Study of pressure effects on laser induced plasma in bulk seawater
Huaming
Hou
,
Ye
Tian
,
Ying
Li
and
Ronger
Zheng
*
Optics and Optoelectronics Laboratory, Ocean University of China, Qingdao, P. R. China 266100. E-mail: rzheng@ouc.edu.cn
First published on 25th October 2013
Abstract
It is a big challenge to apply laser induced breakdown spectroscopy (LIBS) to ocean in situ detection, but there are ample opportunities for LIBS development too. In the present work, laboratory investigations of LIBS on natural seawater at different pressures from 0.1 to 40 MPa were carried out. Pressure and laser pulse energy effects on LIBS emission were investigated. The result showed that enhanced LIBS emission can be obtained under elevated ambient pressure conditions. The line broadening of lines increases as a function of pressure. The time resolved LIBS emission results demonstrated that plasma emission is weakly dependent on the ambient pressure during the early stage of plasma and the pressure has a significant influence on the plasma form during plasma evaluation at a later stage of plasma. The obtained results suggested that the LIBS technique has the potential to be developed as an in situ chemical sensing technique for ocean applications.
A Introduction
Geochemical studies in the deep ocean have traditionally relied upon sample recovery by bottles, cores and dredges deployed from surface ships or collected by manned submersibles and remotely operated vehicles (ROVs).1 However, although sample recovery is reliable and allows for accurate analysis of chemical composition, it is time consuming and the information obtained sometimes is discrete and not available for real-time analysis. The application of sea-going vehicles2 and ocean observation networks3 provides new ocean observation platforms which allow for real-time feedback and long-time observation. The development of in situ chemical sensors has been identified as a major priority for understanding the ocean.4–7 The determination of chemical composition of seawater or hydrothermal vent fluids has great importance in the research of marine chemistry, marine geochemistry, and understanding the hydrothermal vent ecosystems.7–11LIBS is a type of atomic emission spectroscopy using a high energy laser pulse as the excitation source.12–14 The laser is focused to form plasma, and the plasma emits light containing atomic and ionic spectral lines, which allows qualitative or quantitative determination of the elemental sample composition.15–17 It is an attractive technique for in situ multi-elemental analysis because its result is available in real time.18,19 Several studies have proved that LIBS is a technique suitable for in situ or in-field elemental analysis of sea minerals.13,20,21 When using LIBS for in situ marine applications, the pressure effect is a key parameter that should be considered because it has great effects on the plasma expansion, collision, and the quenching process, and the moving breakdown phenomenon22 makes this process more complex. High density effect induced by high pressure plays a key role for laser ablated particles.23 Studies on the submerged solid target demonstrated that well resolved emission spectra can be obtained even at 30 MPa,24,25 and suggested a possibility that chemical reactions and physical states (pressure and temperature) of liquid-phase laser ablation plasma can be controlled by adding external pressure to an ambient liquid.26 In comparison with solid sediments samples, determination of seawater and hydrothermal vent fluid chemical composition is more practical because it is very hard to control the sub-millimetre distance precision between the focal point and the sample surface for in situ applications, which is very important and has great influence on the LIBS signal.27,28 The pioneering work by Lawrence-Snyder et al.29,30 showed that several Group I and II elements as well as Mn were detected in bulk aqueous solution at a pressure exceeding 2.76 × 107 Pa, and H and Zn were detected using sequential laser pulses for excitation at pressures up to ∼1.38 × 107 Pa.31,32 It has previously been reported that when comparing dual pulse LIBS with single pulse LIBS, the use of dual pulse LIBS for analyte detection in high pressure aqueous solution did not improve the limit of detection,33 although a better performance is achieved using dual pulse LIBS under 1 atm conditions in several studies.12,34–36 In the work in ref. 33, the limits of detection of Mg, K, Ca, Mn and Na could reach 5000, 500, 500, 1000 and 50 ppm, respectively, in aqueous solutions at 2.76 × 107 Pa.
In this study, we performed spectroscopic analysis of K, Ca, Na and Mg in seawater to demonstrate pressure and laser energy effects on elemental detection. We also performed a mechanism investigation with the aim of providing some insight into the laser ablation process at a pressure of up to 40 MPa.
B Experimental set-up
The pulsed Nd:YAG laser (Quantel Brilliant B; wavelength λ = 1064 nm; pulse duration 10 ns; repetition frequency 10 Hz) was employed for ablation of bulk solution. The 180° backscattering geometry was used in plasma emission sampling, as shown in Fig. 1. A dichroic mirror (Thorlabs, DMLP 900, transmission 920–1300 nm and reflectivity 390–872 nm) was used to transmit ablation laser and reflect plasma emission. A lens with 38 mm focal length was used as a focal lens. The distance between the focal point and the inner surface of the sapphire window was calculated to be 24 mm. Another lens with 38 mm focal length was used to couple plasma emission into optical fibers. LIBS emission was dispersed with a C-T spectrometer (Acton sp2500, f = 500 mm, f# = 6.5, and g = 1200 mm−1). The spectrometer was integrated with an intensified CCD (Andor, DH734i). The lid with a hole (10 mm) used to fix the sapphire was the actual aperture in the optical set-up, and the effective collection solid angle was ∼0.2. | ||
| Fig. 1 Schematic representation of ablation and radiation collection geometries of the experimental set-up. | ||
High pressure liquid conditions were provided by a homemade high pressure system (HPS). It consists of a reciprocating plunger pulsation-free pump (RPPP) (Lab Alliance Series III), pressurization equipment, and a titanium chamber. The core part of the HPS is the chamber (60 mL) equipped with a sapphire window (diameter 12 mm, thick 9 mm), in which plasma is generated. The maximum pressure we can obtain in the HPS is 40 MPa with a precision of 0.1 MPa. Natural clean seawater from 4000 m depth was used as the analyte.
C Results and discussion
Overall analysis of typical LIBS spectra under different pressures
Typical LIBS spectra were taken at three different pressures, atmosphere pressure (0.1 MPa), 20 MPa and 40 MPa. The results are shown in Fig. 2. Ten lines of four elements (Na, Mg, K, Ca) were detected under three different pressures. The spectra of Ca and K were of high quality because of their high single to noise ratio (SNR) and we found no self-absorption on these lines. Mg lines were very weak but still detectable under 40 MPa conditions. The spectral intensities of Na were strong but with severe self-absorption because the concentration of Na in seawater is high. No other elements were detected. In natural seawater, the concentrations of Na, Mg, K and Ca are 10770, 1290, 380 and 412 ppm, respectively. Besides these four elements, the concentrations of other metal elements are less than 10 ppm.37 In an extreme ocean environment, such as hydrothermal vent, the concentrations of Fe, Mn, Cu, Zn can increase up to 103 ppm.11 The LIBS technique can be used for in situ measurement of these elements. To develop an in situ LIBS instrument, two key parameters must be considered. One is the pressure effects on the LIBS signal because the pressure is quite different at different depths. Another one is laser energy influence on the LIBS signal. This is important to increase the ablation efficiency and simplify the design of an oceanographic LIBS instrument.32 Both parameters were studied in detail in the following sections.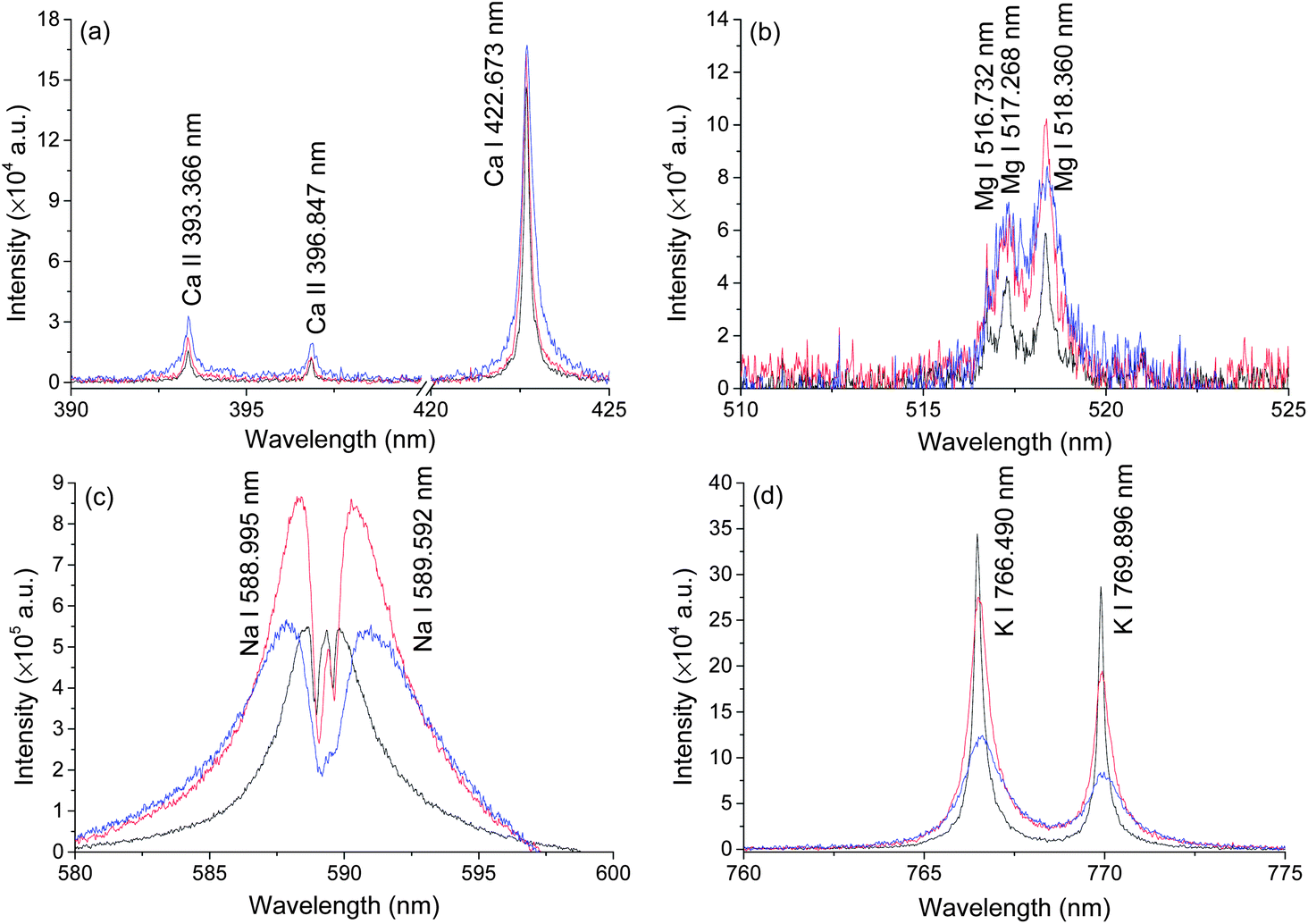 | ||
| Fig. 2 Typical LIBS spectra of natural seawater obtained at a delay of 100 ns with a gate width of 5 μs under the pressure of 0.1 MPa (black), 20 MPa (red), and 40 MPa (Blue). The spectra of Ca(a), Na(c) and K(d) are taken with a laser energy of 40 mJ on 100 pulses average, and atomic emission of Mg(b) is recorded on 1000 pulses average. | ||
Pressure effects on LIBS spectra
In order to investigate the pressure effect on laser induced plasma emission, the pressure effects on peak width, intensity and integrated intensity of Ca, K and Mg lines were studied in the pressure range of 0.1 to 40 MPa. LIBS signals were obtained at a delay of 100 ns with a gate width of 5 μs. Lorentz fitting was used to extract the line broadening, intensity and integrated intensity. The fitted peak area is defined as the integrated intensity, and the peak height is defined as the intensity. Three atomic lines K I 766 nm, Ca I 422 nm, Mg I 518 nm and one single ionic line Ca II 393 nm were selected for analysis. The line intensity and integrated intensity were normalized by the highest values of data obtained at selected nine different pressures. Fig. 3 shows the dependence of line broadening on pressure. An obvious increase of line broadening was observed under elevated ambient pressure conditions. The line broadening dependence on pressure for K I 766 nm and Ca I 422 nm is quite similar. The peak width at 5 MPa is very close to that at atmosphere pressure, but there is an approximately linear increase of peak width with the increase of pressure. The peak width of calcium ionic lines at 393 nm increases as the pressure increases. The line width of Mg I 518 nm increases gradually as pressure increases from 0.1 to 30 MPa, after 30 MPa, a dramatic increase is observed. The possible reason for the slight difference of line width over pressure between Mg I 518 nm and K I 766 nm, Ca I 422 nm is that the variation of broadening coefficients of different elements with plasma temperature is different. The line broadening coefficient is plasma temperature dependent and the plasma temperature should be different under different pressure conditions. The line width of Mg I 518 nm is obtained by multi-peak fitting of two Mg I lines at 517 nm and 518 nm. Moreover, the emission of Mg I 518 nm is very weak, as shown in Fig. 2(b). These can introduce larger measurement error in line width detection. The electron density is directly proportional to the line broadening.38,39 The result shown in Fig. 3 suggested that the electron density is higher under higher pressure conditions. The variations of the line width of K I 766 nm and Ca I 422 nm can reflect the electron density changes with higher accuracy.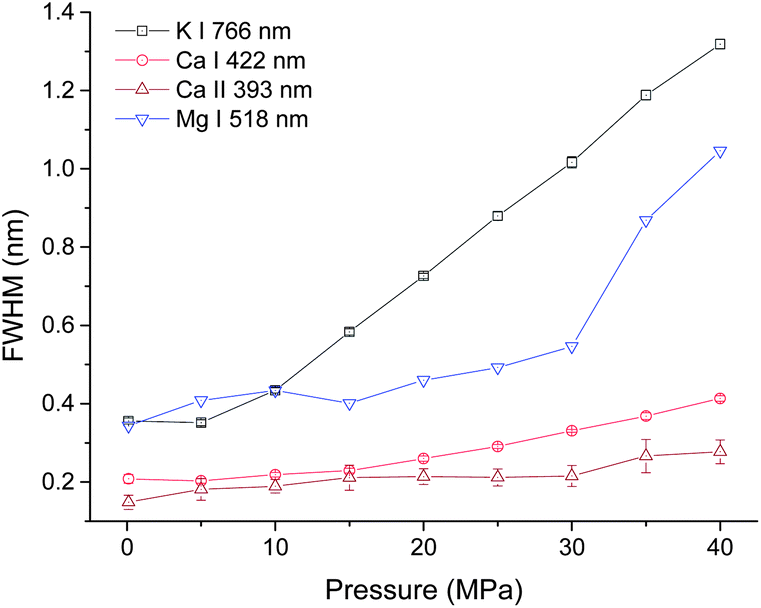 | ||
| Fig. 3 Pressure dependence of peak broadening of four spectral lines with a laser energy of 40 mJ on 100 pulses average. | ||
The dependences of intensity of three atomic lines investigated on pressure are similar, as seen in Fig. 4. As the pressure increases, a corresponding increase in intensity occurs until a maximum intensity was reached at 15 MPa. Above this value, the emission intensity decreases. But for the intensity of the ionic line of Ca II 393 nm, an increasing trend with pressure was obtained besides a marginal decrease observed from 15 to 20 MPa. The different pressure dependence of ionic and atomic calcium emissions implies that the ionization degree is higher under higher pressure conditions.
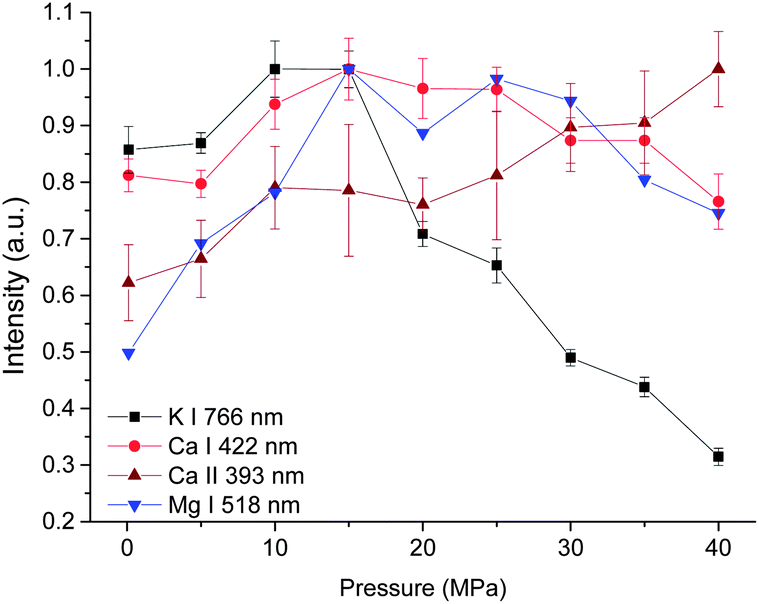 | ||
| Fig. 4 Pressure dependence of intensity of four spectral lines with a laser energy of 40 mJ on 100 pulses average. | ||
Fig. 5 shows the integrated intensity dependence of four lines on pressure. A gradually increasing trend was observed for both atomic and ionic calcium lines and atomic magnesium lines with pressure. But for line K I 766 nm, as the pressure increases, the integrated intensity increases sharply up to 15 MPa, above which, a gradual decrease trend with pressure is observed. The possible reason for the different behavior of K I 766 nm is that the upper energy levels of K I 766 nm (1.62 eV) are low compared to the other three lines (2.93 eV for Ca I, 3.15 eV for Ca II and 5.11 eV for Mg I). Moreover, K is easy to be ionized because of its lower ionization energy (4.34 eV for K, 6.11 eV for Ca and 7.65 eV for Mg). More potassium atoms are ionized at higher pressure.
 | ||
| Fig. 5 Pressure dependence of integrated intensity of four spectral lines with a laser energy of 40 mJ on 100 pulses average. | ||
The results presented in Fig. 3, 4 and 5 indicate that the pressure has two main influences on plasma. (1) Higher pressure leads to higher electron density. (2) With suitable higher pressure, the integrated intensity can be enhanced up to 3 times. There are two possible reasons to explain the signal enhancement phenomenon. (1) An increment of collision probability among atoms, ions and electrons. The results in this work proved that the electron density is higher at higher pressure as a result of the compression effect. The densification of plasma particles can increase the collision probability. (2) An increment of plasma temperature. A simulated result in ref. 23 showed that the effect of the external pressure can lead to the increase of plasma temperature during its reduced oscillation period as a consequence of faster dynamics.
Time resolved analysis of LIBS emission
To better understand the mechanism of how pressure influences the laser induced plasma, time resolved spectra were recorded with 200 ns gate width and 200 ns step. The time resolved K I line broadening, intensity and integrated intensity are shown in Fig. 6. Here we use the trend of line broadening as an indication of electron density changes. As seen in Fig. 6, the lifetime of K I emission is in the range of 700 to 1700 ns under different pressures conditions. Fig. 6(a) shows the time resolved line broadening of K I 766 nm at different pressures. When the pressure is less than 10 MPa, the line broadening decreases approximately exponential over time; when the pressure is equal to or over 10 MPa, as time increases, a decreasing first then increasing trend occur. At higher pressure, the line broadening is wider. Similar results were obtained from Ca I 422 nm, as shown in Fig. 7. The data of 35 MPa at 700 ns in Fig. 7(a) has larger measurement error because of the fitting of the weak peak. The difference of line broadening under different pressure conditions is larger at longer delays over 300 ns compared to the initial 100 ns, which implies that the pressure effect has greater influence on the late stage of laser induced plasma than that on the initial stage of laser induced plasma. The reason for the increase of line broadening under elevated ambient pressure conditions is attributed to the fast shrink of plasma under the compression effect resulted from high ambient pressure, which can densify the particles (atoms, ions and electrons) inside plasma and increase the Stark broadening of spectral lines.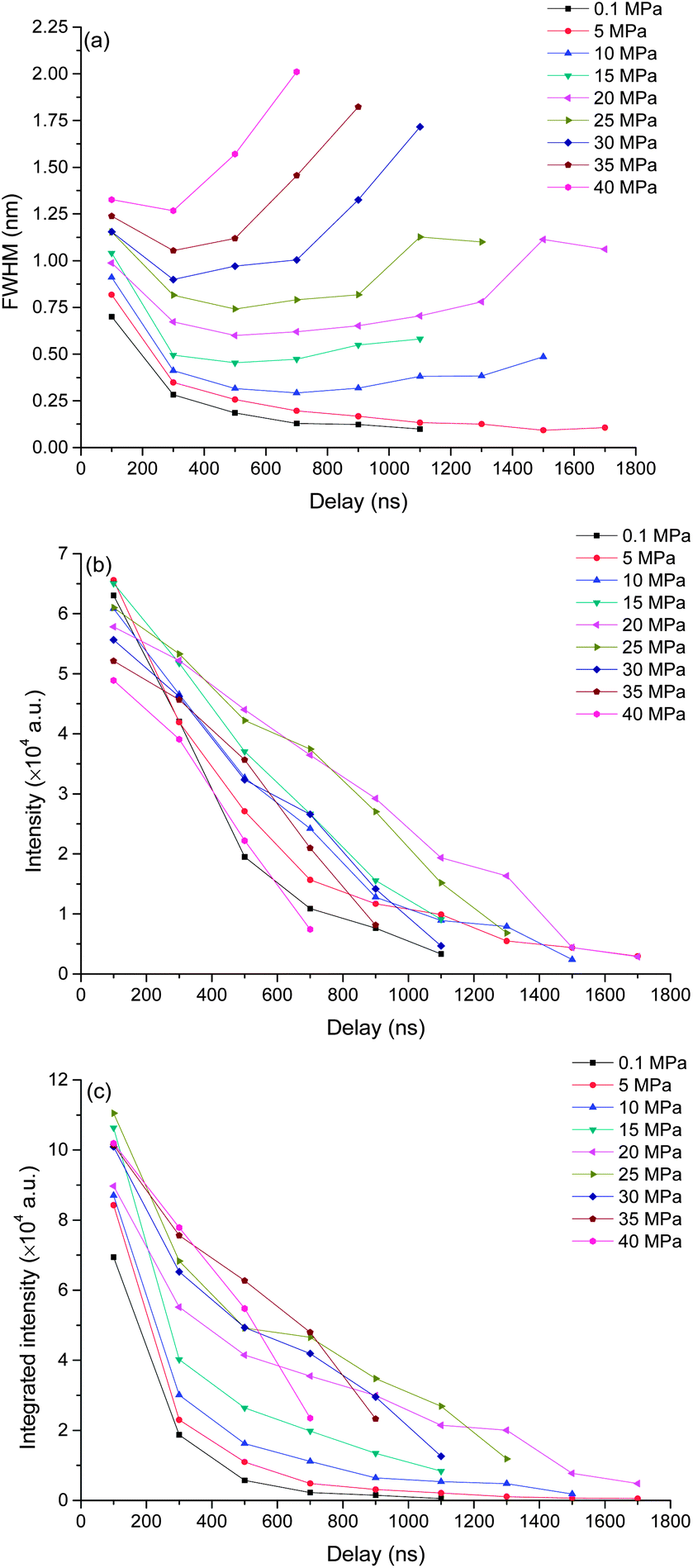 | ||
| Fig. 6 Time resolved intensity, integrated intensity and line broadening of K I 766 nm under different pressures as a function of delay. The spectra were taken with a laser energy of 40 mJ on 100 pulses average. | ||
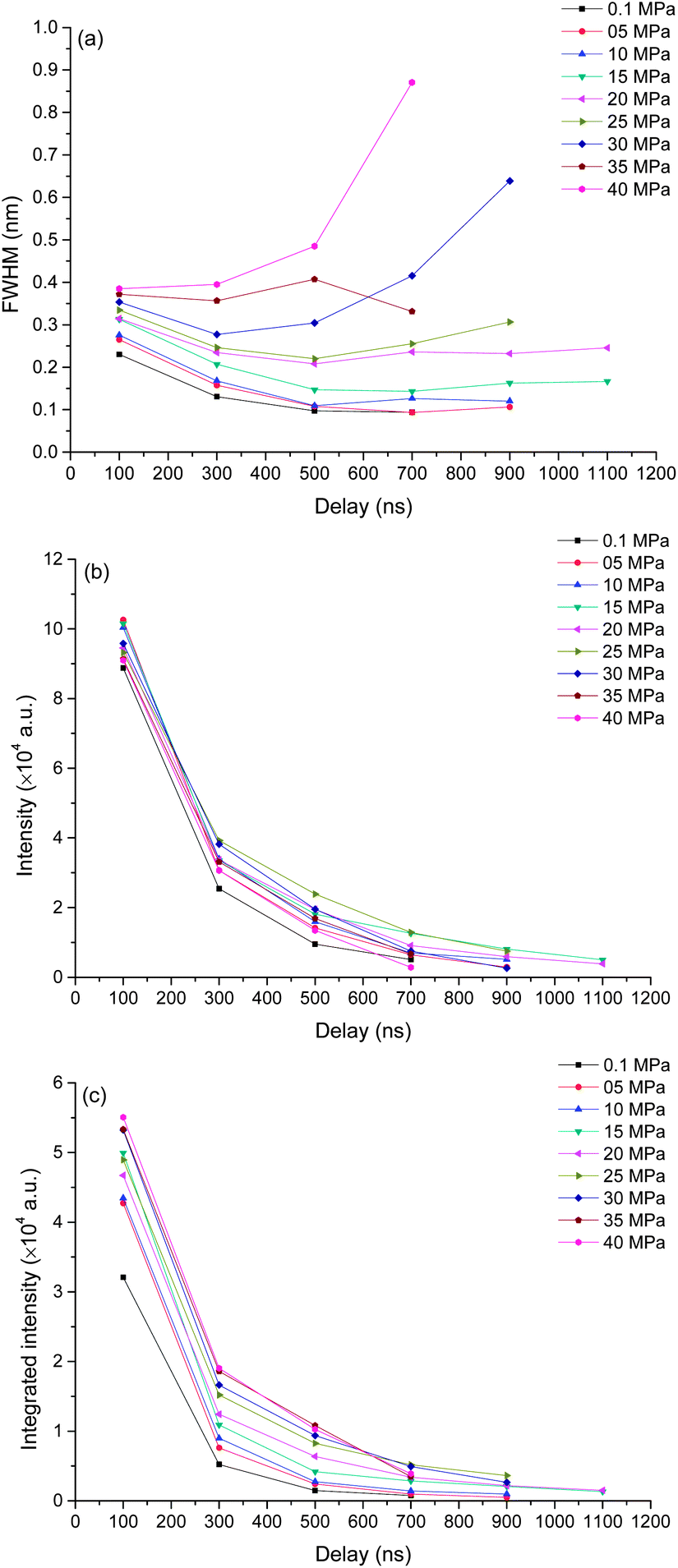 | ||
| Fig. 7 Time resolved intensity, integrated intensity and line broadening of Ca I 422 nm under different pressures as a function of delay. The spectra were taken with a laser energy of 40 mJ on 100 pulses average. | ||
Fig. 6(b) and (c) show the time resolved intensity and integrated intensity of K I 766 nm. At lower pressure (≤10 MPa), the intensity and integrated intensity decrease with time approximately exponentially, but at higher pressure (≥20 MPa), the intensity and integrated intensity approximately linearly decrease with time. The integrated intensities obtained under different pressure conditions have a larger difference at a delay over 300 ns compared to the initial emission at 100 ns. The weak dependence of plasma emission on pressure at the initial stage of laser induced plasma further implies that the pressure has limited influence on the initial stage of laser induced plasma; instead pressure has greater influence on the late stage of plasma during plasma evaluation. Fig. 7(b) and (c) show the time resolved intensity and integrated intensity of Ca I 422 nm. Under all different pressure conditions, the trends of both intensity and integrated intensity are approximately exponential as a function of time for all pressures. But under higher pressure conditions, the intensity or integrated intensity tend to decrease faster. The time resolved intensity and integrated intensity trends of Ca I and K I are a little different at a pressure over 20 MPa. The possible reason is that the evaluation of the outcome of g × exp(−Ei/kT)/U(T) is different during the plasma evaluation because the energy levels of Ca and K are quite different. Ei and g are the upper energy level and its statistical weight, k is the Boltzmann constant and U(T) is the internal partition function.
Actually, the pressure inside plasma is much higher than 40 MPa at the initial stage of laser induced plasma. A calculation of pressure using the Gilmore model of cavitation bubble dynamics and the Kirkwood–Bethe hypothesis showed that the plasma rim pressure ranges from 1300 MPa (50 μJ, 30 ps) to 7150 MPa (10 mJ, 6 ns).40 So the pressure effect has no great influence on the initial emission of plasma. Laser induced plasma is a dynamic process. When the plasma pressure decreases to the same order as the surrounding pressure, the surrounding pressure will have great influence on the plasma behavior. After a transient equilibrium between the plasma pressure and the ambient pressure, the plasma begins to shrink. During the shrinking process, the plasma will lose energy because of its optical radiation, thermal transmission, shockwave generation and other processes. However, plasma will gain energy from the compression process, and it can lead to an increase of plasma emission. The compression mechanism also can lead to an increase of electron density because of the decrease of plasma volume. This is under the hypothesis that it is the compression process dominated the electron density other than the recombination of ions and electrons, and it agrees with the results shown in Fig. 6(a) and 7(a). For a typical LIP under air or reduced pressure conditions, the electron density decreases over time at the late stage of LIP for the recombination of ions and electrons.41,42
For a typical laser induced plasma in bulk transparent liquid, the geometry of plasma is filamentous because of the moving breakdown phenomenon.22 The pressure effect has been verified that it has great influence on the plasma size, where plasma was induced at a submerged solid target.24,25 Further studies using fast imaging techniques are needed to elucidate the effect of pressure on the dynamic morphology of plasma.
Laser energy effects on LIBS spectra
The effect of laser pulse energy on the LIBS signal for analyte under elevated ambient pressure conditions was investigated with the goal of optimizing powder consumption. The peak intensity, integrated intensity and line broadening of analytes (Ca I, K I) were measured at laser energies ranging from 20 to 80 mJ at three pressures (0.1 MPa, 20 MPa and 40 MPa). Fig. 8 and 9 show the dependence of the K I 766 nm and Ca I 422 nm emission lines on laser pulse energy. Under all three different pressure conditions, as laser pulse energy increases, a corresponding increase in peak intensity and integrated intensity until a maximum intensity is reached at 30 or 40 mJ. Above these values, peak intensity and integrated intensity decrease with energy up to 80 mJ. Higher laser pulse energy does not lead to a higher LIBS signal. The trends of peak intensity and integrated intensity are similar as a function of laser energy because the peak width is weakly dependent on laser pulse energy. Moreover, the relationship between line intensity and laser energy is similar at different pressures, and it agrees with the results in ref. 32. The results shown in Fig. 8 and 9 are obtained with natural seawater, and the result presented in ref. 32 was obtained with a laboratory made solution. The consistency of results means that lots of ions such as Na+, Cl−, etc. have a marginal effect on the LIBS signal. The data for K and Ca suggest that the best LIBS signal can be obtained with 30 to 40 mJ energy under different pressures in the range of 0.1 to 40 MPa. At higher laser energy, the plasma can be formed in front of the focal point, and the plasma morphology could be changed greatly.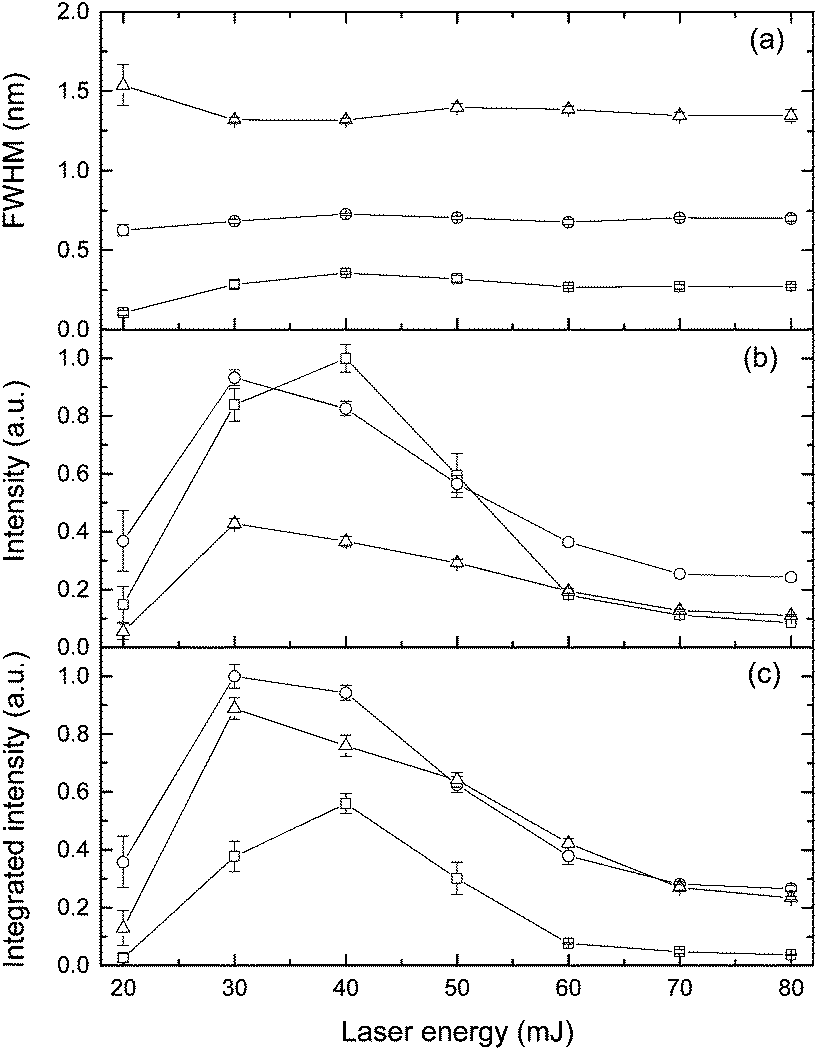 | ||
| Fig. 8 Influence of laser energy on the characteristics of K I 766 nm under the pressure of 0.1 MPa (square), 20 MPa (circle), and 40 MPa (triangle). The spectra were recorded at a delay of 100 ns with the gate width of 5 μs. | ||
 | ||
| Fig. 9 Influence of laser energy on the characteristics of Ca I 422 nm under the pressure of 0.1 MPa (square), 20 MPa (circle), and 40 MPa (triangle). The spectra were recorded at a delay of 100 ns with the gate width of 5 μs. | ||
D Conclusions
This study has demonstrated that the LIBS signal can be obtained under 40 MPa bulk solution conditions, which indicates that LIBS can be used at 4000 m depth of the ocean. The time resolved LIBS emission results demonstrated that plasma emission is weakly dependent on the external pressure during the early stage but external pressure has a significant effect on the plasma form during the late stage of plasma evaluation. The electron density tends to be higher under the effect of higher pressure, especially at the late stage of plasma. But the laser pulse energy has a marginal effect on electron density. The study of laser pulse energy on the Ca and K signal at three different pressures proved that the effects of laser pulse energy on the LIBS signal is minimally pressure dependent. The results presented here suggested that the LIBS technique has the potential to be developed into a new kind of in situ chemical sensing technique.Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant no. 41106080 and 41376107) and Innovation and Research Foundation of Ocean University of China.Notes and references
- P. G. Brewer, G. Malby, J. D. Pasteris, S. N. White, E. T. Peltzer, B. Wopenka, J. Freeman and M. O. Brown, Deep Sea Res., Part I, 2004, 51, 739–753 CrossRef CAS PubMed.
- K. R. Newman, M.-H. Cormier, J. K. Weissel, N. W. Driscoll, M. Kastner, E. A. Solomon, G. Robertson, J. C. Hill, H. Singh, R. Camilli and R. Eustice, Earth Planet. Sci. Lett., 2008, 267, 341–352 CrossRef CAS PubMed.
- S. Martin Taylor, Nucl. Instrum. Methods Phys. Res., Sect. A, 2009, 602, 63–67 CrossRef CAS PubMed.
- R. D. Prien, Mar. Chem., 2007, 107, 422–432 CrossRef CAS PubMed.
- S. N. White, R. M. Dunk, E. T. Peltzer, J. J. Freeman and P. G. Brewer, Geochem., Geophys., Geosyst., 2006, 7, Q05023 Search PubMed.
- X. Zhang, P. M. Walz, W. J. Kirkwood, K. C. Hester, W. Ussler, E. T. Peltzer and P. G. Brewer, Deep Sea Res., Part I, 2010, 57, 297–306 CrossRef CAS PubMed.
- L. L. Demina, N. G. Holm, S. V. Galkin and A. Y. Lein, J. Mar. Syst., 2013, 126, 94–105 CrossRef PubMed.
- K. L. Daly, R. H. Byrne, A. G. Dickson, S. M. Gallager, M. J. Perry and M. K. Tivey, Mar. Technol. Soc. J., 2004, 38, 121–143 CrossRef.
- E. Douville, J. L. Charlou, E. H. Oelkers, P. Bienvenu, C. F. Jove Colon, J. P. Donval, Y. Fouquet, D. Prieur and P. Appriou, Chem. Geol., 2002, 184, 37–48 CrossRef CAS.
- W. E. Seyfried, N. J. Pester, K. Ding and M. Rough, Geochim. Cosmochim. Acta, 2011, 75, 1574–1593 CrossRef CAS PubMed.
- M. K. Tivey, Oceanography, 2007, 20, 50–65 CrossRef.
- A. De Giacomo, M. Dell'Aglio, O. De Pascale and M. Capitelli, Spectrochim. Acta, Part B, 2007, 62, 721–738 CrossRef PubMed.
- V. Lazic, F. Colao, R. Fantoni, V. Spizzichino and S. Jovićević, Spectrochim. Acta, Part B, 2007, 62, 30–39 CrossRef PubMed.
- C. Aragón and J. A. Aguilera, Spectrochim. Acta, Part B, 2008, 63, 893–916 CrossRef PubMed.
- E. Tognoni, G. Cristoforetti, S. Legnaioli and V. Palleschi, Spectrochim. Acta, Part B, 2010, 65, 1–14 CrossRef PubMed.
- R. Fantoni, L. Caneve, F. Colao, L. Fornarini, V. Lazic and V. Spizzichino, Spectrochim. Acta, Part B, 2008, 63, 1097–1108 CrossRef PubMed.
- M. Pouzar, T. Kratochvíl, L. Čapek, L. Smoláková, T. Černohorský, A. Krejčová and L. Hromádko, Talanta, 2011, 83, 1659–1664 CrossRef CAS PubMed.
- B. Sallé, P. Mauchien and S. Maurice, Spectrochim. Acta, Part B, 2007, 62, 739–768 CrossRef PubMed.
- A. A. Bol'shakov, J. H. Yoo, C. Liu, J. R. Plumer and R. E. Russo, Appl. Opt., 2010, 49, C132–C142 CrossRef CAS.
- S. Guirado, F. J. Fortes, V. Lazic and J. J. Laserna, Spectrochim. Acta, Part B, 2012, 74–75, 137–143 CrossRef CAS PubMed.
- A. De Giacomo, M. Dell'Aglio, F. Colao and R. Fantoni, Spectrochim. Acta, Part B, 2004, 59, 1431–1438 CrossRef PubMed.
- P. K. Kennedy, D. X. Hammer and B. A. Rockwell, Prog. Quantum Electron., 1997, 21, 155–248 CrossRef CAS.
- A. De Giacomo, A. De Bonis, M. Dell'Aglio, O. De Pascale, R. Gaudiuso, S. Orlando, A. Santagata, G. S. Senesi, F. Taccogna and R. Teghil, J. Phys. Chem. C, 2011, 115, 5123–5130 CAS.
- B. Thornton, T. Masamura, T. Takahashi, T. Ura, T. Sakka and K. Ohki, Proceedings of IEEE/MTS Oceans, 2011, Hawaii, 110416-001 Search PubMed.
- B. Thornton and T. Ura, Applied Physics Express, 2011, 4, 022702 CrossRef.
- N. Takada, T. Nakano and K. Sasaki, Appl. Surf. Sci., 2009, 255, 9572–9575 CrossRef CAS PubMed.
- J. A. Aguilera and C. Aragón, Spectrochim. Acta, Part B, 2008, 63, 793–799 CrossRef PubMed.
- J. T. Schiffern, D. W. Doerr and D. R. Alexander, Spectrochim. Acta, Part B, 2007, 62, 1412–1418 CrossRef PubMed.
- M. Lawrence-Snyder, J. Scaffidi, S. M. Angel, A. P. M. Michel and A. D. Chave, Appl. Spectrosc., 2006, 60, 786–790 CrossRef CAS PubMed.
- M. Lawrence-Snyder, J. Scaffidi, S. M. Angel, A. P. M. Michel and A. D. Chave, Appl. Spectrosc., 2007, 61, 171–176 CrossRef CAS PubMed.
- A. P. M. Michel and A. D. Chave, Appl. Opt., 2008, 47, G122–G130 CrossRef CAS.
- A. P. M. Michel, M. Lawrence-Snyder, S. M. Angel and A. D. Chave, Appl. Opt., 2007, 46, 2507–2515 CrossRef CAS.
- A. P. M. Michel and A. D. Chave, Appl. Opt., 2008, 47, G131–G143 CrossRef CAS.
- A. D. Giacomo, M. Dell'Aglio, F. Colao, R. Fantoni and V. Lazic, Appl. Surf. Sci., 2005, 247, 157–162 CrossRef PubMed.
- D. A. Cremers, L. J. Radziemski and T. R. Loree, Appl. Spectrosc., 1984, 38, 721–729 CrossRef CAS.
- V. I. Babushok, F. C. DeLucia, J. L. Gottfried, C. A. Munson and A. W. Miziolek, Spectrochim. Acta, Part B, 2006, 61, 999–1014 CrossRef PubMed.
- G. Bearman, Ocean Chemistry and Deep-Sea Sediments, Pergamon Press, Sydney, 1989 Search PubMed.
- B. Verhoff, S. S. Harilal, J. R. Freeman, P. K. Diwakar and A. Hassanein, J. Appl. Phys., 2012, 112, 093303 CrossRef.
- H. R. Griem, Plasma Spectroscopy, McGraw-Hill, 1964 Search PubMed.
- A. Vogel, S. Busch and U. Parlitz, J. Acoust. Soc. Am., 1996, 100, 148–165 CrossRef PubMed.
- A. De Giacomo, R. Gaudiuso, M. Dell'Aglio and A. Santagata, Spectrochim. Acta, Part B, 2010, 65, 385–394 CrossRef PubMed.
- W. Lei, V. Motto-Ros, M. Boueri, Q. Ma, D. Zhang, L. Zheng, H. Zeng and J. Yu, Spectrochim. Acta, Part B, 2009, 64, 891–898 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |