Characterizing the laulimalide–peloruside binding site using site-directed mutagenesis of TUB2 in S. cerevisiae†
Reem
Hanna
a,
David R.
Maass
a,
Paul H.
Atkinson
a,
Peter T.
Northcote
b,
Paul H.
Teesdale-Spittle
a,
David S.
Bellows
a and
John H.
Miller
*a
aCentre for Biodiscovery, School of Biological Sciences, Victoria University of Wellington, Kelburn Parade, Kelburn, Wellington, 6012, New Zealand. E-mail: john.h.miller@vuw.ac.nz; Fax: +64 4 463 5331; Tel: +64 4 463 6082
bCentre for Biodiscovery, Chemical and Physical Sciences, Victoria University of Wellington, PO Box 600, Wellington, 6140, New Zealand
First published on 21st October 2013
Abstract
Baker's yeast, Saccharomyces cerevisiae, has significant sequence conservation with a core subset of mammalian proteins and can serve as a model for disease processes. The aim of this study was to determine whether yeast could be used as a model system to identify new agents that interact with the laulimalide–peloruside binding site on β-tubulin. Agents that bind to this site cause stabilization of microtubules and interfere with cell division. Based on the location of the proposed laulimalide–peloruside binding site and of previously identified mutations shown to cause resistance in mammalian cells, we made the corresponding mutations in yeast and tested whether they conferred resistance to laulimalide and peloruside. Mutations A296T and R306H, which cause 6-fold and 40-fold increased resistance in human 1A9 ovarian carcinoma cells, respectively, also led to resistance in yeast to these compounds. Similarly, other mutations led to resistance or, in one case, increased sensitivity. Thus, we conclude that yeast is an appropriate model to screen for small molecule drugs that may be efficacious in cancer therapy in humans through the newly characterised laulimalide–peloruside binding site.
Introduction
Yeast is a versatile and robust model system for studying eukaryotic cell function. The genetic flexibility of yeast and the high degree of conservation between its cellular processes and those of human cells has made it a popular model organism for new research in molecular and cell biology,1 drug discovery,2,3 and disease processes.4 Rapid genome-wide screens can be readily carried out in yeast, leading to the identification of networks of genetic interactions. These networks can provide clues to a novel compound's mechanism of action5 and identify the complex interactions involved in the action of a drug.6 In many cases where yeast lack a human homolog, the gene can be easily introduced into the yeast genome and the ‘humanized’ strain used to search for compounds that reduce the toxicity of a human disease gene, thus providing information on drugs that may interfere with the disease process.4Microtubule-targeting drugs have been extremely useful in the treatment of cancer, in part because of the obligatory requirement for microtubule function during mitosis in the segregation of the chromosomes.7 A diverse array of chemical compounds have been identified that either stabilize the microtubule (microtubule-stabilizing agent (MSA)) or destabilize the polymer, microtubule-destabilizing agent (MDA).8 Two types of MSA are currently used clinically, the taxanes and the epothilones. The taxanes include paclitaxel (Taxol®) and docetaxel (Taxotere®), and the epothilones include ixabepilone (Ixempra®), patupilone, and sagopilone.9 Because of poor solubilities, susceptibility to cancer cell resistance mechanisms, and/or unwanted side effects of these front line agents, second generation drugs are being sought with improved efficacy alone or in combination with the current drugs for longer-term control of tumour growth.
Given the favourable properties of yeast for use in drug discovery,2 its value as a model organism for examining drug interactions opens the possibility of screening for novel microtubule-targeting agents in the yeast genetic system. In tests of the three classically used MSAs, only epothilone A and B were shown to stabilize yeast microtubules in the test tube, but not in intact cells.10 However, substitution of five amino acids in the taxane binding site of yeast Tub2 with their human counterparts conferred MSA activity for paclitaxel in vitro, but, like epothilone, had no effect on intact cells.11 It was subsequently shown that if the binding site mutations were made in a pleiotropic drug efflux pump knockout background, then paclitaxel and docetaxel were able to inhibit yeast cell proliferation and induce apoptosis in the cells.12–14 Epothilone B was not investigated in the Tub2p-modified, pump knockout strain, but a derivative of epothilone B, ixabepilone, failed to inhibit yeast growth in a pdr-deficient strain in our laboratory at concentrations as high as 300 μM (unpublished results).
Two other MSAs, laulimalide and peloruside A,15,16 that bind to a different site on β-tubulin to the taxoid site drugs (reviewed by Field et al.17) were shown to be active in inhibiting yeast cell growth,18,19 although at low μM concentrations compared to the low nM concentrations needed for mammalian cells.20,21
The aim of the present study was to use site-directed mutagenesis to alter single amino acids in the laulimalide–peloruside binding site to investigate the interaction between the MSA and yeast microtubules. Mutations that are known to cause resistance in human ovarian cancer cells, A296T and R306H22 would be tested, as well as mutations predicted by molecular modeling to have an effect on laulimalide and peloruside interactions with microtubules, based on the proposed β-tubulin binding site for these MSAs.23 This information would validate the utility of yeast in preliminary screens for novel, small molecule MSAs or related compounds that could be translated to the clinic and used in place of or in combination with the front line chemotherapeutic MSAs or MDAs. Both laulimalide24 and peloruside25,26 are known to synergise with paclitaxel and epothilone in cancer cells, indicating the potential for their use in combination therapy with the front line anticancer drugs.
Results
Growth inhibitory effects of laulimalide and peloruside in WT, mad2Δ, and efflux pump deficient mutant strains
The growth inhibitory activity of laulimalide and peloruside was tested in wild type (WT) yeast compared with several haploid deletion mutants. The mad2Δ haploid deletion strain previously showed increased sensitivity to peloruside compared to the WT strain.18 At the highest concentrations of MSA (50 μM), deletion of the mitotic checkpoint gene MAD2 increased the growth inhibition by both MSAs, presenting as a decreased % residual growth (Table 1) and a reduced IC50 (Table 1, Fig. 1). Deletion of the main yeast pleiotropic drug efflux pump gene, PDR5, (strain pdr5Δ) or the master transcription factor genes for the yeast efflux pump network, PDR1 and PDR3, (strain pdr1Δ + pdr3Δ) conferred additive sensitivity to laulimalide and peloruside even further. Laulimalide was more potent than peloruside in all the strains tested, being up to 10-fold more active in the efflux pump-deficient strains. The S. cerevisiae drug efflux pump interactome has recently been mapped and shown to consist of a complex network of interactions across the yeast proteome.27| Drug | Laulimalide | Peloruside | ||
|---|---|---|---|---|
| Strains | % Residual growth (at 50 μM) | IC50 (μM) | % Residual growth (at 50 μM) | IC50 (μM) |
| Different strains were treated for 18 h with laulimalide or peloruside at concentrations up to 50 μM. The % residual growth at 50 μM was calculated relative to growth with DMSO at an equivalent concentration. IC50 values were calculated from concentration–response curves using SigmaPlot (v10.0). Data are presented as the mean ± SD of three independent experiments. | ||||
| WT | 35 ± 4 | 36 ± 1 | 49.6 ± 0.3 | 49 ± 1 |
| mad2Δ | 8 ± 4 | 24 ± 1 | 6 ± 1 | 36 ± 1 |
| pdr5Δ | 6 ± 2 | 3 ± 1 | 8 ± 1 | 29 ± 1 |
| pdr1Δ & pdr3Δ | 7 ± 2 | 2 ± 1 | 7 ± 1 | 22 ± 1 |
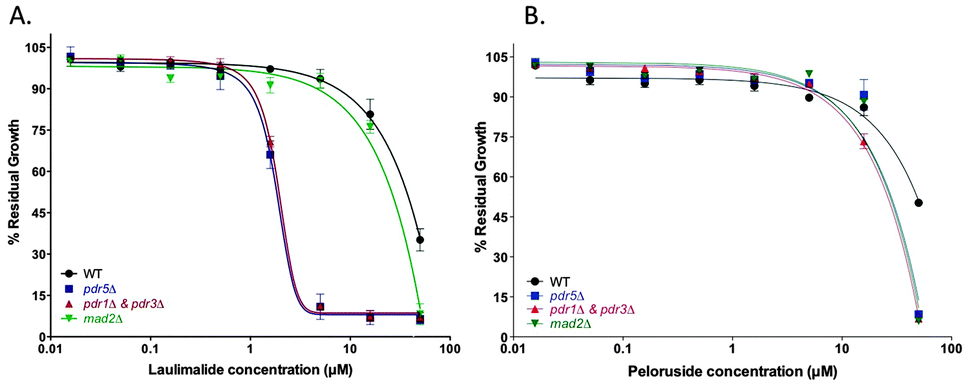 | ||
| Fig. 1 Half-log concentration–response curves for laulimalide (A) and peloruside (B). Concentration–response curves are presented for the haploid wild type BY4741 (WT) and the deletion mutant strains pdr5Δ, PDR-deficient strain (pdrΔ & pdr3Δ), and madΔ. Cells were treated with laulimalide or peloruside at concentrations up to 50 μM for 18 h. All mutant curves for a single compound were run simultaneously. Data are presented as the mean ± SD of three independent experiments. | ||
Testing of site-directed mutations in TUB2 that confer resistance in mammalian cells
Single amino acid mutations in the TUB2 gene of S. cerevisiae that are known to confer laulimalide and/or peloruside22,28 resistance on human 1A9 ovarian carcinoma cells were generated by overlap PCR in the haploid starting strain mad2Δ. A296T (resistance ratio of 6 in 1A9-R1 cells for peloruside but no resistance to laulimalide) and R306H (resistance ratio of 39 in 1A9-L4 cells for both laulimalide and peloruside) and the combined double mutant A296T + R306H all led to resistance to laulimalide and peloruside in yeast (Fig. 2). In the three TUB2 mutant strains, there was no inhibition of growth by the MSAs even at 50 μM, consistent with the effects of these mutations in mammalian cancer cell lines.22,28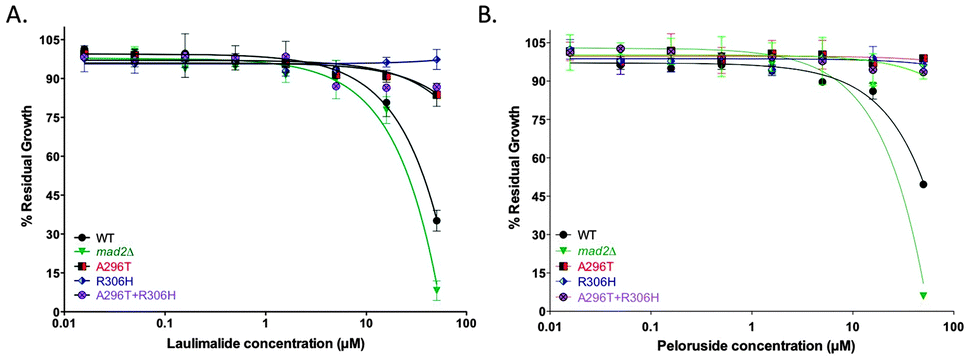 | ||
| Fig. 2 Growth inhibition curves of mad2Δ and tub2 mutant strains treated with laulimalide (A) and peloruside (B). The three mutant strains generated on a mad2Δ background (A296T, R306H, and A296T + R306H), the haploid deletion mutant background strain mad2Δ (positive control), and WT strain were treated with up to 50 μM laulimalide for 18 h. The WT and mad2Δ growth curves are taken from Fig. 1. All mutant curves for a single compound were run simultaneously. Data are presented as the mean ± SD of three independent experiments. | ||
Testing of predicted mutations of TUB2
Using the proposed binding site for laulimalide and peloruside originally suggested from hydrogen–deuterium exchange mass spectrometry (HDX-MS) by David Schriemer23 and later others,29–31 specific amino acids that were predicted to have an effect on binding of the MSA to the microtubule (ESI,† Fig. S1 and Tables S1 and S2) were mutated, and the strains were tested for sensitivity to laulimalide and peloruside. Two of the three mutant strains, N337L and V333W, as expected conferred resistance on the cells to both MSAs. Interestingly, the third mutation, Q291M, increased the sensitivity of the cells to both laulimalide and peloruside. The fourth mutation, R282Q, was chosen as a negative control because of its location outside the laulimalide–peloruside binding pocket; however, although it had no major effect on laulimalide sensitivity as expected, it led to major resistance to peloruside.Discussion
The laulimalide–peloruside binding site in yeast
Best et al.19 previously showed that laulimalide and peloruside exerted their growth inhibitory effects on yeast by targeting the microtubule, a well known mode of action in mammalian cells for these two MSAs.20,21 By mutating yeast tubulin amino acids in the proposed Huzil et al.23 binding site, it was expected that the action of the MSA would be altered, providing indirect evidence that the MSA interacted with β-tubulin in this region. Thus, mutations A296T and R306H, previously shown to confer resistance to human ovarian carcinoma cells22 also gave resistance in yeast, suggesting similar binding of laulimalide and peloruside to yeast tubulin and human tubulin. Whereas the prediction of effects of altering specific amino acids generally held true for four of the six mutations tested, two of the six mutations gave unexpected results (Fig. 3). | ||
| Fig. 3 Growth inhibition curves of the predicted tub2 point mutation strains treated with laulimalide (A) and peloruside (B). Four predicted mutant strains generated on a mad2Δ background (Q291M, N337L, V333W and R282Q), the haploid deletion mutant background strain mad2Δ (positive control), and WT strain were treated with up to 50 μM laulimalide or peloruside for 18 h. The WT and mad2Δ growth curves are taken from Fig. 1. All mutant curves for a single compound were run simultaneously. Data are presented as the mean ± SD of three independent experiments. | ||
There are a number of binding sites on β-tubulin that show varying degrees of conservation relative to human tubulin (Fig. 4). Studies in the laboratory of Richard Himes showed that five amino acids needed to be ‘humanized’ before paclitaxel could stabilize purified yeast tubulin in vitro.10,11 Other binding sites on yeast tubulin show varying degrees of sequence homology to mammalian tubulin, with 72% conservation in the laulimalide–peloruside site (Fig. 4).
 | ||
| Fig. 4 Sequence alignment of S. cerevisiae β-tubulin gene (TUB2) versus human brain tubulin (TUB1). Sequence homology between yeast and human tubulin is about 75%. Red highlighted amino acids are not conserved. Yellow highlights the five amino acids that when ‘humanized’ allow paclitaxel to stabilize yeast tubulin (Gupta et al. 2003).11 Pink highlights the amino acid mutations in the laulimalide–peloruside site, two of which confer resistance in mammalian cell lines. The binding sites for various microtubule-targeting agents are illustrated by coloured boxes as indicated at the bottom of the figure. | ||
The expected increase in resistance when mutations were made to amino acid residues 296 and 306 were based on biological evidence that these sites are important in the action of laulimalide and peloruside in human cancer cells.22,28 Properties of amino acids that were mutated in this study can be found in Table S2 (ESI†). The predicted single amino acid mutations involved suspected disruption of hydrogen bonding or hydrophobic interactions of the MSA at the proposed binding site. The predictions were primarily based on modeling with peloruside, yet laulimalide closely followed the peloruside effects of the mutations, although there were a few differences.
The WT amino acid at position 291 is Gln which contains a hydrophilic side chain that may form a H-bond between the amide group and peloruside. In order to examine the importance of the H-bonding at this site, we changed Gln to an amino acid with a hydrophobic side chain. The Q291M substitution resulted in a strain that was hypersensitive to both laulimalide and peloruside. This suggests that there is no H-bond at this site and that substitution with Met allows for improved binding, potentially through a hydrophobic interaction.
Mutation of Asn337 to Leu was predicted to interfere with the H-bond between Asn and an OH group of peloruside since Leu cannot form H-bonds. This change gave the expected resistance, suggesting that this H-bond is important for activity. Begaye et al.28 also showed this amino acid was important in binding since conversion of the Asn to Asp gave resistance to 1A9 cells.
For the other amino acid change that gave the predicted resistance, V333W, it was suggested that peloruside may make a contact with Val333, and the change to Trp would create a steric clash due to the larger size of the amino acid.
Mutation R282Q was initially included in this study as a negative control for laulimalide and peloruside, but a positive control for epothilone derivatives, as it is an epothilone B-resistant point mutation in mammalian cells.32 It has been shown in mammalian cells that laulimalide and peloruside do not bind to the taxoid site on β-tubulin where epothilone binds.15,16 Amino acid Arg282 sits on the M loop in β-tubulin. This loop, located near the taxane binding site, is believed to be important for lateral contacts between protofilaments. This specific point mutation unexpectedly caused significant resistance to peloruside, but not laulimalide. Although interesting, the mechanism is unknown. This is the first direct experimental evidence that laulimalide and peloruside may bind differently to tubulin. Previous binding competition studies in mammalian cells by Gaitanos et al.16 showed that peloruside and laulimalide bound to a similar or overlapping site on the microtubule. It is worthwhile to keep in mind that point mutations may cause global conformational changes in a protein or may alter the distributions of the conformational sub-states in a way similar to that of an external agent, such as temperature, pH, ionic strength, or pressure, or they may mimic a change in the binding conditions of a drug.33,34 Thus, effects of the mutations on drug sensitivities do not prove the amino acid involved is located at the binding site, as seems the case for mutation R282Q and peloruside sensitivity. Yet, these global changes would be expected to affect laulimalide in a similar manner to peloruside, but this was not the case. In addition, peloruside in human ovarian cancer cells is unaffected by mutation R282Q,16 despite it being affected in yeast.
Despite these differences, we have demonstrated that yeast provides a useful model for studying binding site interactions of drugs like laulimalide and peloruside, as proposed by others and recently reviewed by Botstein and Fink.35
Materials and methods
Chemicals and drugs
Laulimalide and peloruside A were isolated from the marine sponges Cacospongia mycofijiensis36 and Mycale hentscheli,37 respectively, and stored at −80 °C at a concentration of 10 mM in dimethyl sulphoxide (DMSO). As both drugs were in limited supply, only a small number of experimental repeats could be undertaken. All chemicals were obtained from Sigma Chemical Company (St. Louis, MO) unless otherwise indicated.Saccharomyces cerevisiae strains
The BY4741 haploid yeast strain was used as the WT. The mutated yeast strains used in the study are summarized in Table 2. The % residual growth was calculated as:| [(meanexp abs − background)/(meancontrol abs − background)] × 100 |
| ORF | Strain | Genotype |
|---|---|---|
| All haploid deletion mutant strains, as well as the haploid wild type (BY4741), were obtained from a −80 °C glycerol freezer stock, and streaked out on 10 cm yeast extract peptone dextrose plates or synthetic complete plates supplemented with the appropriate antibiotics for growth selection (200 μg mL−1 G418 antibiotic (Geneticin, Gibco, Invitrogen) and/or 100 μg mL−1 nourseothricin (NAT) antibiotic (Werner BioAgents)). Once strains were streaked, the plates were incubated at 30 °C (Contherm Digital Incubator) for 48 h to allow the yeast to grow, and then the plates were kept at 4 °C. The background strain of the haploid deletion mutant strains listed above is the haploid wild type (BY4741). The deletion mutant strains pdr5Δ and mad2Δ were part of the yeast genome deletion collection obtained from Open Biosystems, Thermo Scientific, Auckland, N.Z. | ||
| YCG160 | BY4741 | MAT a ura3Δ0 leu2Δ0his3Δ1 met15Δ1 |
| YJL030W | mad2Δ | MAT a mad2A::G418 ura3Δ0 leu2Δ0his3Δ1 met15Δ1 |
| YCG141 | pdr1Δ | MAT alpha pdr1Δ::NAT can1Δ::STE2pr-HIS5 lypΔ1::STE3prLEU2 ura3Δ0 leu2Δ0 |
| YOR153W | pdr5Δ | MAT a pdr5::G418 ura3Δ0 leu2Δ0his3Δ1 met15Δ1 |
| YCG198 | pdr1Δ & pdr3Δ | MAT alpha pdr1Δ::NAT pdr3Δ::URA3 can1Δ::STE2pr-Sp_HIS5 lyp1Δ::STE3pr LEU2 his3Δ1 leu2Δ0 ura3Δ0 |
Primer design of tubulin (TUB2) mutants by overlap PCR
Overlap PCR was used to introduce the desired point mutation into the yeast genome at the TUB2 locus. A schematic view of the overlap PCR design is shown in Fig. S3 (ESI†). Seven site-directed point mutations were inserted into the yeast genome: A296T, R306H, A296T + R306H (double mutant), R282Q, Q291M, V333W, and N337L. For each point mutation, a 5′ → 3′ primer A (primer sequences are in the ESI,† Table S3) was designed to contain the nucleotide changes and include a 45 bp homology with the TUB2 gene. Primer B (complementary to primer C) was designed to have 21 bp homology to the NatMX cassette (Fig. S2, ESI†) plus 25 bp homology to genomic DNA downstream of the TUB2 gene. Primer C and D were designed to have sequence homology to the NatMX cassette (encoding a nourseothricin antibiotic resistance marker for selection of clones) plus sequence homology to genomic DNA downstream of the TUB2 gene. PCR amplification with primer A and B was carried out using genomic DNA as template; whereas, PCR amplification with primer C and D was carried out using purified plasmid p4339 (refer to Fig. S2, ESI†) containing the NatMX cassette as a template. The PCR products were used to transform the haploid starting strain mad2Δ.PCR was carried out with Qiagen HotstarTaq polymerase using the method of Janke et al.38 involving 15 min of initial activation at 95 °C, followed by 10 cycles of: 1 min DNA denaturation at 97 °C, 30 s of annealing at 56 °C, and 2 min and 40 s of extension at 68 °C. This was followed by another 25 cycles of: 1 min of DNA denaturation at 97 °C, 30 s of annealing at 56 °C, and 2 min and 40 s of (with the addition of 20 s per cycle) extension at 68 °C and a final 10 min 72 °C extension. Following PCR, 5 μL of each PCR product was electrophoresed on a 1% agarose gel for 1 h at 100 V.
Transformation
The PCR products were used to transform the haploid starting strain mad2Δ using a high efficiency transformation protocol described by Gietz and Woods.39 Transformed cells were plated on a YPD + ClonNAT (200 mg mL−1) plate and incubated at 30 °C for 2 days.Confirmation PCR primer design
Colony PCR was performed to confirm that the correct point mutation was inserted (Fig. 5). Two 18 bp reverse primers were designed (Table S3, ESI†): one primer (P.μ) ended with the point mutation nucleotide sequence while the other primer (WT) ended with the WT nucleotide sequence. A forward primer was designed to anneal approximately 400 bp up-stream of these reverse primers.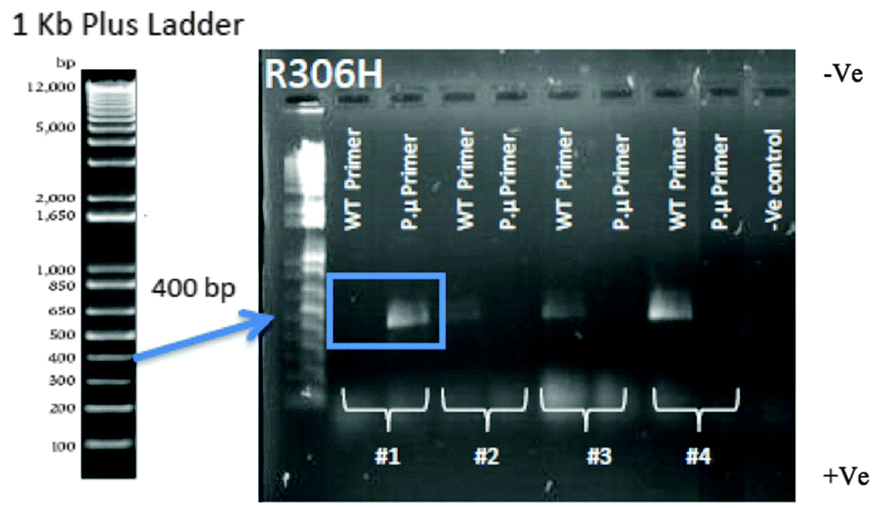 | ||
| Fig. 5 Colony PCR confirmation of the R306H transformation. Using the p.μ. primer set, only colony 1 shows the correct R306H point mutation (blue box). Colonies 2–4 show only product with the wild type primer set, indicating the mutation failed to insert into the DNA. Two primers were used for the confirmation of mutant colonies. Along with the forward primer that was designed to be 400 bp upstream of the desired point mutation, the reverse primer was designed to have either the WT sequence or the point mutation. Two PCR reactions were performed for each colony picked at random from an agar transformation plate containing geneticin. Thus, if transformation occurred, a band would only be present if the colony had the expected WT sequence or the expected point mutation for the primer set used. Only colony #1 has the successful point mutation (blue box), with a product for the mutant primer set but not the wild type primer set. | ||
DNA template was prepared in which yeast cells from a colony were transferred into an Eppendorf tube containing 10 μL of 1 mg mL−1 of Zymolyase 20T (1 mg mL−1 Zymolyase dissolved in 1 mL of 2 M sorbitol (AMS Biotechnology Ltd, UK)) and incubated at 30 °C for 15 min. The PCR cycling times were as follows: 15 min of polymerase activation at 95 °C, followed by 30 cycles of: 1 min of DNA denaturation at 95 °C, 30 s of annealing at 54 °C and 1 min of extension at 72 °C, followed by 10 min of extension at 72 °C. For each colony, two reactions were prepared, one with the WT primer and one with the point mutation primer (P.μ primer) using 2 μL of template for each reaction.
Following PCR, 5 μL of each PCR product was electrophoresed on a 1% agarose gel for 40 min at 100 V. Colonies that showed the correct band size (Fig. S4, ESI†) were subjected to a second colony PCR to confirm their status (see Fig. 5 for example of R306H mutation). The fragment sizes of the PCR products were the expected size of 650–775 bp for TUB2 mutations (sizes varied depending on the position of the amino acid) and 1200 bp for NatMX cassette (Fig. S4, ESI†).
Conclusions
Of the six different point mutations generated in TUB2, four (A296T, R306H, V333W, and N337L) located in the peloruside binding site on β-tubulin gave resistance to yeast as expected. Only mutation Q291M failed to confer resistance in yeast and in fact actually increased the sensitivity to both laulimalide and peloruside. Though unexpected, this result may provide important information for medicinal chemists to increase the potency of this MSA class. Another mutation in the region of the taxane binding pocket of β-tubulin (R282Q) that was not expected to confer resistance to peloruside gave resistance to peloruside but not to laulimalide. Overall, these results suggest that peloruside binds to yeast tubulin in the region predicted by Huzil et al.23 Therefore, investigating the binding site on yeast tubulin could provide useful information about the mammalian binding site for peloruside and laulimalide, suggesting substantial evolutionary conservation of sequence in these areas despite clear divergence at the taxoid site. Laulimalide and peloruside showed a number of differences in yeast, with laulimalide being more potent than peloruside, and responding differently to a mutation R282Q outside the proposed binding pocket that confers resistance to epothilone in human cancer cells. The resistance to peloruside generated by R282Q may involve a global change to the protein. Finally, the efflux pump mutations increased the potency of laulimalide much more than that of peloruside, suggesting that the two compounds may have differential affinity for the efflux pumps. We conclude that the mechanism of binding and stabilization of microtubules in mammalian cells can be effectively modeled in yeast, a cell type ideally suited for genetic analysis and genome-wide screening techniques and also having the advantage of lacking any β-tubulin isotypes that can complicate interpretation of experiments in mammalian cells.Acknowledgements
The authors thank Jim Snyder and Pahk Thepchatri of Emory University for predictions of amino acids that might interfere with the action of peloruside, and ESR Ltd. for financial support and employment of David Maass. This research was supported by grants to J. H. Miller from the Foundation of Research, Science and Technology, the Cancer Society of New Zealand, the Wellington Medical Research Foundation, and Victoria University of Wellington.Notes and references
- M. Menacho-Márquez and J. R. Murguía, Clin. Transl. Oncol., 2007, 9, 221–228 CrossRef PubMed.
- A. Barberis, T. Gunde, C. Berset, S. Audetat and U. Lüthi, Drug Discov. Today: Technologies., 2005, 2, 187–192 CrossRef CAS PubMed.
- K. Baetz, L. McHardy, K. Gable, T. Tarling, D. Rebérioux, J. Bryan, R. J. Andersen, T. Dunn, P. Hieter and M. Roberge, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4525–4530 CrossRef CAS PubMed.
- W. H. Mager and J. Winderickx, Trends Pharmacol. Sci., 2005, 26, 265–273 CrossRef CAS PubMed.
- P. Yibmantasiri, D. C. Leahy, B. P. Busby, S. A. Angermayr, A. G. Sorgo, K. Boeger, R. Heathcott, J. M. Barber, G. Moraes, J. H. Matthews, P. T. Northcote, P. H. Atkinson and D. S. Bellows, Mol. Biosyst., 2012, 8, 902–912 RSC.
- M. Costanzo, A. Baryshnikova, J. Bellay, Y. Kim, E. D. Spear, C. S. Sevier, H. Ding, J. L. Y. Koh, K. Toufighi, S. Mostafavi, J. Prinz, R. P. St. Onge, B. VanderSluis, T. Makhnevych, F. J. Vizeacoumar, S. Alizadeh, S. Bahr, R. L. Brost, Y. Chen, M. Cokol, R. Deshpande, Z. Li, Z.-Y. Lin, W. Liang, M. Marback, J. Paw, B.-J. San Luis, E. Shuteriqi, A. H. Y. Tong, N. van Dyk, I. M. Wallace, J. A. Whitney, M. T. Weirauch, G. Zhong, H. Zhu, W. A. Houry, M. Brudno, S. Ragibizadeh, B. Papp, C. Pál, F. P. Roth, G. Giaever, C. Nislow, O. G. Troyanskaya, H. Bussey, G. D. Bader, A.-C. Gingras, Q. D. Morris, P. M. Kim, C. A. Kaiser, C. L. Myers, B. J. Andrews and C. Boone, Science, 2010, 327, 425–431 CrossRef CAS PubMed.
- M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef CAS PubMed.
- C. Dumontet and M. A. Jordan, Nat. Rev. Drug Discovery, 2010, 9, 790–803 CrossRef CAS PubMed.
- L. A. Amos, Semin. Cell Dev. Biol., 2011, 22, 916–926 CrossRef CAS PubMed.
- C. J. Bode, M. L. Gupta, E. A. Reiff, K. A. Suprenant, G. I. Georg and R. H. Himes, Biochemistry, 2002, 41, 3870–3874 CrossRef CAS PubMed.
- M. L. Gupta, C. L. Bode, G. I. Georg and R. H. Himes, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6394–6397 CrossRef CAS PubMed.
- T. B. Foland, W. L. Dentler, K. A. Suprenant, M. L. Gupta Jr. and R. H. Himes, Yeast, 2005, 22, 971–978 CrossRef CAS PubMed.
- R. D. Winefield, R. A. Entwistle, T. B. Foland, G. H. Lushington and R. H. Himes, ChemMedChem, 2008, 3, 1844–1847 CrossRef CAS PubMed.
- R. A. Entwistle, R. D. Winefield, T. B. Foland, G. H. Lushington and R. H. Himes, FEBS Lett., 2008, 582, 2467–2470 CrossRef CAS PubMed.
- D. E. Pryor, A. O'Brate, G. Bilcer, J. F. Díaz, Y. Wang, M. Kabaki, M. K. Jung, J. M. Andreu, A. K. Ghosh, P. Giannakakou and E. Hamel, Biochemistry, 2002, 41, 9109–9915 CrossRef CAS PubMed.
- T. N. Gaitanos, R. M. Buey, J. F. Díaz, P. T. Northcote, P. Teesdale-Spittle, J. M. Andreu and J. H. Miller, Cancer Res., 2004, 64, 5063–5067 CrossRef CAS PubMed.
- J. J. Field, J. F. Díaz and J. H. Miller, Chem. Biol., 2013, 20, 301–315 CrossRef CAS PubMed.
- A. Wilmes, R. Hanna, R. Heathcott, P. T. Northcote, P. H. Atkinson, D. S. Bellows and J. H. Miller, Gene, 2012, 497, 140–146 CrossRef CAS PubMed.
- H. A. Best, J. H. Matthews, R. W. Heathcott, R. Hanna, D. C. Leahy, N. V. C. Coorey, D. S. Bellows, P. H. Atkinson and J. H. Miller, Mol. Biosyst., 2013, 9, 2842–2852 RSC.
- S. L. Mooberry, G. Tien, A. H. Hernandez, A. Plubrukarn and B. S. Davidson, Cancer Res., 1999, 59, 653–660 CAS.
- K. A. Hood, L. M. West, B. Rouwé, P. T. Northcote, M. V. Berridge, S. J. Wakefield and J. H. Miller, Cancer Res., 2002, 62, 3356–3360 CAS.
- A. Kanakkanthara, A. Wilmes, A. O'Brate, D. Escuin, A. Chan, A. Gjyrezi, J. Crawford, P. Rawson, B. Kivell, P. T. Northcote, E. Hamel, P. Giannakakou and J. H. Miller, Mol. Cancer Ther., 2011, 10, 1419–1429 CrossRef CAS PubMed.
- J. T. Huzil, K. Chik, G. W. Slysz, H. Freedman, J. Tuszynski, R. E. Taylor, D. L. Sackett and D. C. Schriemer, J. Mol. Biol., 2008, 378, 1016–1030 CrossRef CAS PubMed.
- E. A. Clark, P. M. Hills, B. S. Davidson, P. A. Wender and S. L. Mooberry, Mol. Pharm., 2006, 3, 457–467 CrossRef CAS PubMed.
- A. Wilmes, K. Bargh, C. Kelly, P. T. Northcote and J. H. Miller, Mol. Pharm., 2007, 4, 269–280 CrossRef CAS PubMed.
- A. Wilmes, D. O'Sullivan, A. Chan, C. Chandrahasen, I. Paterson, P. T. Northcote, A. C. La Flamme and J. H. Miller, Cancer Chemother. Pharmacol., 2011, 68, 117–126 CrossRef CAS PubMed.
- J. Snider, A. Hanifl, M. E. Lee, K. Jin, A. R. Yu, C. Graham, M. Chuk, D. Damjanovic, M. Wierzbicka, P. Tang, D. Balderes, V. Wong, M. Jessulat, K. D. Darowski, B.-J. San Luis, I. Shevelev, S. L. Sturley, C. Boonel, J. F. Greenblatt, Z. Zhang, C. M. Paum, M. Babu, H.-O. Park, S. Michaelis and I. Stagljar, Nat. Chem. Biol., 2013, 9, 565–572 CrossRef CAS PubMed.
- A. Begaye, S. Trostel, Z. M. Zhao, R. E. Taylor, D. C. Schriemer and D. L. Sackett, Cell Cycle, 2011, 10, 3387–3396 CrossRef CAS PubMed.
- J. Bennett, K. Barakat, J. T. Huzil, J. Tuszynski and D. C. Schriemer, Cell, 2010, 17, 725–734 Search PubMed.
- T. L. Nguyen, X. M. Xu, R. Gussio, A. K. Ghosh and E. J. Hamel, Chem. Info. Model., 2010, 50, 2019–2028 CrossRef CAS PubMed.
- M. Khrapunovich-Baine, V. Menon, C. P. H. Yang, P. T. Northcote, J. H. Miller, R. H. Angeletti, A. Fiser, S. B. Horwitz and H. Xiao, J. Biol. Chem., 2011, 286, 11765–11778 CrossRef CAS PubMed.
- P. Giannakakou, R. Gussio, E. Nogales, K. H. Downing, D. Zaharevitz, B. Bollbuck, G. Poy, D. Sackett, K. C. Nicolaou and T. Fojo, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 2904–2909 CrossRef CAS PubMed.
- C. J. Tsai, B. Ma and R. Nussinov, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9970–9972 CrossRef CAS.
- S. Kumar, B. Ma, C. J. Tsai, N. Sinha and R. Nussinov, Protein Sci., 2000, 9, 10–19 CrossRef CAS PubMed.
- D. Botstein and G. R. Fink, Genetics, 2011, 189, 695–704 CrossRef CAS PubMed.
- J. J. Field, A. J. Singh, A. Kanakkanthara, T. Halafihi, P. T. Northcote and J. H. Miller, J. Med. Chem., 2009, 52, 7328–7332 CrossRef CAS PubMed.
- L. M. West, P. T. Northcote and C. N. Battershill, J. Org. Chem., 2000, 65, 445–449 CrossRef CAS PubMed.
- C. Janke, M. M. M. Magiera, N. Rathfelder, C. Taxis, S. Reber, H. Maekawa, A. Moreno-Borchart, G. Doenges, E. Schwob, E. Schiebel and M. Knop, Yeast, 2004, 21, 947–962 CrossRef CAS PubMed.
- R. D. Gietz and R. A. Woods, Methods Enzymol., 2002, 350, 87–96 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary methods, results and discussion.doc. See DOI: 10.1039/c3mb70380k |
| This journal is © The Royal Society of Chemistry 2014 |