 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Glia co-culture with neurons in microfluidic platforms promotes the formation and stabilization of synaptic contacts†
Mingjian
Shi‡
a,
Devi
Majumdar‡
a,
Yandong
Gao‡
b,
Bryson M.
Brewer
b,
Cody R.
Goodwin
c,
John A.
McLean
c,
Deyu
Li
b and
Donna J.
Webb
*ad
aDepartment of Biological Sciences and Vanderbilt Kennedy Center for Research on Human Development, Vanderbilt University, Nashville, TN 37235, USA. E-mail: donna.webb@vanderbilt.edu; Fax: +1 615-343-6707; Tel: +1 615-936-8274
bDepartment of Mechanical Engineering, Vanderbilt University, Nashville, TN 37235, USA
cDepartment of Chemistry, Vanderbilt University, Nashville, TN 37235, USA
dDepartment of Cancer Biology, Vanderbilt University, Nashville, TN 37235, USA
First published on 21st May 2013
Abstract
Two novel microfluidic cell culture schemes, a vertically-layered set-up and a four chamber set-up, were developed for co-culturing central nervous system (CNS) neurons and glia. The cell chambers in these devices were separated by pressure-enabled valve barriers, which permitted us to control communication between the two cell types. The unique design of these devices facilitated the co-culture of glia with neurons in close proximity (∼50–100 μm), differential transfection of neuronal populations, and dynamic visualization of neuronal interactions, such as the development of synapses. With these co-culture devices, initial synaptic contact between neurons transfected with different fluorescent markers, such as green fluorescent protein (GFP) and mCherry-synaptophysin, was imaged using high-resolution fluorescence microscopy. The presence of glial cells had a profound influence on synapses by increasing the number and stability of synaptic contacts. Interestingly, as determined by liquid chromatography-ion mobility-mass spectrometry, neuron-glia co-cultures produced elevated levels of soluble factors compared to that secreted by individual neuron or glia cultures, suggesting a potential mechanism by which neuron-glia interactions could modulate synaptic function. Collectively, these results show that communication between neurons and glia is critical for the formation and stability of synapses and point to the importance of developing neuron-glia co-culture systems such as the microfluidic platforms described in this study.
Introduction
Neurons are specialized cells that communicate through complex adhesive junctions called synapses. Synapses are structurally comprised of presynaptic axonal terminals and postsynaptic regions from the dendrites. In the CNS, postsynaptic terminals of excitatory synapses are composed of dendritic spines, which are actin-rich protrusions from dendritic shafts.1,2 Structurally, dendritic spines typically consist of a thin elongated neck connected to a bulbous head.3 Dendritic spines are highly dynamic structures whose morphologies can vary from filopodia-like protrusions to mature stubby, thin, or mushroom-shaped spines, depending upon their stage of development and on neuronal activity.4,5 Abnormalities in the density, morphology, and development of dendritic spines is thought to underlie many neurological disorders, including Alzheimer's disease, epilepsy, Fragile-X syndrome, schizophrenia, Down's syndrome, Parkinson's disease, and intellectual disorders,6–11 which indicates a critical role for spines in cognitive processes. Despite the central importance of spines and synapses in higher brain function, the molecular signals that regulate their formation and maturation are currently not well understood. Advancement in this area has been hindered by the difficulty in dynamically imaging the development of dendritic spines and synapses with most of the available data on this process generated from fixed cell preparations.12,13Glia are another cell type that are found in large numbers in the CNS. While once thought to serve only a passive role, glia are now believed to be active neuronal partners in forming the complex circuitry in the developing brain.14–16 The development of astrocytes, a class of glia, parallels the formation of synapses, which suggest that glia are involved in this process.17 In addition, soluble molecules secreted by astrocytes, such as tumor necrosis factor-α, thrombospondins, and activity dependent neurotrophic factors have been suggested to influence synaptogenesis and regulate synaptic strength.18–20 Although these results point to the possible contribution of glia to synaptic function, additional studies are needed to better understand the role of glia in modulating the initial formation and development of dendritic spines and synapses. Moreover, these data indicate the importance of developing neuron-glia co-culture systems for examining synaptic processes. Dynamic observation of spine and synaptic contacts in the presence of glia and glia-derived factors can provide important information about the rates of formation, stabilization, and maturation of dendritic spines and synapses. The ability to dynamically image neurons and glia under optimal culture conditions also opens up new avenues to test pharmacologically relevant substances that influence the health and development of these cells.
To date, a few microfluidic devices have been described where neurons and glia can be cultured with each other. One co-culture platform has a multi-compartment design with a central neuronal chamber, which holds the cell bodies (soma), and six axonal-glial chambers that contain axons of neurons and oligodendrocytes, a type of glia.21,22 The chambers in this design are separated by microchannels that enable fluidic isolation between the soma and the axon-glia compartments. Another configuration that permits interaction between axons and glia uses a circular microfluidic platform to isolate soma from axons.23 The above-mentioned devices and others reported in the literature allow for association between axon and glia, but do not permit reversible separation of glia from neuronal cultures.21,23–26 More importantly, these devices are not suited for investigating the effects of glia on neuron-neuron interactions, such as synapse formation. We have recently described a microfluidic co-culture device that is very useful for studying neuron-glia interactions in an isolated, controlled environment,27 but it was not designed to capture neuron–neuron interactions. To overcome this limitation, we generated two microfluidic schemes that allow co-culture of neurons and glia as well as live-cell imaging of neuronal (synaptic) contact.
Materials and methods
Fabrication of the microfluidic devices
The microfluidic cell co-culture platforms were fabricated using standard soft-lithography techniques with replica molding poly(dimethylsiloxane) (PDMS) as previously described.27–30 For both the vertically-layered set-up and four chamber devices, the SU-8 mold was constructed for replica-fabrication of cell culture chambers that were 5.4–6.4 mm long, 800 μm wide, and 100–200 μm in height, which were connected by microchannels 100–150 μm wide and 5 μm high. The separation distance between the culture chambers was ∼50–100 μm. In both devices, the cell culture chambers were connected to media reservoirs through channels that were 200 μm wide and 100–200 μm high. We observed successful device performance across the range of dimensions listed.To form the first layer of each device, a pre-polymer PDMS solution was mixed with a curing agent (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) and poured over the molds. After degassing for 1 h, the PDMS layers were then cured for 2 h at 70 °C over their respective molds to permit them to solidify. The PDMS layers were subsequently peeled from their respective molds, and holes for the media wells were made using a sharp metal puncher. The surfaces of microscopic glass coverslips (No. 1, VWR Vista Vision, Suwanee, GA) and the PDMS layers were treated with oxygen plasma and then bonded together to complete the first layer for each device. The second PDMS layer, defining a pressure chamber for each device, was fabricated by gluing a 4–8 mm × 12 mm × 1 mm (width, length, height) glass slab to a standard glass slide. PDMS was then poured onto the mold and cured as described above for the first PDMS layer. The resulting pressure chamber was bonded on top of the first PDMS layer after oxygen plasma treatment (Supplementary† Fig. 1 and 2). The microfluidic devices were finished by attaching cloning cylinders (Fisher Scientific, Pittsburg, PA), which served as media reservoirs, to the punched holes and by gluing two microbore tubes (Cole-Parmer, Vermon Hills, IL) to the pressure chamber with liquid PDMS.
:1 ratio) and poured over the molds. After degassing for 1 h, the PDMS layers were then cured for 2 h at 70 °C over their respective molds to permit them to solidify. The PDMS layers were subsequently peeled from their respective molds, and holes for the media wells were made using a sharp metal puncher. The surfaces of microscopic glass coverslips (No. 1, VWR Vista Vision, Suwanee, GA) and the PDMS layers were treated with oxygen plasma and then bonded together to complete the first layer for each device. The second PDMS layer, defining a pressure chamber for each device, was fabricated by gluing a 4–8 mm × 12 mm × 1 mm (width, length, height) glass slab to a standard glass slide. PDMS was then poured onto the mold and cured as described above for the first PDMS layer. The resulting pressure chamber was bonded on top of the first PDMS layer after oxygen plasma treatment (Supplementary† Fig. 1 and 2). The microfluidic devices were finished by attaching cloning cylinders (Fisher Scientific, Pittsburg, PA), which served as media reservoirs, to the punched holes and by gluing two microbore tubes (Cole-Parmer, Vermon Hills, IL) to the pressure chamber with liquid PDMS.
The PDMS-based microfluidic valve is effective due to the softness and flexibility of the material. Relatively low actuation forces can produce large deflections in PDMS.31 This property allows the application of pressure via the pressure control chamber to collapse the roofs of the connecting microgrooves to the glass substrate and subsequently block the flow. To separate the cell culture chambers, the pressure chamber was filled with 0.2–0.3 ml of either air or water by injection with a syringe. Water was used to hydraulically pressurize the chambers when separation was needed for more than 3–4 h. In order to retain fluid within the pressure chamber, the microbore tubes, which were used to inject air or water, were clamped with two standard metal pinch clamps (Fisher Scientific, Pittsburgh, PA). The valve barriers were deactivated (opened) by removing air or water from the pressure chamber and applying a slight negative pressure to ensure lifting of the barriers.
Loading of glia and neurons into the microfluidic co-culture devices
For the vertically-layered co-culture scheme, the devices were UV sterilized and prepared for co-culture by coating the cell chambers with 1 mg ml−1 poly-L-lysine (PLL) (Sigma-Aldrich, St. Louis, MO) in 0.1 M borate buffer (pH 8.5) for 12 h at 37 °C. Excess PLL was washed away by flowing sterilized, deionized water through the chambers for at least 2 h. The coated chambers were then equilibrated with Minimum Essential Medium (MEM) (Invitrogen, Carlsbad, CA) containing 10% horse serum and 0.6% glucose (glia media) for 20–30 min. The devices were taped to 100 mm tissue culture dishes prior to loading glial cells. Glia were isolated from rat brains of 2 day old pups as previously described.32 Rats were housed in the Vanderbilt University vivarium and were cared for according to national and international guidelines. The protocol for isolation of glial and neuronal cells was approved by the Institutional Animal Care and Use Committee. Glial cells at a density of 5 × 105 cells ml−1 were loaded into the device (25000 cells/chamber). After 3–5 min, the devices were inverted and incubated for an additional 2 h at 37 °C. Due to inversion of the devices, the glial cells deposited on the roofs of the cell culture chambers and attached to the PDMS surfaces. We consistently found that 2 h were sufficient for the suspended glia to attach and spread on the PDMS roofs of the cell chambers. Following the return of the devices to their upright position, the attached glia were incubated for 4–5 days until they reached confluence. Media in the reservoirs were replaced with fresh glia media every 36 h, removing collected media from the waste wells at the same time.
Devices were then prepared for loading of hippocampal neurons by removing glia media from the reservoirs and equilibrating them with B27-supplemented NeurobasalTM media (GIBCOTM Invitrogen, Carlsbad, CA). Hippocampal neurons were isolated from dissected brains of E19 rat embryos as previously described32 and loaded at a density of 5 × 105 cells ml−1 into the microfluidic platforms (50000 cells/chamber). The devices were incubated for 3 h at 37 °C to allow neurons to adhere to the PLL-coated glass surfaces of the cell culture chambers. After neurons attached, the reservoirs were filled with 300 μl of B27-supplemented NeurobasalTM media (neuronal media). The monolayer of confluent glial cells on the roofs of the chambers conditioned the B27-Neurobasal™ media in situ, providing nutrients for the neurons. Every 36 h, media in the reservoirs were replenished, and collected media from the downstream waste reservoirs were removed. Microfluidic co-culture devices were kept in a standard cell culture incubator (Fisher Scientific, Hampton, NH), which was injected with 5% CO2, and maintained at 37 °C.
The UV sterilized four chamber devices were prepared for coating by activating the valve barriers with hydraulic pressure as described above. The two outer chambers, designated as the glial chambers, were coated with 200 μl of type I collagen (10 μg ml−1) in sterile phosphate-buffered saline (PBS) (GIBCOTM Invitrogen, Carlsbad, CA). The two inner neuronal chambers were coated with 200 μl of 1 mg ml−1 PLL in borate buffer. Excess collagen and PLL were removed by flowing sterile PBS and water, respectively, through the chambers for 2 h. The cell culture chambers were then equilibrated in glia and neuron media, respectively. 50 μl of glial cell suspension (5 × 105 cells ml−1) were loaded into each of the glial chambers (25000 cells/chamber), and the devices were incubated for 2 h at 37 °C to permit cell attachment. After 2 h, the chambers were filled with 400 μl of glia media and incubated for 4–5 days until cells reached confluence. During this time, neuron media were flowed through the PLL-coated neuronal chambers to keep them hydrated. After glia reached confluency, neurons were loaded (5 × 105 cells ml−1) into the two central neuronal chambers (50000 cells/chamber) as described above. After neurons attached, the pressure chamber of each device was deflated by releasing the clamps from the tubes and removing the water. A slight negative pressure inside the pressure chamber ensured that the valve barriers underneath were fully deactivated and the neuronal and the glial chambers were connected. Then, 400 μl of fresh neuron media were added to the glial reservoirs and 200 μl of the same media were added to the neuronal reservoirs. The unequal volumes in the reservoirs created a passive pressure difference, which caused a steady flow of media from the glial chambers to the adjacent neuronal chambers, providing fresh glia-conditioned media for the neurons. Fresh neuron media were added to the devices every 36 h, and the collected media were removed from the waste reservoirs at the same time.
In some experiments, neurons were loaded into microfluidic devices as described above and cultured with media, which were conditioned from glia that were plated on tissue culture dishes (externally generated glia-conditioned media). To prepare externally generated glia-conditioned media, glial cells were plated in 60 mm tissue culture dishes (1 × 105 glia/dish) and incubated for 14 days in glia media. The glia media were then replaced with fresh neuronal media, which were conditioned by incubation with glial cells for 15–18 h. Fresh externally generated glia-conditioned media were added to the microfluidic devices every 2–3 days.
Neuronal transfection in the microfluidic platforms
GFP and mCherry cDNAs used for transfection were cloned into an expression vector containing a neuronal specific α1-tubulin promoter, which was provided by Freda Miller.33 mCherry-synaptophysin cDNA was kindly provided by Anne Marie Craig (University of British Columbia, Vancouver, Canada). Neurons in both vertically-layered and four chamber configurations were transfected as previously described using a modified calcium phosphate method with minor modifications.27 Briefly, GFP or mCherry-tagged cDNAs (3–6 μg) were added to a tube containing 30 μl of 120 mM CaCl2. Calcium phosphate-DNA precipitate formation was initiated by slowly bubbling 30 μl of HEPES-buffered solution containing 42 mM HEPES, 274 mM NaCl, 9.5 mM KCl, 1.4 mM Na2HPO4, 15 mM glucose (HBS), pH 7.3 into the tube. Following addition of 740 μl of transfection media, the calcium phosphate-DNA mixture was allowed to stand for ∼5 min at room temperature to facilitate precipitate formation. Transfection media were prepared by mixing neuronal media 1:1 with MEM containing N2 supplement (GIBCOTM Invitrogen, Carlsbad, CA) and conditioning on glial cells for 15–18 h. Neuronal chambers in both vertically-layered and four chamber set-ups were separated by injecting 0.2–0.3 ml of air into the pressure chambers, activating the valve barriers underneath. The calcium phosphate-DNA mixtures were flowed through the neuronal chambers for 1.5 h at 37 °C. After washing, the valve barriers were deactivated and the media reservoirs were filled with fresh neuron media.
Microscopy and live-cell imaging in the microfluidic co-culture devices
At days 12–13 in culture, neurons in both vertically-layered and four chamber set-ups were prepared for live-cell imaging by replacing neuron media with 50 mM HEPES containing B27-supplemented NeurobasalTM media without phenol red, pH 7.4. Live-cell imaging was performed on a Quorum WaveFX spinning disk confocal system equipped with a Nikon Eclipse Ti microscope, a PlanApo 60× TIRF objective (NA 1.49), and a Hamamatsu ImageEM-CCD camera. Images were acquired at 15 min intervals for 12 h using MetaMorph software (Molecular Devices, Sunnyvale, CA). During imaging, cells were maintained at 37 °C using a temperature-controlled chamber (Live Cell Instrument, Seoul, Korea). GFP and mCherry were excited with 491 nm and 561 nm laser lines, respectively.Fluorescence recovery after photobleaching (FRAP) was performed on the Quorum WaveFX spinning disk confocal system with the 60× TIRF objective as previously described.34 Briefly, regions of interest were photobleached using a 405-nm diode laser for 2000 ms with 100% laser power. mCerulean-actin images were acquired at 10 s intervals with a Hamamatsu ImageEM-CCD camera and MetaMorph software. For analysis of FRAP, the background subtracted fluorescence intensity at every time point was corrected for the fluorescence loss that resulted from image acquisition. The corrected data were normalized to the base-line fluorescence (Ipre), defined as 100%, and graphed according to the following equation: FI(t) = (It × Inf, pre)/(Ipre × Inf,t) where nf denotes a region not subjected to FRAP.
Immunostaining cells in microfluidic co-culture platforms
Fixed samples were immunostained as previously described.27,35 Briefly, glia were fixed by flowing 4% paraformaldehyde and 4% sucrose in PBS through the devices for 30 min at room temperature. Fixed cells were permeabilized by flowing 0.2% Triton X-100 in PBS through the devices for 15 min. Following permeabilization, devices were washed for 30 min in PBS and incubated for 1 h with 20% goat serum in PBS to block non-specific binding. Glia were stained for glial fibrillary acidic protein (GFAP) by flowing a GFAP antibody (1:1000 dilution in 5% goat serum in PBS) through the devices for 1 h at room temperature. The GFAP monoclonal antibody was purchased from Cell Signaling Technology, Inc. (Danvers, MA). Unbound antibody was removed by washing with PBS for 30 min, and secondary anti-mouse Alexa Fluor® 647 (Molecular Probes, Eugene, OR) at a dilution of 1:500 in 5% goat serum in PBS was then flowed through the devices for 45 min at room temperature. After three 15 min washes with PBS, devices were filled with Aqua Poly/Mount (Polysciences, Inc., Warrington, PA), and cells were visualized using the Quorum WaveFX spinning disk confocal system with a 10× ADL (NA 0.25) or 20× PlanFluor (NA 0.75) objective. Alexa Fluor 647 was excited with a 642 laser line.
Ultra performance liquid chromatography-ion mobility-mass spectrometry analysis
Metabolites were isolated from 60 μL of effluent from the waste channel of the microfluidic co-culture devices by first precipitating proteins with 3:1 (v:v) cold methanol kept on dry ice. Samples were vortexed for 10 s and centrifuged at 14000 rpm for 10 min at 4 °C. Supernatants (150 μl) from the samples, which contained the metabolites, were collected and dried down in a SpeedVac. The samples were then reconstituted in 100 μL of solvent A (water with 0.1% formic acid), and 10 μL of each sample was combined to create a quality control.
For UPLC analysis, a 25 min method was applied with mobile phase A (water with 0.1% formic acid) and mobile phase B (acetonitrile with 0.1% formic acid). A constant flow rate of 75 μL min−1 was used, with an initial composition of 100% A, which was held for 1 min. Over the next 11 min, a linear gradient from 100% A to 100% B was performed, and the mobile phase was held at 100% B for 2 min. Over the next 0.1 min, the mobile phase was returned to 100% A at a linear gradient. For the next 10.9 min, 100% A was flowed at 75 μL min−1 to re-equilibrate the column. A 5 μL loop was used for injections. Ten column conditions were performed prior to analysis, which injected 10 μL of quality control (doubled-filled sample loop). Following column conditions, technical triplicates of prepared samples were analyzed at random intervals, with quality controls performed after every 5 injections. To increase reproducibility, a 10 μL bolus was injected into the 5 μL sample loop, overfilling the loop and insuring reproducible injection volumes.
In all cases the UPLC effluent was characterized using a state-of-the-art ion mobility-mass spectrometer (IM-MS), Synapt G2 (Waters Corp., Milford, MA). Calibration was performed on date of acquisition with sodium formate clusters in a mass range of 50-2000 Da and a mass accuracy tolerance of 1 ppm. Data were acquired at 3 Hz in resolution continuum mode. The capillary voltage was set at 3.00 kV, the source temperature was held constant at 110 °C, and the sampling cone was held at a setting of 35 with the extraction cone at a setting of 5.0. The nitrogen desolvation gas flow was held at a temperature of 150 °C and a flow rate of 400.0 L h−1. The nitrogen cone gas flow was set to 10.0 L h−1. The helium cell prior to the mobility chamber had a gas flow setting of 180.0, and the ion-mobility nitrogen gas flow was set to 90.0 mL min−1. The ion mobility wave velocity was held constant at 550 m s−1 at a wave height of 40.0 V. Leucine enkephalin was used as a lockmass compound ([M + H]+ equal to 556.2771 Da) and infused at a flow rate of 10 μL min−1. Calibration corrections were performed during data acquisition. Low and high energy data were collected for each scan with a ramped energy profile performed post mobility separation from 10–45 eV for the high energy function.
Data were peak-picked using XCMS36 in R, which is a software environment for statistical computing. Default parameters were used, performing retention correction using the obiwarp method. Multivariate statistical analyses were performed using the Ezinfo 2.0 (Umetrics, Umeå, Sweden).
Results
Development of a vertically-layered microfluidic neuron-glia co-culture scheme for studying neuronal interactions
In the vertically-layered configuration, glia and neurons were co-cultured in a top-bottom configuration with a monolayer of glial cells on the PDMS roofs over the cell chambers, while neurons were cultured on PLL-coated bottom glass surfaces of the cell chambers within the same device (Supplementary† Fig. 1a–d). To achieve this, cell chambers were coated with 1 mg ml−1 PLL to promote cell attachment (Fig. 1a). Following washing and equilibrating the chambers with glia media, glia suspended in culture media were loaded into each of the loading reservoirs connected to the cell culture chambers (Fig. 1b). After 3–5 min, devices were inverted to allow glia to attach to the PDMS roofs (Fig. 1c). Due to the low volume of suspended glia and the inherent surface tension within the loading reservoirs, the device inversion did not cause significant spillage of the glial suspension from the reservoirs. After glial cells attached to the roofs, devices were reverted to their upright position, which caused unattached glia to be washed away by the flow of culture media. Glia were maintained in the co-culture devices until they reached confluence.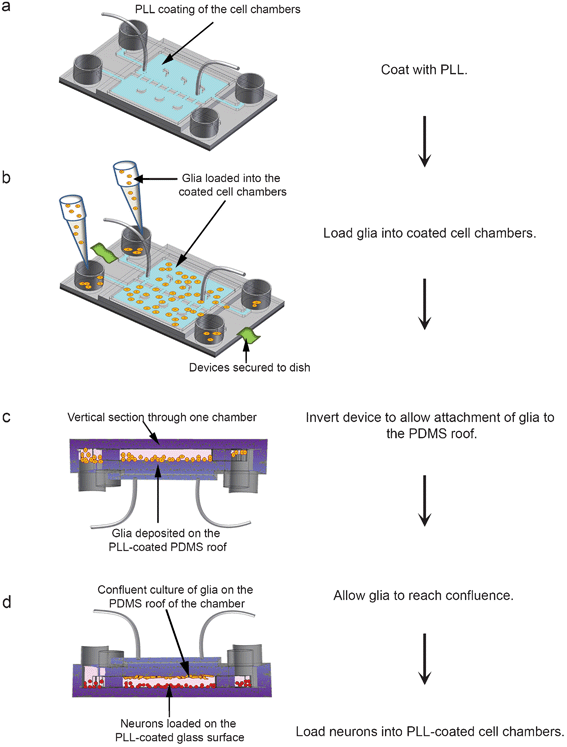 | ||
| Fig. 1 Flow chart for cell loading in vertically-layered microfluidic platforms. (a) The cell culture chambers (blue chambers) of the microfluidic devices were coated for 12 h with 1 mg ml−1 PLL. (b) After coating, devices were secured to a tissue culture dish, and glia (orange spheres) were loaded into the cell chambers. (c) The devices containing the cells were incubated for 3–5 min, inverted, and incubated for an additional 2 h for cells to attach. A cross-section through an inverted device is depicted with attached glia on the PDMS roof of a cell culture chamber. (d) As depicted, devices were then reverted to their upright position and incubated for 4–5 days in glia media until cells reached confluence. Neurons (red spheres) suspended in neuron media were loaded into the devices and allowed to attach to the PLL-coated glass surfaces of the cell chambers. | ||
To prepare for neuronal loading, glia media were removed from the devices, and the cell chambers were equilibrated with fresh neuron media. Dissociated rat hippocampal neurons suspended in neuron media were loaded into the co-culture devices and allowed to attach to the PLL-coated glass surfaces of the cell chambers (Fig. 1d). Neurons in this co-culture configuration could be maintained in a healthy state (>90% cell viability) for over a month.
The vertically-layered configuration could also sustain healthy glia (>90% cell viability) for over a month. After neurons were loaded, only neuron media flowed through the chambers. It was surprising that this did not adversely affect the general health of the glial monolayer, as evident by the sustained normal morphology of glia immunostained for the glia specific marker GFAP (Fig. 2a). Moreover, neurons in the co-culture configuration had a normal morphology that consisted of elaborate branching of neuronal arbors (Fig. 2b).
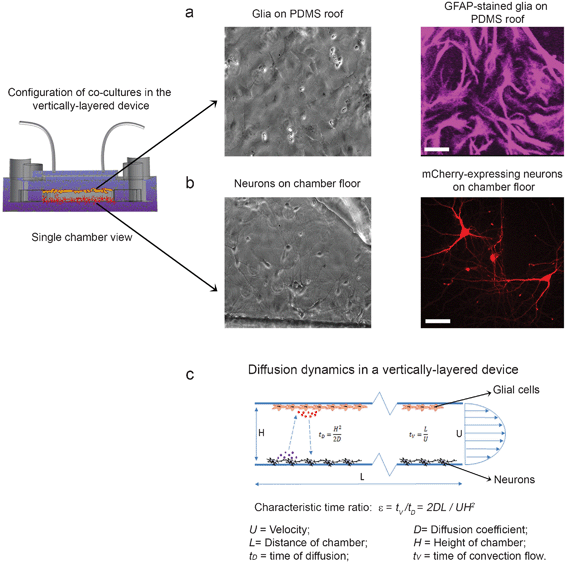 | ||
| Fig. 2 Co-cultures of neurons and glia in the vertically-layered configuration. Neurons in microfluidic platforms were transfected with mCherry at day 3 in culture. Neuron and glia co-cultures were then maintained in the devices and fixed at day 13 in culture. (a) Glial cells were either imaged with DIC (left panel) or immunostained for the glia-specific marker GFAP (right panel). (b) Neurons were imaged with DIC (left panel) or viewed in fluorescence (right panel). (c) Diagram of the diffusion dynamics within the vertically-layered device is shown. For panels a and b, scale bar = 25 μm. | ||
The vertically-layered devices present several benefits for maintaining healthy neuronal cultures. In these devices, both cell types are provided with a continuous source of fresh and adequately aerated media to ensure optimal health. In addition, an even separation distance of ∼100 μm is kept between neurons and glia in the cell chambers. We perceive this as an advantage over the traditional plate method,32 in which the volume of neuron media usually determines the separation distance between the two populations of cells in culture. Moreover, the short separation distance (∼100 μm) should allow for rapid exchange of growth factors and nutrients between the cells. To confirm this, we calculated the time of molecular diffusion (tD) between the top and bottom of the cell chambers using the following equation: tD = H2/2D where H is the height of the chamber and D is the diffusion coefficient (Fig. 2c). For example, in the case of nerve growth factor (NGF), an ∼130 kDa protein complex, with a D of 126 μm2 s−1,37 it would only take ∼40 s (assuming a chamber height of 100 μm) to diffuse across the ∼100 μm distance between the neurons and glia. Due to the short time period required for molecules to diffuse between the two cell types, they should reach their targets before flowing out of the chamber. To verify this, a characteristic time ratio (ε) was determined using the equation: ε = tV/tD where time of convection flow (tV) is defined as tV = L/U (L is length of the chamber and U is velocity of fluid flow)38 (Fig. 2c). For a typical flow velocity of 2–8 μm s−1, the time ratio is between ∼20 and ∼80 for NGF. This means that it takes much less time for the molecules to diffuse across the chamber vertically than for them to flow out of the chamber. Therefore, this microfluidic platform could maintain an effective molecular exchange between the two cell types.
Neurons form dendritic spines and synaptic contacts with the vertically-layered co-culture scheme
These microfluidic devices are uniquely suited to visualize synapses as they develop between neuronal populations, since neurons in the cell culture chambers can be separated using the valve-enabled barrier29 and transfected with different cDNAs encoding fluorescently-tagged proteins (Fig. 3a). After the barrier is raised, neuronal axons and dendrites extend toward the adjacent chamber and form synaptic contacts. This design allowed us to examine the contribution of glia to synapse formation. To begin, we transfected neurons with GFP at day 5 in culture and subjected them to fluorescence microscopy at day 8 in culture. GFP expression permitted us to visualize and subsequently quantify the density of dendritic protrusions, which are precursors to dendritic spines. The number of dendritic protrusions was significantly greater in the vertically-layered configuration with neuron-glia co-culture compared with those observed in which externally generated glia-conditioned media were flowed through the neuronal chambers in the absence of glia (Fig. 3b,c). These results indicate that the presence of glia increased the number of dendritic protrusions and raised the question as to whether these protrusions formed synaptic contacts.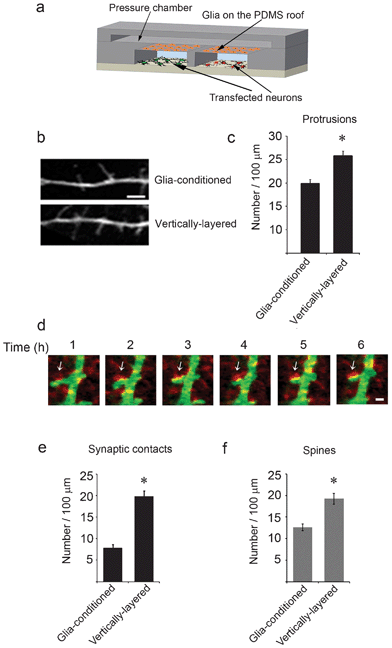 | ||
| Fig. 3 Synaptic contacts form between neurons in adjacent cell chambers. (a) A schematic of a cross-section through a microfluidic device with the vertically-layered co-culture set-up depicting glia on the PDMS roof and differentially transfected neurons in the cell chambers. (b) Neurons in the vertically-layered configuration of the co-culture platform were transfected with GFP at day 5 in culture and imaged at day 8 in culture. Dendritic protrusions, which are precursors to dendritic spines were observed along the dendrites. (c) Quantification of the number of protrusions is shown for neurons co-cultured with glia and for neurons treated with externally-conditioned glia media. Error bars represent S.E.M. for 40–45 dendrites from three independent experiments (* p < 0.0001). (d) At day 3 in culture, neurons in the cell chambers were differentially transfected with GFP (green) and mCherry-synaptophysin (red), maintained in co-culture with glia, and subjected to live-cell imaging at day 13. Time-lapse images show that GFP and mCherry-synaptophysin expressing neurons form contacts. Sites of synaptic contact are observed as yellow puncta along the dendrite (arrows). Scale bar = 2 μm (e,f) Quantification of the number of synaptic contacts and spine density is shown for neurons co-cultured with glia in vertically-layered devices and for neurons cultured alone in two chamber microfluidic devices with externally-conditioned glia media. Error bars represent S.E.M. from five separate experiments (* p < 0.0001). | ||
To assess the number of synaptic contacts, neurons in adjacent cell chambers were transfected with GFP and the presynaptic protein mCherry-synaptophysin, respectively, and subjected to live-cell fluorescence imaging at days 12–13 in culture. Synaptic contacts were observed when dendrites from GFP-expressing neurons contacted mCherry-synaptophysin puncta on axons from adjacent neurons (Fig. 3d). Interestingly, the number of synaptic contacts was increased by approximately 2.5-fold in the vertically-layered devices compared with those observed with neurons cultured alone with glia-conditioned media (Fig. 3e). Moreover, the number of dendritic spines was increased by about 1.5 fold in the vertically-layered co-culture devices compared with those observed when neurons were cultured alone with glia-conditioned media (Fig. 3f). These results suggest that labile factors secreted by glia and/or the presence of glial cells themselves are critical for development of spines and synapses. In the microfluidic co-culture devices, the presence of glia in close proximity (<100 μm) to neurons may ensure a continuous supply of critical factors to neurons that positively contribute to dendritic spine and synaptic development.
Development of a four chamber microfluidic co-culture platform for studying neuron and glia communication
The four chamber co-culture device consists of four cell culture chambers that are separated by three valve-enabled PDMS barriers. A single pressure chamber constructed on top of the barriers controls all three valve barriers, allowing for reversible separation of the cell chambers (Supplementary† Fig. 2a–d). Since our goal was to examine synaptic contact between adjacent neuronal populations, neurons were cultured in the two inner chambers and glia in the two outer cell chambers. To confirm that the valve mechanism was effective with the four chamber design, the valve barriers were activated, which should effectively separate the four cell chambers and prevent fluid exchange between them. Then, 200 μl of Alexa Fluor 488 conjugated avidin (green) were added to one glia chamber, and 200 μl of Alexa Fluor 647 conjugated ovalbumin (red) were added to the other glia chamber (Supplementary† Fig. 3a). In both cases, we did not observe cross-flow of the two fluorescent proteins into the neuronal chambers. However, when the barriers were released, the fluorescent proteins flowed into the adjacent neuronal chambers (Supplementary† Fig. 3b). Similar results were obtained when the fluorescent markers were added to the neuronal chambers. Collectively, these results show that the valve barriers can reversibly separate the four cell culture chambers of the microfluidic platforms. Compared with the vertically-layered scheme, the four chamber design provides additional control for media exchange between different cell populations by manipulating the pressure in different chambers.To facilitate cell loading, the valve barriers were activated to isolate the four cell chambers in the microfluidic devices. The glial and neuronal chambers were coated with type I collagen and PLL, respectively (Fig. 4a,b). After washing to remove uncoated type I collagen/PLL, glia were loaded into each of the two outer chambers and allowed to attach and subsequently reach confluence (Fig. 4c). Once the glia reached confluence, neurons were loaded into the neuronal chambers as described for the vertically-layered co-culture scheme (Fig. 1d and 4d). After neurons attached, the chambers were connected by deactivating the valve barriers.
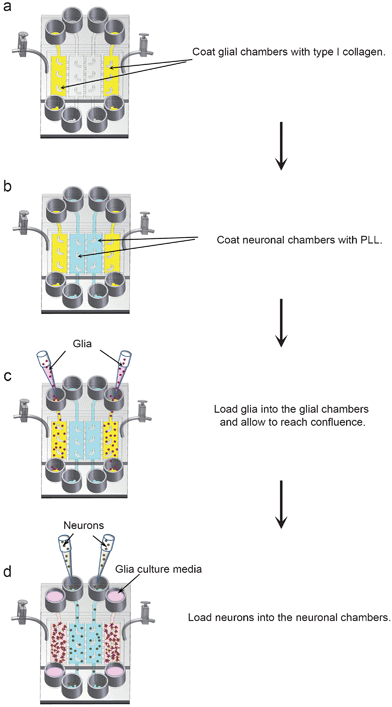 | ||
| Fig. 4 Flow chart for loading glia and neurons into four chamber microfluidic devices. (a,b) The four cell chambers were separated by activating the valve barriers. The glial chambers were coated with type I collagen (yellow chambers) and the neuronal chambers were coated with PLL (blue chambers). (c) Glia (purple spheres) were loaded into the glial chambers and incubated for 4–5 days until they reached confluence. (d) After glia reached confluence, neurons (grey spheres) were loaded into the neuronal chambers. All cells were incubated in neuron media after loading neurons into the devices. | ||
Visualization of synaptic contact within four chamber co-culture devices
As with the vertically-layered co-culture scheme, cells in the four chamber platform were healthy (>90% cell viability) and could be differentially transfected with fluorescent markers (Fig. 5a,b), which permitted us to determine if glia similarly affected the development of synapses in the four chamber device. When neurons and glia were co-cultured in the four chamber device, the number of dendritic protrusions was significantly increased compared to those observed with neurons cultured alone with glia-conditioned media (Supplementary† Fig. 4a,b). Indeed, the increase in the density of dendritic protrusions was similar to that seen when neurons and glia were co-cultured in the vertically-layered configuration (Supplementary† Fig. 4a,b). These results further show that the presence of glia enhances protrusion density, which could lead to increased synaptic contact.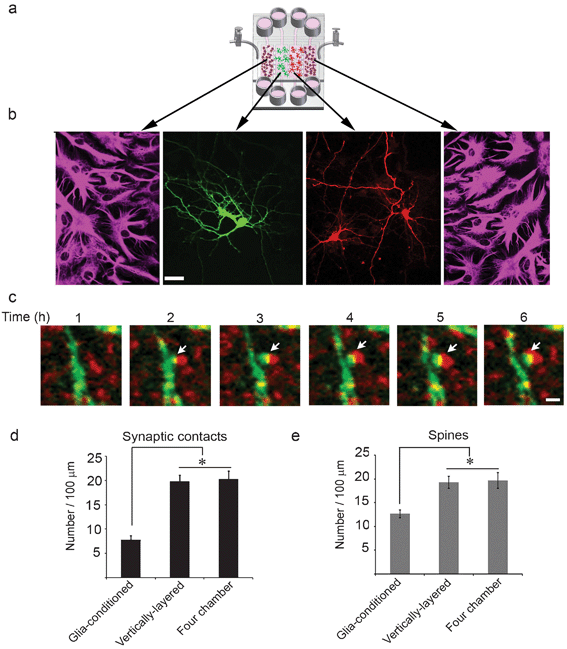 | ||
| Fig. 5 Neuronal interaction within four chamber devices. (a) A schematic of glia and neurons co-cultured in the four chamber microfluidic device. (b) Neurons, at day 3 in culture, were differentially transfected with GFP (green) and mCherry (red), maintained in co-culture with glia, and fixed at day 13. Glia were immunostained for GFAP and visualized in fluorescence (outer panels). GFP- and mCherry-expressing neurons were visualized in fluorescence (middle panels). Scale bar = 25 μm. (c) Neurons in the adjacent cell chambers were transfected with GFP and mCherry-synaptophysin, respectively, at day 3 in culture and subjected to live-cell imaging at day 13. Time-lapse images show that synaptic contacts (yellow puncta, arrows) formed between neurons expressing GFP and mCherry-synaptophysin. Scale bar = 2 μm. (d,e) Quantification of the number of synaptic contacts and dendritic spines are shown for neurons co-cultured with glia in vertically-layered and four chamber microfluidic devices. Neurons incubated with externally-conditioned glia media in two chamber microfluidic platforms are also shown for comparison. Error bars represent S.E.M. from five separate experiments (* p < 0.0001). | ||
To examine synaptic contact in the four chamber devices, the cell chambers were separated and neurons were differentially transfected with mCherry-synaptophysin and GFP, respectively. Following transfection, the valve barriers were opened, allowing neuronal processes in the adjacent chambers to extend toward each other. Even though opening of the valve barriers connected the glial and neuronal chambers, significant infiltration of glia into the neuronal chambers was not observed. This is most likely because the glass surfaces underneath the barriers between the chambers were not coated with type I collagen or PLL and/or further glia proliferation was inhibited by addition of cytosine β-D-arabinofuranoside (Ara-C) to the glial cultures after they reached confluence. mCherry-synaptophysin expressing neurons formed contacts with neurons expressing GFP (Fig. 5c). The number of synaptic contacts was increased by approximately 2.5-fold in four chamber co-culture devices as compared to those observed when neurons were cultured alone with glia-conditioned media (Fig. 5d). In addition, the spine density was increased by about 1.5-fold as compared to those in which neurons were cultured alone with glia-conditioned media (Fig. 5e). Moreover, both the number of synaptic contacts as well as dendritic spines were similar in the vertically-layered and four chamber co-culture schemes (Fig. 5d,e). Since comparable results were obtained with both types of devices, subsequent experiments were performed with only the four chamber devices. Collectively, our data indicate that glia increase the number of dendritic protrusions, which leads to more synaptic contacts and dendritic spines.
Co-culture of glia with neurons promotes the stabilization of synaptic contacts
Since glia enhanced the number of synaptic contacts, we asked whether co-culture of neurons with glia affected the stability of synaptic contacts. To address this, the stability of synapses after initial contact was assessed as a function of time for neurons co-cultured with glia and for neurons cultured alone with glia-conditioned media. The synaptic contacts which formed when neurons were co-cultured with glia were maintained for longer periods of time than those that formed with neurons cultured alone with glia-conditioned media, suggesting the synaptic contacts were more stable in neuron-glia co-cultures (Fig. 6a). Indeed, quantification demonstrated that the percentage of unstable (lost) synaptic contacts for neurons cultured alone with glia-conditioned media were approximately 1.5-fold greater than with neuron-glia co-cultures (Fig. 6b), indicating glia significantly enhanced the stabilization of synapses after initial contact.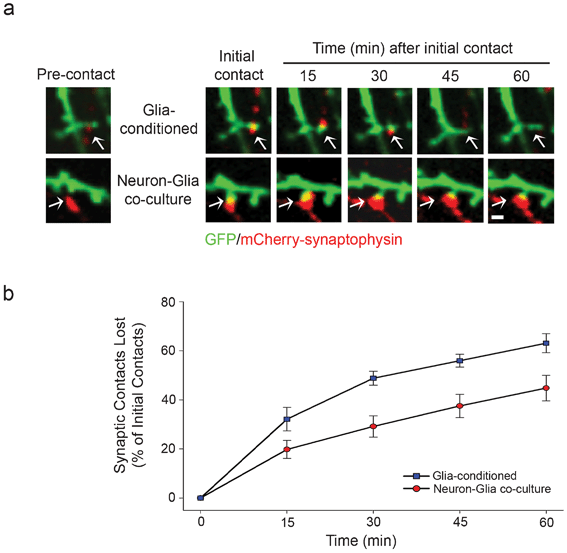 | ||
| Fig. 6 Co-culture of neurons with glia increases the stability of synaptic contacts. (a) Neurons in adjacent chambers were transfected with GFP and mCherry-synaptophysin at day 5 in culture and subjected to live-cell imaging at day 12 in culture. GFP and mCherry-synaptophysin expressing neurons formed synaptic contacts, which were imaged over 60 min. (b) The stability of synaptic contacts was quantified and expressed as the percentage of initial contacts that were lost over the indicated time. | ||
Synaptic contact enhances the stability of the actin cytoskeleton
Since actin remodeling is thought to underlie the morphological plasticity of dendritic spines and synapses,39 we hypothesized that synaptic contact, which stabilizes pre and postsynaptic terminals, leads to a decrease in actin dynamics. To test our hypothesis, we transfected neurons in adjacent chambers with mCerulean-actin and mCherry-synaptophysin, respectively, and subjected dendritic protrusions from mCerulean-actin expressing neurons to FRAP (Fig. 7a). Recovery of mCerulean-actin in protrusions, which were in contact with mCherry-synaptophysin puncta (synaptic contact), was significantly slower than in those that lacked synaptic contact (Fig. 7b,c). Since recovery of the photobleached mCerulean-actin depends on the rate of actin assembly and disassembly, these results suggest that synaptic contact reduces actin dynamics and leads to increased stabilization of the actin cytoskeleton.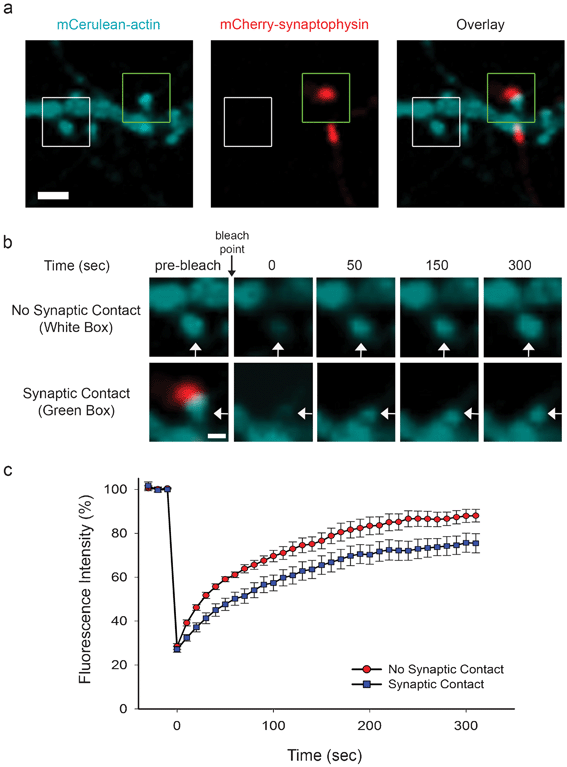 | ||
| Fig. 7 Synaptic contact increases actin stability. (a) Neurons in adjacent chambers were transfected with mCerulean-actin and mCherry-synaptophysin at day 5 in culture. At day 10 in culture, protrusions expressing mCerulean-actin were subjected to FRAP. A mCerulean-actin-containing protrusion, which was in contact with mCherry-synaptophysin puncta (Overlay, green box), and a protrusion that lacked synaptic contact (Overlay, white box) are shown. Scale bar = 2 μm. (b) Prebleach and subsequent recovery images of mCerulean-actin in protrusions (white arrows) from boxed regions in panel a are shown. The bleach point is indicated (black arrow). Scale bar = 1 μm. (c) The graph shows the recovery of mCerulean-actin, which is expressed as a percentage of the fluorescence intensity prior to bleaching. | ||
Metabolite expression levels in neuron-glia co-cultures differ from those found in neuron or glia cultures alone
Our data demonstrate that glia increase the number and stabilization of synaptic contacts, raising the question as to how this is accomplished by glia. One possibility is that when neurons are co-cultured with glia, they up- or down-regulate expression of distinct molecules, which enhance the formation and maturation of synapses. To determine if expression levels of soluble factors in neuron-glia co-cultures differed from those produced by neurons or glia individually, we cultured neurons alone, glia alone, or neurons and glia together in microfluidic devices and collected the media (effluent). As a control, media from microfluidic devices that did not contain cells was collected and used as background. The effluents were subjected to ultra performance liquid chromatography-ion mobility-mass spectrometry (UPLC-IM-MS), and principal component analysis was performed. Principal component analysis distils multi-dimensional data sets (in this case, the ∼1100 detected molecules) to those components that best represent the differences among samples. As a result, samples that are similar cluster together and are separated from samples that are different. As shown in Fig. 8a, separately cultured glial and neuronal samples have distinct profiles, which were different from neuron-glia co-cultures. This indicates that from a global metabolomic perspective each experimental condition fosters a distinct cellular response.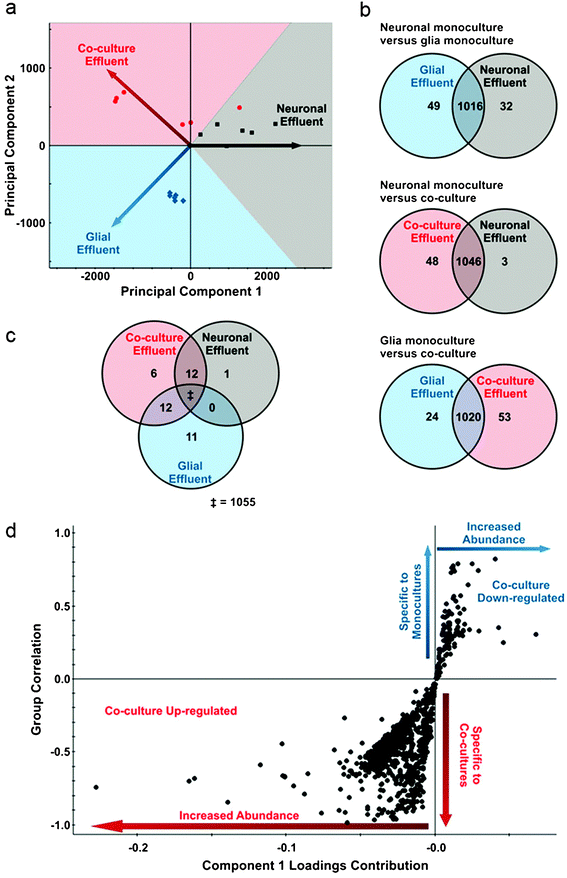 | ||
| Fig. 8 Co-culture of neurons with glia alters expression of distinct metabolites. (a) Principal component analysis of processed UPLC-IM-MS data from neurons alone (gray), glia alone (blue), and neuron-glia co-cultures (red). Each of these cultures exhibit distinct metabolic profiles giving rise to the vectored separation as depicted. Each point shows a discrete analysis where in a total of two biological and three technical replicates are represented. In each point, approximately 1100 distinct metabolites are represented in calculating the coordinates in principal component space. (b) Venn diagrams representing pairwise comparisons of each culture condition. Labeling within the circles indicate the number of significant metabolites (p < 0.05, Bonferroni-corrected) over-expressed relative to the other experimental condition. Labeling in shared regions correspond to metabolites that are not significantly altered between the two conditions. (c) Venn tripartite comparison of the metabolites observed for all experimental conditions. Uniquely over-expressed metabolites when compared to both other experimental conditions are labeled within each circle (p < 0.05, Bonferroni-corrected). Overlapping regions correspond to metabolites that are over-expressed in both experimental conditions when compared to the third. For example, there are zero metabolites that are over-expressed as a result of culturing glia alone or neurons alone that are shared when compared to co-culture conditions. This allows us to understand what is specific to the co-culture conditions and not an artifact of an individual culture. The double-dagger (“‡”) corresponds to the 1055 metabolites that are not significant (p > 0.05, Bonferroni-corrected) in a tripartite comparison of the three experimental conditions. To ensure that isotopic overlap did not influence these comparisons, the data was manually deisotoped. (d) Feature correlation and intensity are depicted for an orthogonal partial least squares-discriminant analysis of media from cultures of neurons alone, glia alone, and neuron-glia co-cultures. Each point corresponds to a detected feature through UPLC-IM-MS analysis. Quadrant III metabolites (lower left quadrant) are highly up-regulated in neuron-glia co-cultures, and quadrant I metabolites (upper right quadrant) are significantly down-regulated in co-cultures. | ||
To determine which metabolites are up- and down-regulated in response to culturing conditions, supervised multivariate statistical analysis methods were applied, namely orthogonal partial least squares-discriminate analysis. This method describes the greatest differences between two groups, and then determines which molecules contribute the most to these differences. Fig. 8d shows plots of metabolites that are up-regulated and down-regulated in the neuron-glia co-cultures and the magnitude of the change. Molecules in the lower left quadrant of the plot are up-regulated as a result of neuron-glia co-culturing, while those in the upper right quadrant are either down-regulated or absent in the neuron-glia co-cultures.
Fig. 8b shows Venn diagrams for binary comparisons of significant metabolites (Bonferroni-corrected p-value < 0.05) from glia cultures, neuron cultures, and neuron-glia co-cultures. Generally, the metabolites observed (∼1100) do not significantly vary between the experimental groups. However, in each experimental group, a distinct subset of metabolites were expressed at high levels. For example, 3 metabolites from the neuron alone cultures were significantly up-regulated, while 53 metabolites were expressed at high levels in the neuron-glia co-cultures. Fig. 8c shows a tripartite comparison of all three experimental groups. In this comparison, 1055 metabolites are shared among all groups. Notably, 6 metabolites are significantly elevated only in the neuron-glia co-culture. Putatively, these 6 molecules represent effectors of neuron-glia signaling.
Discussion
In this study, we developed microfluidic devices for co-culturing neurons and glia. These devices offer several advantages over traditional methods used to co-culture these cell types. By virtue of the design, glial and neuronal chambers in the microfluidic co-culture devices can be separated or connected, which allows us to control communication between these cell types. This is an important advantage because it provides a platform for differentially transfecting neuronal populations and dynamically imaging initial synaptic contact between neurons with high spatial and temporal resolution. Traditional co-culture methods do not provide the control and organization necessary to reproducibly image the development of synapses. Another advantage of our microfluidic devices is that neuron-glia co-cultures can be treated separately with drugs, trophic factors, or other molecules to determine their effects on cells. This improves upon other microfluidic designs that employ fluidic isolation to separate compartments, as the isolation is only effective in one direction, preventing the simultaneous independent treatment of both populations. Finally, the microfluidic co-culture devices allow for the possibility of media exchange control through regulating the amount of media loaded into the reservoirs connected to the different chambers. A possible limitation of PDMS-based microfluidic devices is the potential for small molecule absorption and non-specific protein adsorption to the PDMS.40,41 However, several methods have been developed to prevent molecular absorption and protein adsorption to PDMS,42–45 which permits this limitation to be overcome.The results obtained with these devices show that glia enhance the formation and stabilization of synaptic contacts. How do glia regulate these processes? Communication between neurons and glia could modulate the secretion of molecules that are critical for synapses to form and mature. Indeed, our results indicate that expression levels of metabolites in neuron-glia co-cultures differ from those found in neuron or glia cultures alone, suggesting these culture conditions elicit distinct cellular responses. The soluble factors may initiate signals that induce and/or stabilize contact between pre and postsynaptic terminals. Future studies will be needed to determine the specific factors that contribute to the formation and maturation of synapses.
Our results underscore an emerging view that glia are an important, active participant in forming the complex, neuronal circuitry found in the brain.46,47 This points to a need to develop new methodologies to investigate interactions between neurons and glia. The microfluidic devices described in this study provide a technology for co-culturing CNS neurons and glia that can be used to generate significant insight into the mechanisms by which these cells communicate.
Conclusions
In this study, we designed two novel microfluidic co-culture schemes, a vertically-layered and a four chamber configuration, for studying communication between neurons and glia, in which these cell types are co-cultured in close proximity (∼50–100 μm). Neurons co-cultured with glia showed an increased number of dendritic protrusions/spines and made significantly more synaptic contact with neighboring neurons, compared to those neurons cultured alone in glia-conditioned media. Moreover, the presence of glia enhanced the stability of synaptic contacts as well as the underlying actin cytoskeleton. Interestingly, expression levels of soluble factors secreted by neuron-glia co-cultures were different from those found in individual neuron or glia cultures. These results suggest that communication between neurons and glia is critical for the development of spines and synapses and point to the importance of co-culturing glia with neurons when examining these processes. The microfluidic devices described here are well-suited for culturing neurons with glia and can be easily adapted for the co-culture of different populations of other cell types.Acknowledgements
We thank Anne Marie Craig and Freda Miller for generously providing us with reagents. We thank Lan Hu for providing technical assistance in preparing neuronal and glial cultures. This work was supported by NIH grants GM092914 (D.J.W.), MH093903 (D.J.W. and D.L.), and UH2TR000491 (D.J.W., D.L., and J.A.M), NSF grant CBET0643583 (D.L.), DTRA grant HDTRA-09-1-0013 (J.A.M.), National Science Foundation Graduate Research Fellowship DGE0909667 (B.B.), the Vanderbilt Institute of Chemical Biology, and the Vanderbilt Institute for Integrative Biosystems Research and Education. The Quorum spinning disk confocal system was purchased from grant S10RR025524 from the National Center for Research Resources at NIH.References
- A. Matus, M. Ackermann, G. Pehling, H. R. Byers and K. Fujiwara, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 7590–7594 CrossRef CAS.
- E. G. Gray, J. Anat., 1959, 93, 420–433 CAS.
- K. M. Harris and S. B. Kater, Annu. Rev. Neurosci., 1994, 17, 341–371 CrossRef CAS.
- A. Peters and I. R. Kaiserman-Abramof, Am. J. Anat., 1970, 127, 321–355 CrossRef CAS.
- K. E. Sorra and K. M. Harris, Hippocampus, 2000, 10, 501–511 CrossRef CAS.
- Y. Smith, R. M. Villalba and D. V. Raju, Parkinsonism Relat. Disord., 2009, 15 Suppl 3, S156–161 CrossRef CAS.
- M. Chechlacz and J. G. Gleeson, Pediatr. Neurol., 2003, 29, 11–17 CrossRef.
- A. W. Grossman, G. M. Aldridge, I. J. Weiler and W. T. Greenough, J. Neurosci., 2006, 26, 7151–7155 CrossRef CAS.
- J. W. Swann, S. Al-Noori, M. Jiang and C. L. Lee, Hippocampus, 2000, 10, 617–625 CrossRef CAS.
- I. Ferrer and F. Gullotta, Acta Neuropathol., 1990, 79, 680–685 CrossRef CAS.
- M. Suetsugu and P. Mehraein, Acta Neuropathol., 1980, 50, 207–210 CrossRef CAS.
- J. L. Chen and E. Nedivi, Curr. Opin. Neurobiol., 2010, 20, 557–562 CrossRef CAS.
- D. H. Bhatt, S. Zhang and W. B. Gan, Annu. Rev. Physiol., 2009, 71, 261–282 CrossRef CAS.
- R. C. Paolicelli, G. Bolasco, F. Pagani, L. Maggi, M. Scianni, P. Panzanelli, M. Giustetto, T. A. Ferreira, E. Guiducci, L. Dumas, D. Ragozzino and C. T. Gross, Science, 2011, 333, 1456–1458 CrossRef CAS.
- S. Doretto, M. Malerba, M. Ramos, T. Ikrar, C. Kinoshita, C. De Mei, E. Tirotta, X. Xu and E. Borrelli, PLoS One, 2011, 6, e19849 CAS.
- C. Eroglu and B. A. Barres, Nature, 2010, 468, 223–231 CrossRef CAS.
- E. M. Ullian, S. K. Sapperstein, K. S. Christopherson and B. A. Barres, Science, 2001, 291, 657–661 CrossRef CAS.
- E. C. Beattie, D. Stellwagen, W. Morishita, J. C. Bresnahan, B. K. Ha, M. Von Zastrow, M. S. Beattie and R. C. Malenka, Science, 2002, 295, 2282–2285 CrossRef CAS.
- K. S. Christopherson, E. M. Ullian, C. C. Stokes, C. E. Mullowney, J. W. Hell, A. Agah, J. Lawler, D. F. Mosher, P. Bornstein and B. A. Barres, Cell, 2005, 120, 421–433 CrossRef CAS.
- O. Blondel, C. Collin, W. J. McCarran, S. Zhu, R. Zamostiano, I. Gozes, D. E. Brenneman and R. D. McKay, J. Neurosci., 2000, 20, 8012–8020 CAS.
- J. Park, H. Koito, J. Li and A. Han, J. Vis. Exp., 2009, 31, e1399 Search PubMed.
- J. Park, H. Koito, J. Li and A. Han, Lab Chip, 2012, 12, 3296–3304 RSC.
- S. Hosmane, I. H. Yang, A. Ruffin, N. Thakor and A. Venkatesan, Lab Chip, 2010, 10, 741–747 RSC.
- J. Park, H. Koito, J. Li and A. Han, Biomed. Microdevices, 2009, 11, 1145–1153 CrossRef CAS.
- I. H. Yang, R. Siddique, S. Hosmane, N. Thakor and A. Hoke, Exp. Neurol., 2009, 218, 124–128 CrossRef CAS.
- S. Hosmane, M. A. Tegenge, L. Rajbhandari, P. Uapinyoying, N. G. Kumar, N. Thakor and A. Venkatesan, J. Neurosci., 2012, 32, 7745–7757 CrossRef CAS.
- D. Majumdar, Y. Gao, D. Li and D. J. Webb, J. Neurosci. Methods, 2011, 196, 38–44 CrossRef.
- G. M. Whitesides, E. Ostuni, S. Takayama, X. Jiang and D. E. Ingber, Annu. Rev. Biomed. Eng., 2001, 3, 335–373 CrossRef CAS.
- Y. Gao, D. Majumdar, B. Jovanovic, C. Shaifer, P. C. Lin, A. Zijlstra, D. J. Webb and D. Li, Biomed. Microdevices, 2011, 13, 539–548 CrossRef CAS.
- J. C. McDonald and G. M. Whitesides, Acc. Chem. Res., 2002, 35, 491–499 CrossRef CAS.
- M. A. Unger, H. P. Chou, T. Thorsen, A. Scherer and S. R. Quake, Science, 2000, 288, 113–116 CrossRef CAS.
- K. Goslin, H. Asmussen and G. Banker, Rat hippocampal neurons in low-density culture, MIT Press, Cambridge, MA, 1998 Search PubMed.
- A. Gloster, H. El-Bizri, S. X. Bamji, D. Rogers and F. D. Miller, J. Comp. Neurol., 1999, 405, 45–60 CrossRef CAS.
- W. H. Lin, C. A. Nebhan, B. R. Anderson and D. J. Webb, J. Biol. Chem., 2010, 285, 36010–36020 CrossRef CAS.
- A. M. Wegner, C. A. Nebhan, L. Hu, D. Majumdar, K. M. Meier, A. M. Weaver and D. J. Webb, J. Biol. Chem., 2008, 283, 15912–15920 CrossRef CAS.
- C. A. Smith, E. J. Want, G. O'Maille, R. Abagyan and G. Siuzdak, Anal. Chem., 2006, 78, 779–787 CrossRef CAS.
- M. Stroh, W. R. Zipfel, R. M. Williams, W. W. Webb and W. M. Saltzman, Biophys. J., 2003, 85, 581–588 CrossRef CAS.
- H. Wu, B. Huang and R. N. Zare, J. Am. Chem. Soc., 2006, 128, 4194–4195 CrossRef CAS.
- M. Fischer, S. Kaech, D. Knutti and A. Matus, Neuron, 1998, 20, 847–854 CrossRef CAS.
- M. W. Toepke and D. J. Beebe, Lab Chip, 2006, 6, 1484–1486 RSC.
- I. Wong and C. M. Ho, Microfluid. Nanofluid., 2009, 7, 291–306 CrossRef CAS.
- R. Gomez-Sjoberg, A. A. Leyrat, B. T. Houseman, K. Shokat and S. R. Quake, Anal. Chem., 2010, 82, 8954–8960 CrossRef CAS.
- G. T. Roman and C. T. Culbertson, Langmuir, 2006, 22, 4445–4451 CrossRef CAS.
- J. Zhou, H. Yan, K. Ren, W. Dai and H. Wu, Anal. Chem., 2009, 81, 6627–6632 CrossRef CAS.
- J. Zhou, A. V. Ellis and N. H. Voelcker, Electrophoresis, 2010, 31, 2–16 CrossRef CAS.
- W. S. Chung and B. A. Barres, Curr. Opin. Neurobiol., 2012, 22, 438–445 CrossRef CAS.
- M. Santello, C. Cali and P. Bezzi, Adv. Exp. Med. Biol., 2012, 970, 307–331 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3lc50249j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2013 |