecules with their additional hydroxyl substituent (Fig. 14).23,94
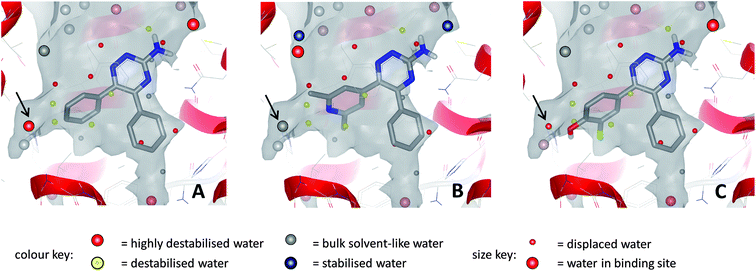 | ||
| Fig. 14 Relative energies of water molecules within the binding site networks for compound 13 (A), compound 17 (B) and compound 18 (C); calculations performed with SZMAP. In all cases, several destabilised and highly destabilised water molecules are displaced by the ligands. For waters indicated by arrows: (A) water is highly destabilised relative to bulk solvent; (B) water is stabilised by pyridyl N and is comparable in energy to bulk solvent; (C) water is displaced by phenolic OH. | ||
1,2,4-Triazine derivatives for the treatment of Parkinson's disease
The lead series exemplified by 1,2,4-triazines13–19 showed a very favourable in vitro ADME profile and good physicochemical properties. Representative examples were therefore selected for further analysis in in vivo pharmacokinetic experiments. An example dataset is shown in Table 5. Compound 19 showed an estimated oral bioavailability of 100% in rats, rapid absorption (Tmax = 0.4 h) and good oral exposure (AUCinf = 846 ng h mL−1). Following intravenous administration, plasma clearance and volume of distribution were found to be moderate (42 mL min−1 kg−1 and 4.6 L kg−1, respectively), leading to an acceptable half-life of 1.2 hours. Compound 19 was found to have good brain penetration and a moderate free fraction in the brain 30 minutes after administration (brain:plasma = 3.2, 3.6% unbound).85On the basis of their in vitro ADME and in vivo pharmacokinetic profiles, compounds such as 19 were progressed into a haloperidol-induced catalepsy model of PD in rats. In this pharmacodynamic model a loss of striatal dopamine receptor function is induced by blockade of the receptors with the antagonist haloperidol, causing a Parkinsonian like-state which can be reversed by A2AR antagonism. Istradefylline was used as a control during these studies as it has previously shown efficacy in various models of PD as well in clinical trials with human patients suffering from PD.95,96 An example dataset for the effect of compound 19 measured in this model is shown in Fig. 15. Compound 19 was found to potently reverse the cataleptic state induced in the rats, with measured ED50 values of 0.2 mg kg−1 at both 1 and 2 hour time points after administration. Indeed, 0.3 mg kg−1 doses of compound 19 were found to have comparable efficacy to 1.0 mg kg−1 doses of istradefylline.
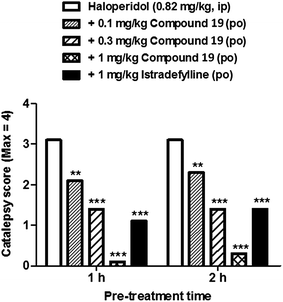 | ||
| Fig. 15 The effects of compound 19 in the haloperidol-induced catalepsy model. A dose-dependent reversal of catalepsy was observed with compound 19 in rats; istradefylline was used as a positive control. | ||
Conclusions
Structural information can play a key role in drug discovery, enabling hit identification and improving efficiency during lead optimisation. This has been well established with soluble targets such as kinases and proteases but, until relatively recently, little structural data has been available for GPCRs, hampering SBDD on this important superfamily of receptors. This article has delineated the use of StaRs® in a drug discovery programme on a GPCR target: StaRs® have enabled the use of biophysical screening techniques and facilitated the crystallisation of ligand–receptor complexes to allow, for the first time, true SBDD on a GPCR.Biophysical Mapping of hits identified through virtual screening against homology models of A2AR allowed their rapid progression into lead series. Molecular modelling, BPM and X-ray crystallography were then used in synergy to optimise the potency, selectivity and receptor kinetics of a series of 1,2,4-triazine derivatives, culminating in the nomination of a low molecular weight and orally bioavailable candidate for the treatment of CNS disease. The characterisation of this agent will be the subject of future publications.
Abbreviations
| PD | Parkinson's disease |
| GPCR | G protein-coupled receptor |
| SBDD | Structure-based drug design |
| BPM | Biophysical Mapping |
| PO | Oral route of administration |
| IV | Intravenous route of administration |
| T ½ | Half-life |
| RLM | Rat liver microsomes |
| rPPB | Rat plasma protein binding |
| V d (ss) | Volume of distribution at steady state |
| AUCinf | Total drug exposure (area under the curve to infinite time) |
| CSF | Cerebral spinal fluid |
| C max | Maximum drug concentration |
| T max | Time at which maximum drug concentration is reached |
| F po | Oral bioavailability |
| XAC | Xanthine amine congener |
| NECA | 5′-N-Ethylcarboxamidoadenosine |
| DPCPX | 8-Cyclopentyl-1,3-dipropylxanthine |
| WT | Wild-type. |
Acknowledgements
The authors wish to thank Andrea Bortolato and Jon Mason for insightful discussions about GRID maps and mapping water molecules, and for producing the graphic for Fig. 14. Fiona Marshall, Miles Congreve and Malcolm Weir are thanked for their stimulating discussion & suggestions for improving this manuscript.References
- K. Palczewski, T. Kumasaka, T. Hori, C. A. Behnke, H. Motoshima, B. A. Fox, T. Le, I. D. C. Teller, T. Okada, R. E. Stenkamp, M. Yamamoto and M. Miyano, Science, 2000, 289, 739–745 CrossRef CAS.
- S. G. Rasmussen, H. J. Choi, D. M. Rosenbaum, T. S. Kobilka, F. S. Thian, P. C. Edwards, M. Burghammer, V. R. Ratnala, R. Sanishvili, R. F. Fischetti, G. F. Schertler, W. I. Weis and B. K. Kobilka, Nature, 2007, 450, 383–387 CrossRef CAS.
- T. Warne, M. J. Serrano-Vega, J. G. Baker, R. Moukhametzianov, P. C. Edwards, R. Henderson, A. G. Leslie, C. G. Tate and G. F. Schertler, Nature, 2008, 454, 486–491 CrossRef CAS.
- V. Cherezov, D. M. Rosenbaum, M. A. Hanson, S. G. Rasmussen, F. S. Thian, T. S. Kobilka, H. J. Choi, P. Kuhn, W. I. Weis, B. K. Kobilka and R. C. Stevens, Science, 2007, 318, 1258–1265 CrossRef CAS.
- V. P. Jaakola, M. T. Griffith, M. A. Hanson, V. Cherezov, E. Y. Chien, J. R. Lane, A. P. Ijzerman and R. C. Stevens, Science, 2008, 322, 1211–1217 CrossRef CAS.
- B. Wu, E. Y. Chien, C. D. Mol, G. Fenalti, W. Liu, V. Katritch, R. Abagyan, A. Brooun, P. Wells, F. C. Bi, D. J. Hamel, P. Kuhn, T. M. Handel, V. Cherezov and R. C. Stevens, Science, 2010, 330, 1066–1071 CrossRef CAS.
- E. Y. Chien, W. Liu, Q. Zhao, V. Katritch, G. W. Han, M. A. Hanson, L. Shi, A. H. Newman, J. A. Javitch, V. Cherezov and R. C. Stevens, Science, 2010, 330, 1091–1095 CrossRef CAS.
- T. Shimamura, M. Shiroishi, S. Weyand, H. Tsujimoto, G. Winter, V. Katritch, R. Abagyan, V. Cherezov, W. Liu, G. W. Han, T. Kobayashi, R. C. Stevens and S. Iwata, Nature, 2011, 475, 65–70 CrossRef.
- K. Haga, A. C. Kruse, H. Asada, T. Yurugi-Kobayashi, M. Shiroishi, C. Zhang, W. I. Weis, T. Okada, B. K. Kobilka, T. Haga and T. Kobayashi, Nature, 2012, 482, 547–551 Search PubMed.
- A. C. Kruse, J. Hu, A. C. Pan, D. H. Arlow, D. M. Rosenbaum, E. Rosemond, H. F. Green, T. Liu, P. S. Chae, R. O. Dror, D. E. Shaw, W. I. Weis, J. Wess and B. K. Kobilka, Nature, 2012, 482, 552–556 CrossRef CAS.
- M. A. Hanson and R. C. Stevens, Structure, 2009, 17, 8–14 CrossRef CAS.
- S. Granier, A. Manglik, A. C. Kruse, T. S. Kobilka, F. S. Thian, W. I. Weis and B. K. Kobilka, Nature, 2012, 485, 400–404 Search PubMed.
- H. Wu, D. Wacker, M. Mileni, V. Katritch, G. W. Han, E. Vardy, W. Liu, A. A. Thompson, X. P. Huang, F. I. Carroll, S. W. Mascarella, R. B. Westkaemper, P. D. Mosier, B. L. Roth, V. Cherezov and R. C. Stevens, Nature, 2012, 485, 327–332 Search PubMed.
- A. Manglik, A. C. Kruse, T. S. Kobilka, F. S. Thian, J. M. Mathiesen, R. K. Sunahara, L. Pardo, W. I. Weis, B. K. Kobilka and S. Granier, Nature, 2012, 485, 321–326 Search PubMed.
- A. A. Thompson, W. Liu, E. Chun, V. Katritch, H. Wu, E. Vardy, X. P. Huang, C. Trapella, R. Guerrini, G. Calo, B. L. Roth, V. Cherezov and R. C. Stevens, Nature, 2012, 485, 395–399 Search PubMed.
- J. A. Salon, D. T. Lodowski and K. Palczewski, Pharmacol. Rev., 2011, 63, 901–937 Search PubMed.
- J. H. Park, P. Scheerer, K. P. Hofmann, H. W. Choe and O. P. Ernst, Nature, 2008, 454, 183–187 CrossRef CAS.
- G. Lebon, K. Bennett, A. Jazayeri and C. G. Tate, J. Mol. Biol., 2011, 409, 298–310 Search PubMed.
- F. Xu, H. Wu, V. Katritch, G. W. Han, K. A. Jacobson, Z. G. Gao, V. Cherezov and R. C. Stevens, Science, 2011, 332, 322–327 CrossRef CAS.
- S. G. Rasmussen, H. J. Choi, J. J. Fung, E. Pardon, P. Casarosa, P. S. Chae, B. T. DeVree, D. M. Rosenbaum, F. S. Thian, T. S. Kobilka, A. Schnapp, I. Konetzki, R. K. Sunahara, S. H. Gellman, A. Pautsch, J. Steyaert, W. I. Weis and B. K. Kobilka, Nature, 2011, 469, 175–180 CrossRef CAS.
- T. Hino, T. Arakawa, H. Iwanari, T. Yurugi-Kobayashi, C. Ikeda-Suno, Y. Nakada-Nakura, O. Kusano-Arai, S. Weyand, T. Shimamura, N. Nomura, A. D. Cameron, T. Kobayashi, T. Hamakubo, S. Iwata and T. Murata, Nature, 2012, 482, 237–240 Search PubMed.
- S. G. Rasmussen, B. T. DeVree, Y. Zou, A. C. Kruse, K. Y. Chung, T. S. Kobilka, F. S. Thian, P. S. Chae, E. Pardon, D. Calinski, J. M. Mathiesen, S. T. Shah, J. A. Lyons, M. Caffrey, S. H. Gellman, J. Steyaert, G. Skiniotis, W. I. Weis, R. K. Sunahara and B. K. Kobilka, Nature, 2011, 477, 549–555 CrossRef CAS.
- J. S. Mason, A. Bortolato, M. Congreve and F. H. Marshall, Trends Pharmacol. Sci., 2012, 33, 249–260 Search PubMed.
- B. K. Kobilka, Biochim. Biophys. Acta, 2007, 1768, 794–807 Search PubMed.
- S. Costanzi, I. G. Tikhonova, T. K. Harden and K. A. Jacobson, J. Comput.-Aided Mol. Des., 2009, 23, 747–754 CrossRef CAS.
- J. Wess, S. J. Han, S. K. Kim, K. A. Jacobson and J. H. Li, Trends Pharmacol. Sci., 2008, 29, 616–625 CrossRef CAS.
- C. G. Tate and G. F. Schertler, Curr. Opin. Struct. Biol., 2009, 19, 386–395 CrossRef CAS.
- D. M. Rosenbaum, S. G. Rasmussen and B. K. Kobilka, Nature, 2009, 459, 356–363 CrossRef CAS.
- K. A. Jacobson and S. Costanzi, Mol. Pharmacol., 2012, 82, 361–371 Search PubMed.
- V. Katritch, V. Cherezov and R. C. Stevens, Trends Pharmacol. Sci., 2012, 33, 17–27 CrossRef CAS.
- M. Congreve and F. Marshall, Br. J. Pharmacol., 2010, 159, 986–996 Search PubMed.
- M. Congreve, C. Langmead and F. H. Marshall, Adv. Pharmacol., 2011, 62, 1–36 Search PubMed.
- M. Congreve, C. W. Murray and T. L. Blundell, Drug Discovery Today, 2005, 10, 895–907 CrossRef CAS.
- N. Robertson, A. Jazayeri, J. Errey, A. Baig, E. Hurrell, A. Zhukov, C. J. Langmead, M. Weir and F. H. Marshall, Neuropharmacology, 2011, 60, 36–44 Search PubMed.
- J. P. Overington, B. Al-Lazikani and A. L. Hopkins, Nat. Rev. Drug Discovery, 2006, 5, 993–996 CrossRef CAS.
- C. E. Muller and K. A. Jacobson, Biochim. Biophys. Acta, 2011, 1808, 1290–1308 Search PubMed.
- K. P. Garnock-Jones and M. P. Curran, Am. J. Cardiovasc. Drugs, 2010, 10, 65–71 Search PubMed.
- B. C. Shook and P. F. Jackson, ACS Chem. Neurosci., 2011, 2, 555–567 Search PubMed.
- S. P. Alexander and P. J. Millns, Eur. J. Pharmacol., 2001, 411, 205–210 Search PubMed.
- A. Uustare, A. Vonk, A. Terasmaa, K. Fuxe and A. Rinken, Life Sci., 2005, 76, 1513–1526 Search PubMed.
- W. Dauer and S. Przedborski, Neuron, 2003, 39, 889–909 CrossRef CAS.
- N. Singh, V. Pillay and Y. E. Choonara, Prog. Neurobiol., 2007, 81, 29–44 Search PubMed.
- U. Shah and R. Hodgson, Curr. Opin. Drug Discovery Dev., 2010, 13, 466–480 Search PubMed.
- C. V. Gomes, M. P. Kaster, A. R. Tome, P. M. Agostinho and R. A. Cunha, Biochim. Biophys. Acta, 2011, 1808, 1380–1399 Search PubMed.
- R. A. Hodgson, P. J. Bedard, G. B. Varty, T. M. Kazdoba, P. T. Di, M. E. Grzelak, A. J. Pond, A. Hadjtahar, N. Belanger, L. Gregoire, A. Dare, B. R. Neustadt, A. W. Stamford and J. C. Hunter, Exp. Neurol., 2010, 225, 384–390 Search PubMed.
- C. G. Tate, Trends Biochem. Sci., 2012, 37(9), 343–352 Search PubMed.
- D. M. Rosenbaum, V. Cherezov, M. A. Hanson, S. G. Rasmussen, F. S. Thian, T. S. Kobilka, H. J. Choi, X. J. Yao, W. I. Weis, R. C. Stevens and B. K. Kobilka, Science, 2007, 318, 1266–1273 CrossRef CAS.
- E. Chun, A. A. Thompson, W. Liu, C. B. Roth, M. T. Griffith, V. Katritch, J. Kunken, F. Xu, V. Cherezov, M. A. Hanson and R. C. Stevens, Structure, 2012, 20, 967–976 Search PubMed.
- Q. Zhao and B. L. Wu, Acta Pharmacol. Sin., 2012, 33, 324–334 Search PubMed.
- F. Magnani, Y. Shibata, M. J. Serrano-Vega and C. G. Tate, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10744–10749 Search PubMed.
- A. S. Dore, N. Robertson, J. C. Errey, I. Ng, K. Hollenstein, B. Tehan, E. Hurrell, K. Bennett, M. Congreve, F. Magnani, C. G. Tate, M. Weir and F. H. Marshall, Structure, 2011, 19, 1283–1293 CAS.
- G. Lebon, T. Warne, P. C. Edwards, K. Bennett, C. J. Langmead, A. G. Leslie and C. G. Tate, Nature, 2011, 474, 521–525 CrossRef CAS.
- J. C. Mobarec, R. Sanchez and M. Filizola, J. Med. Chem., 2009, 52, 5207–5216 CrossRef CAS.
- T. Yarnitzky, A. Levit and M. Y. Niv, Curr. Opin. Drug Discovery Dev., 2010, 13, 317–325 CAS.
- V. Katritch, M. Rueda, P. C. Lam, M. Yeager and R. Abagyan, Proteins, 2010, 78, 197–211 Search PubMed.
- M. Michino, E. Abola, C. L. Brooks, III, J. S. Dixon, J. Moult and R. C. Stevens, Nat. Rev. Drug Discovery, 2009, 8, 455–463 Search PubMed.
- I. Kufareva, M. Rueda, V. Katritch, R. C. Stevens and R. Abagyan, Structure, 2011, 19, 1108–1126 Search PubMed.
- A. Sali and T. L. Blundell, J. Mol. Biol., 1993, 234, 779–815 CrossRef CAS.
- Chemical Computing Group, Molecular Operating Environment (MOE2008.10), Montreal, Quebec, Canada Search PubMed.
- J. D. Thompson, D. G. Higgins and T. J. Gibson, Nucleic Acids Res., 1994, 22, 4673–4680 CrossRef CAS.
- C. J. Langmead, S. P. Andrews, M. Congreve, J. C. Errey, E. Hurrell, F. H. Marshall, J. S. Mason, C. M. Richardson, N. Robertson, A. Zhukov and M. Weir, J. Med. Chem., 2012, 55, 1904–1909 Search PubMed.
- J. Kim, J. Wess, A. M. van Rhee, T. Schoneberg and K. A. Jacobson, J. Biol. Chem., 1995, 270, 13987–13997 CrossRef CAS.
- S. K. Kim, Z. G. Gao, R. P. Van, A. S. Gross, A. Chen, C. S. Van and K. A. Jacobson, J. Med. Chem., 2003, 46, 4847–4859 Search PubMed.
- B. D. Dal, C. Lambertucci, G. Marucci, R. Volpini and G. Cristalli, Curr. Top. Med. Chem., 2010, 10, 993–1018 Search PubMed.
- A. A. Ivanov, D. Barak and K. A. Jacobson, J. Med. Chem., 2009, 52, 3284–3292 CrossRef CAS.
- F. Deflorian, T. S. Kumar, K. Phan, Z. G. Gao, F. Xu, H. Wu, V. Katritch, R. C. Stevens and K. A. Jacobson, J. Med. Chem., 2012, 55, 538–552 Search PubMed.
- D. K. Tosh, K. Phan, Z. G. Gao, A. A. Gakh, F. Xu, F. Deflorian, R. Abagyan, R. C. Stevens, K. A. Jacobson and V. Katritch, J. Med. Chem., 2012, 55, 4297–4308 Search PubMed.
- J. Carlsson, L. Yoo, Z. G. Gao, J. J. Irwin, B. K. Shoichet and K. A. Jacobson, J. Med. Chem., 2010, 53, 3748–3755 Search PubMed.
- V. Katritch, V. P. Jaakola, J. R. Lane, J. Lin, A. P. Ijzerman, M. Yeager, I. Kufareva, R. C. Stevens and R. Abagyan, J. Med. Chem., 2010, 53, 1799–1809 Search PubMed.
- C. de Graaf and D. Rognan, Curr. Pharm. Des., 2009, 15, 4026–4048 CrossRef CAS.
- G. L. Warren, C. W. Andrews, A. M. Capelli, B. Clarke, J. LaLonde, M. H. Lambert, M. Lindvall, N. Nevins, S. F. Semus, S. Senger, G. Tedesco, I. D. Wall, J. M. Woolven, C. E. Peishoff and M. S. Head, J. Med. Chem., 2006, 49, 5912–5931 CrossRef CAS.
- D. C. Rees, M. Congreve, C. W. Murray and R. Carr, Nat. Rev. Drug Discovery, 2004, 3, 660–672 CrossRef CAS.
- J. P. Renaud and M. A. Delsuc, Curr. Opin. Pharmacol., 2009, 9, 622–628 CrossRef CAS.
- I. Navratilova, M. Dioszegi and D. G. Myszka, Anal. Biochem., 2006, 355, 132–139 Search PubMed.
- M. Congreve, R. L. Rich, D. G. Myszka, F. Figaroa, G. Siegal and F. H. Marshall, Methods Enzymol., 2011, 493, 115–136 Search PubMed.
- R. L. Rich, J. Errey, F. Marshall and D. G. Myszka, Anal. Biochem., 2011, 409, 267–272 CrossRef CAS.
- D. Chen, J. Errey, L. Heitman, F. Marshall, A. P. Ijzerman and G. Siegal, ACS Chem. Biol., in press.
- Dallas Hughes, WO2011036476A1, 2011.
- C. Austin, S. N. Pettit, S. K. Magnolo, J. Sanvoisin, W. Chen, S. P. Wood, L. D. Freeman, R. J. Pengelly and D. E. Hughes, J. Biomol. Screening, 2012, 17(7), 868–876 Search PubMed.
- M. K. Walker, unpublished results.
- A. Zhukov, S. P. Andrews, J. C. Errey, N. Robertson, B. Tehan, J. S. Mason, F. H. Marshall, M. Weir and M. Congreve, J. Med. Chem., 2011, 54, 4312–4323 Search PubMed.
- T. Warne, M. J. Serrano-Vega, C. G. Tate and G. F. Schertler, Protein Expression Purif., 2009, 65, 204–213 Search PubMed.
- E. M. Landau and J. P. Rosenbusch, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14532–14535 CrossRef CAS.
- V. Cherezov, Curr. Opin. Struct. Biol., 2011, 21, 559–566 CrossRef CAS.
- M. Congreve, S. P. Andrews, A. S. Dore, K. Hollenstein, E. Hurrell, C. J. Langmead, J. S. Mason, I. W. Ng, B. Tehan, A. Zhukov, M. Weir and F. H. Marshall, J. Med. Chem., 2012, 55, 1898–1903 Search PubMed.
- H. Piirainen, Y. Ashok, R. T. Nanekar and V. P. Jaakola, Biochim. Biophys. Acta, 2011, 1808, 1233–1244 Search PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS.
- T. A. Halgren, R. B. Murphy, R. A. Friesner, H. S. Beard, L. L. Frye, W. T. Pollard and J. L. Banks, J. Med. Chem., 2004, 47, 1750–1759 CrossRef CAS.
- C. Abad-Zapatero and J. T. Metz, Drug Discovery Today, 2005, 10, 464–469 CrossRef.
- A. L. Hopkins, C. R. Groom and A. Alex, Drug Discovery Today, 2004, 9, 430–431 CrossRef.
- P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881–890 CrossRef CAS.
- P. J. Goodford, J. Med. Chem., 1985, 28, 849–857 CrossRef CAS.
- S. Sciabola, R. V. Stanton, J. E. Mills, M. M. Flocco, M. Baroni, G. Cruciani, F. Perruccio and J. S. Mason, J. Chem. Inf. Model., 2010, 50, 155–169 CrossRef CAS.
- A. Bortolato and J. S. Mason, personal communication.
- P. Jenner, Expert Opin. Invest. Drugs, 2005, 14, 729–738 Search PubMed.
- P. Jenner, A. Mori, R. Hauser, M. Morelli, B. B. Fredholm and J. F. Chen, Parkinsonism Relat. Disord., 2009, 15, 406–413 Search PubMed.
- R. J. Gillespie, S. J. Bamford, R. Botting, M. Comer, S. Denny, S. Gaur, M. Griffin, A. M. Jordan, A. R. Knight, J. Lerpiniere, S. Leonardi, S. Lightowler, S. McAteer, A. Merrett, A. Misra, A. Padfield, M. Reece, M. Saadi, D. L. Selwood, G. C. Stratton, D. Surry, R. Todd, X. Tong, V. Ruston, R. Upton and S. M. Weiss, J. Med. Chem., 2009, 52, 33–47 Search PubMed.
- L. E. Collins, T. N. Sager, A. G. Sams, A. Pennarola, R. G. Port, M. Shahriari and J. D. Salamone, Pharmacol., Biochem. Behav., 2012, 100, 498–505 Search PubMed.
- A. G. Sams, G. K. Mikkelsen, M. Larsen, M. Langgard, M. E. Howells, T. J. Schroder, L. T. Brennum, L. Torup, E. B. Jorgensen, C. Bundgaard, M. Kreilgard and B. Bang-Andersen, J. Med. Chem., 2011, 54, 751–764 Search PubMed.
- P. M. Luthra, C. B. Mishra, P. K. Jha and S. K. Barodia, Bioorg. Med. Chem. Lett., 2010, 20, 1214–1218 Search PubMed.
- B. C. Shook, S. Rassnick, N. Wallace, J. Crooke, M. Ault, D. Chakravarty, J. K. Barbay, A. Wang, M. T. Powell, K. Leonard, V. Alford, R. H. Scannevin, K. Carroll, L. Lampron, L. Westover, H. K. Lim, R. Russell, S. Branum, K. M. Wells, S. Damon, S. Youells, X. Li, D. A. Beauchamp, K. Rhodes and P. F. Jackson, J. Med. Chem., 2012, 55, 1402–1417 Search PubMed.
- CE experiments were performed at Selcia. CE conditions: running & injection buffers: Tris–HCl (30 mM, pH 7.4 + 0.1% DM); polarity: normal; separation: 7.5 kV. In the runs with caffeine, this ligand is not observed as it is in the running and injection buffers at the same concentration (100 μM). DMSO was used as a control for the runs without caffeine.
| This journal is © The Royal Society of Chemistry 2013 |