 Open Access Article
Open Access ArticleNanocatalysis in continuous flow: supported iron oxide nanoparticles for the heterogeneous aerobic oxidation of benzyl alcohol†
David
Obermayer
a,
Alina M.
Balu
b,
Antonio A.
Romero
b,
Walter
Goessler
c,
Rafael
Luque
*b and
C. Oliver
Kappe
*a
aChristian Doppler Laboratory for Microwave Chemistry (CDLMC) and Institute of Chemistry, Karl-Franzens-University Graz, Heinrichstrasse 28, A-8010 Graz, Austria. E-mail: oliver.kappe@uni-graz.at; Fax: (+43)-316-380-9840; Tel: (+43)-316-380-5352
bDepartamento de Quimica Organica, Universidad de Cordoba, Edificio Marie-Curie (C-3), Campus de Rabanales, Ctra. Nnal. IV-A, Km 396, E14014, Cordoba, Spain. E-mail: q62alsor@uco.es; Fax: (+34)-957212066; Tel: (+34)-957211050
cInstitute of Chemistry, Analytical Chemistry, Karl-Franzens-University Graz, Stremayrgasse 16/3, 8010 Graz, Austria
First published on 19th March 2013
Abstract
Investigations on heterogeneous iron catalysis in the selective aerobic oxidation of a primary alcohol are presented. Continuous flow technology was used in combination with an iron oxide nanoparticle catalyst stabilized in a mesoporous aluminosilicate support (“flow nanocatalysis”) as a process intensification tool to maximize catalyst efficiency. Using 5 mol% 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) as a co-catalyst, up to 42% benzyl alcohol as a simple model substrate was selectively converted to benzaldehyde in a single pass of the reactor. Full conversion was achieved by continuous recirculation, simulating an extension of the catalyst bed. ICPMS analysis indicated that the catalyst is highly stable and does not leach under the investigated conditions, providing solid evidence for the participation of a heterogeneous iron species in the catalytic cycle.
Introduction
Aldehydes are valuable precursors for the production of pharmaceuticals, dyes and cosmetics. A wide range of transition-metal (Cu, Ru, Pd, Au, Fe, V, Ir, Os, Co) based catalysts are known today to catalyze the aerobic oxidation of alcohols to aldehydes.1,2 Although the use of Dess–Martin periodinane,3 Swern reagents4 or the tetrapropylammonium perruthenate/N-methylmorpholine-N-oxide (TPAP/NMO)5 system is still preferred for the selective oxidation of primary alcohols in the pharmaceutical industry,2 aerobic oxidations are becoming increasingly attractive as more atom-efficient and environmentally friendly alternatives to stoichiometric oxidants. Several industrial processes have been developed involving the homogeneous phosphonomolybdate/2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO)6 or Cu+/TEMPO7 systems as prominent examples.The use of catalytic transition metal oxide nanoparticles in such processes would simplify work-up procedures and has the potential to reduce environmentally problematic reaction waste streams. Since heterogeneous catalytic systems are kinetically constrained by surface availability, it may not surprise that most research groups working on heterogeneously catalyzed aerobic oxidations have mainly focused in the past on highly active noble metals including palladium, ruthenium or gold.2 Apart from cost considerations, noble metal residues (e.g. Pt and Pd) in pharmaceutical and nutritional products are highly problematic, and thus should be kept to a minimum.8a,b,9 Gold nanoparticles are nowadays embraced by many researchers for biomedical applications since cytotoxicity tests have shown a low acute toxicity of gold. On the other hand, the use of gold may not be totally without complication either, since it has been found to be able to act as an allergic sensitizer.8b
Recent work on nanoparticle catalysts demonstrates that also catalytically active and less toxic “base”-metals such as manganese can be used in selective oxidations of alcohols.10 Similarly, Beller and coworkers have reported iron oxide nanopowders as (unsupported) oxidation catalysts using hydrogen peroxide as an oxidant, demonstrating that under suitable conditions, active catalysts on the border between heterogeneous bulk-catalysts and homogeneous iron systems can be achieved.11 Along similar lines, we recently identified low loaded (<1 wt%) mesoporous silica supported iron oxide nanoparticle systems as efficient and selective oxidation nanocatalysts of primary alcohols with hydrogen peroxide.12
Only a very few examples of immobilized catalyst systems for the aerobic oxidation of alcohols in continuous flow are to date found in the literature. Polymer-supported Pd-nanoparticles,13a Au–(Pt/Pd) nanoclusters13b and gold-coated microcapillaries14 have been used as catalysts in the selective oxidation of benzylic and aliphatic alcohols, using molecular oxygen as an oxidant. Ruthenium in the form of perruthenate on Al2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15a as well as Ru–Al2O315b,c has been investigated in dedicated packed-bed microreactors. In addition, perruthenate has also been ionically immobilized on MCM41-mesoporous silica.15d
15a as well as Ru–Al2O315b,c has been investigated in dedicated packed-bed microreactors. In addition, perruthenate has also been ionically immobilized on MCM41-mesoporous silica.15d
In recent years, we have developed a series of low loaded-iron oxide nanoparticles supported on a range of materials (e.g. mesoporous silicas, aluminosilicates and MOFs) which were proved to be catalytically active in a variety of chemistries including acid-catalysed and redox processes.12,16 Particularly, the use of hydrogen peroxide in conjunction with iron oxide nanoparticle systems was found to be highly efficient in the oxidation of a range of substrates from alcohols to alkenes and sulfides under mild batch reaction conditions.12,16,17 Interestingly, iron catalysis in aerobic batch oxidations of alcohols has to date been exclusively reported with homogeneous iron catalysts in the form of inorganic salts, with relatively high iron loadings (5–20 mol%).18a–g This concept has not yet been reported using heterogeneous catalysts. The combination of iron nanocatalysis heterogeneous aerobic-promoted oxidations with continuous flow conditions constitutes a novel and greener approach to conduct oxidation processes with alternative environmentally friendly transition metal nanocatalysts different from conventional aerobic oxidations with noble metals (e.g. Au). In the light of these premises, we report herein our investigations on the use of a heterogeneous iron nanocatalyst system (aluminosilicate supported 1 wt% Fe nanoparticle system (Fe/Al-SBA15) for the selective oxidation of benzyl alcohol to benzaldehyde under continuous flow conditions.
Results and discussion
Iron oxide nanocatalyst
In an initial exploratory study we have reinvestigated some of the previously published results on the use of iron oxide nanoparticles in combination with hydrogen peroxide as an oxidant.12,16,17 Using small-scale microwave batch technology,19 we were aiming to identify suitable support/solvent combinations as a starting point for aerobic oxidations in a three-phase microreactor. These preliminary experiments demonstrated a very similar activity/chemoselectivity of supported Fe2O3 nanoparticles on various types of mesoporous silica supports (see ESI† for details).For our aerobic oxidations in continuous flow, Fe/Al-SBA15 (1 wt% Fe) was selected as a catalyst. This catalyst was prepared using a very simple microwave batch approach: iron(II) chloride in ethanol together with a dispersion of mesoporous Al-SBA15 silica support was heated to 150 °C for some minutes to directly stabilize in situ generated iron oxide nanocrystals on the silica support.20,21 XRD and TEM measurements confirmed that the active sites of the catalyst were predominantly iron oxide nanocrystals in the form of hematite phase (γ-Fe2O3) which were found to be covalently bound to the aluminosilicate framework of the support via Al–O–Fe bridges.12 This structural feature has been reported to be responsible for observed rate enhancements in the oxidation of benzyl alcohol with hydrogen peroxide (as compared to Si–O–Fe) leading to higher lattice oxygen flexibility.12 BET measurements of the catalyst indicate a surface area of ca. 700 m2 g−1, a pore size of ca. 7.5 nm and a pore volume of 0.65 mL g−1. The average size of the deposited nanocrystals is around 6–7 nm.12,21
Flow reactor
Micro-flow systems enable a directed reactant flux through an immobilized catalyst, making them very attractive process intensification tools by increasing mass-transfer, and additionally allow very high temperatures and gas-pressures in three-phase reactions.22 This specialized family of tubular reactors has low hold-up volumes and an excellent thermal transport capability. Combustion and explosion hazards arising from the notoriously dangerous combination of pressurized oxygen with flammable organic solvents are eliminated and exothermicity can be easily controlled.23This study was performed using a commercially available stainless-steel flow reactor-system (H-Cube Pro with Gas Module, Thales Nanotechnology Inc.) equipped with changeable catalyst cartridges and an external gas-module with a mass-flow controller (1–98 mL min−1 oxygen) interfaced with the reactor software (Scheme 1).24 Reaction mixtures are delivered by an HPLC pump (0.3–3 mL min−1) and the gas–liquid mixing ratio and bubble generation can be monitored with a bubble detector followed by a gas-mixing-valve. The gas–liquid mixing ratio is automatically controlled and stabilized by the software. All reactor parameters and valves are controlled via a touchscreen-operated user-interface.
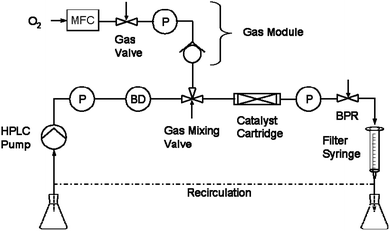 | ||
| Scheme 1 Process schematic of the reactor (H-Cube Pro with Gas Module, Thales Nanotechnology Inc., Hungary).24 P = pressure sensor, BD = bubble detector, MFC = mass flow controller, BPR = back-pressure regulator. | ||
The installed thermostatable (20–150 °C) catalyst cartridge is 70 mm long (4 mm i.d.) and contains ∼200 mg mesoporous Fe/Al-SBA15 (average particle size 1–2 μm). Together with the void volume inside the cartridge of ∼500 μL, the total internal volume of the reactor with all pipe connections is ∼1.5 mL. A motor-controlled variable backpressure-regulator allows conducting reactions at pressures up to 100 bar. The product mixture can either be directly collected for rapid screenings, or continuously recirculated to reach higher conversions. For recirculation, a 10 mL filter-syringe (without plunger) was used in place of the standard porous filter-inlet to provide a vessel for liquid buffering, gas–liquid separation and sample withdrawal.
Continuous flow optimizations using benzyl alcohol as a model substrate
We started our investigation of Fe/Al-SBA15 as a fixed-bed catalyst using a 0.1 M solution of benzyl alcohol in a selection of oxidation-stable aprotic solvents (Table 1). Using dioxane, we initially obtained only traces of benzaldehyde at reaction temperatures up to 120 °C and oxygen pressures up to 100 bar (entry 1, Table 1). A pressure drop of up to 100 bar was observed across the cartridge, making it impossible to vary the oxygen pressure (according to the working principle of the reactor, the liquid backpressure also determines the oxygen pressure in the reaction). In response to this problem, the mesoporous silica/iron oxide catalyst was diluted 3:1 with silica gel (60–100 mesh), which led to a significantly lower pressure drop across the cartridge (30–50 bar; entry 2, Table 1). Diluting the catalyst with inert material is clearly undesirable as the amount of catalyst is reduced and a large part of the catalyst is by-passed via neighboring silica gel particles. The use of toluene as a solvent (entry 5, Table 1) did not furnish any benzaldehyde, but quantitatively gave benzyltoluenes. This side-reaction can be ascribed to the high Lewis acidity of the Fe/Al-SBA15 catalyst, causing Friedel–Crafts alkylation of benzyl alcohol.25
| Entry | T [°C] | p [bar] | Solvent | η [mPa s] | Vapor pressurec [bar] | Catalyst:silica gel (w/w) |
Conv. [%] | Sel. [%] |
|---|---|---|---|---|---|---|---|---|
| a 0.1 M benzyl alcohol, 0.5 mL min−1, X-Cube, Thales Nanotechnology.27 Conversions and selectivities were calculated from HPLC areas at 215 nm referenced against genuine samples of the analytes using o-xylene as an internal standard. Selectivities correspond to benzaldehyde selectivity calculated from calibrated HPLC yields of benzaldehyde and benzoic acid. Byproducts other than benzoic acid (with the exception of entry 5) were not observed. b Viscosity data from ref. 24. c Calculated using the Antoine equation; dioxane values were measured in an autoclave. d Fe/Al-SBA15 (1 wt% Fe); ∼200 mg per cartridge. High cartridge pressure-drop (up to 100 bar). e GC/MS shows almost quantitative formation of benzyltoluenes. | ||||||||
| 1 | 120 | 100 | Dioxane | 1.1 | Pure catalystd | 2 | >99 | |
| 2 | 120 | 50 | Dioxane | 1:3 |
4 | >99 | ||
| 3 | 140 | 50 | Dioxane | 1.3 | 3.7 | 1:3 |
12 | >91 |
| 4 | 160 | 50 | Dioxane | 6.0 | 1:3 |
6 | >99 | |
| 5 | 120 | 50 | Toluene | 0.56 | 1.3 | 1:3 |
0e | — |
| 6 | 120 | 50 | MeCN | 0.37 | 3.1 | 1:3 |
0 | — |
Furthermore, taking the vapor pressure/conversion data from Table 1 into consideration, an inverse dependence of the conversions on solvent vapor pressure inside the reactor could be inferred. Both the utilization of acetonitrile (entry 6, Table 1) as a low boiling solvent or an increase of the reaction temperature to 160 °C using dioxane (entry 4, Table 1) caused a drop in activity. This confirmed our assumption that only solvents with moderate (>100 °C) to high boiling points would be appropriate to be employed at higher temperatures. In the light of these premises, the highest conversion of benzyl alcohol to benzaldehyde obtainable was quite low (<12%) at around 140 °C and 50 bar oxygen pressure in dioxane (entry 3, Table 1).
In order to increase the efficiency of the catalyst-bed, we looked for a suitable co-solvent as auxiliary for pressure-drop management by reducing the solvent viscosity. Both the vapor pressure and viscosity criteria are fulfilled by n-heptane (bp 98 °C, η = 0.39 mPa s).26 Using mixtures of n-heptane with dioxane or n-heptane alone allowed us to conduct all further experiments without the need of catalyst dilution (Table 2). Consequently, we were able to use significantly lower oxygen pressures (12 and 25 bar) solving initial problems with lifetime of the catalyst (Fig. S7†). This drastic modification of reaction conditions had a comparably low impact on conversion rates (4% at 25 bar vs. 12% at 50 bar).
| Entry | Catalyst | Co-catalyst | PhC(O)H [%] | PhCO2H [%] |
|---|---|---|---|---|
|
a 0.1 M in n-heptane–dioxane 2:1, 140 °C, 25 bar O2 at 5 mL min−1 and liquid flow rate 0.22 mL min−1.
|
||||
| 1 | Fe/Al-SBA15 (1 wt% Fe) | None | 4 | <1 |
| 2 | Fe/Al-SBA15 (1 wt% Fe) | 5 mol% NHPI | 10 | — |
| 3 | Fe/Al-SBA15 (1 wt% Fe) | 5 mol% TEMPO | 42 | — |
| 5 | SBA15 | 5 mol% TEMPO | — | — |
| 6 | Al-SBA15 | 5 mol% TEMPO | 4 | — |
In this context it must be stressed that both dioxane and n-heptane are clearly not examples of green solvents. Their use in this laboratory scale investigation can only be justified to obtain proof-of-concept for the anticipated heterogeneous flow oxidation process. For any industrial scale process, more sustainable alternatives will have to be sought.
Stable nitroxyl-radicals such as TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) or PINO (phthalimide-N-oxyl formed in situ from N-hydroxyphthalimide (NHPI)) are known to be effective hydrogen-shuttles in catalytic cycles of transition-metal oxidations but at the same time are potent oxidation inhibitors (e.g., overoxidation of alcohols to carboxylic acids).28 In a recent example, Gao and coworkers demonstrated that iron(III) chloride/TEMPO (8 and 2–5 mol%, respectively) in the presence of silica gel is an efficient catalytic system for the aerobic oxidation of benzyl alcohols.18g The role of silica gel, which was found to significantly enhance the conversion rate, was however not fully understood.18g
Utilizing an optimized set of temperatures/pressures/solvents (140 °C at 25 bar O2 in n-heptane–dioxane 2:1), we examined the effects of adding 5 mol% NHPI in combination with Fe/Al-SBA15 (1 wt% Fe) or 5 mol% TEMPO as a co-catalyst under continuous flow conditions. Very notably, up to 42% (entries 2 and 3, Table 2) of benzyl alcohol was selectively converted to aldehyde using TEMPO as a co-catalyst. NHPI had a comparably small effect on conversion rates (10 vs. 4%). Benzoic acid was not observed to be formed in any case.
After having established that TEMPO can be used to enhance the catalytic activity in this reaction, the iron-catalyst cartridges were replaced by cartridges packed with parent supports including a purely siliceous mesoporous silica (SBA15) and mesoporous aluminosilicate (Al-SBA15) to test for possible catalytic background activity of these two supports. These reference experiments showed that benzyl alcohol is either not converted to aldehyde at all or only in traces in the absence of iron oxide (entries 5 and 6, Table 2).
Transition-metal impurities are unlikely to be responsible for the observed catalytic activities, according to an ICPMS assay of the catalyst [Fe/Al-SBA15 (1 wt% Fe)]. This analysis revealed that manganese was only present in quantities of ca. 1 mg kg−1, while other catalytically active transition metals including Pd, Co and Cu were found only in sub-mg kg−1 to μg kg−1 traces (Table 3). Only trace amounts of iron (<50 ng kg−1vs. ∼30 ng kg−1 in the blanks) were found in subsequent leaching studies of the selective Fe/Al-SBA15/TEMPO oxidation system under optimum reaction conditions (140 °C, 0.22 mL min−1 flow-rate and 25 bar O2 at 5 mL min−1), suggesting a genuine and unprecedented heterogeneous iron oxide nanoparticle catalyzed mechanism for the aerobic oxidation of benzyl alcohol (see ESI† for further details on leaching studies).
| Element | Fe/Al-SBA15 |
|---|---|
| Fe [%] | 1.0 |
| Mn [mg kg−1] | 1.4 |
| Co [mg kg−1] | 0.2 |
| Cu [mg kg−1] | <0.05 |
| Rh [mg kg−1] | <0.005 |
| Pd [mg kg−1] | 0.040 |
| Pt [mg kg−1] | <0.005 |
| Au [mg kg−1] | <0.005 |
Parallel investigations on solvent effects indicated that the choice of the solvent is a remarkably critical parameter that influences the chemoselectivity of this reaction (Table 4). n-Heptane as a non-coordinating solvent leads to a significantly superior oxidation activity of the iron oxide nanocatalyst with respect to dioxane that would most likely compete for catalytic sites with benzyl alcohol. Careful tuning of reaction conditions was necessary to avoid the formation of dibenzyl ether and Friedel–Crafts alkylation products (see Fig. S7 and S12 in the ESI†).
| Entry | Solvent | PhC(O)H [%] | Byproducts [%] |
|---|---|---|---|
| a 140 °C, 0.22 mL min−1, 25 bar O2 at 5 mL min−1, 0.1 M, 5 mol% TEMPO, Fe/Al-SBA15 (1 wt% Fe). b Based on HPLC peak areas at 215 nm (o-xylene as an internal standard was alkylated under these conditions). Byproduct composition: 48% dibenzyl ether, 21% benzyl xylenes and 5% benzyl formate. c Total amount of byproducts (dibenzyl ether/alkylation products, also see Fig. S12 in the ESI) = 100 − ∑(PhC(O)H + BzOH + PhCO2H). | |||
| 1 | n-Heptane | 15 | 73b |
| 2 | Dioxane | 14 | — |
| 3 |
n-Heptane–dioxane 1:1 |
14 | — |
| 4 |
n-Heptane–dioxane 2:1 |
42 | — |
| 5 |
n-Heptane–dioxane 4:1 |
29 | — |
| 6 |
n-Pentane–dioxane 2:1 |
17 | 3c |
The formation of the ether under these conditions was comparatively faster in the triphasic oxidation of benzyl alcohol to benzaldehyde (entry 1, Table 4). In contrast to that, Lewis acid promoted side-reactions were surprisingly suppressed in dioxane. In view of these premises, n-heptane–dioxane mixtures were screened to obtain an improved solvent system for the selective oxidation. A mixture of n-heptane–dioxane 2:1 was observed to deliver the best results (42% conversion to benzaldehyde, selectivity >99%, traces of benzyl formate <1%). To further corroborate our previously made assumptions on the inverse vapor pressure dependency of the reaction, n-heptane was replaced by n-pentane (entry 6, Table 4). As expected, the conversion dropped significantly (17% vs. 42%).
The possibility of continuous recirculation of the reaction mixture through the three-phase reactor to reach full conversion was subsequently investigated. Because all other process parameters were left unchanged, it can be assumed that conversions/purity profiles obtained with the proposed methodology would be reproduced without recirculation by simply using a longer catalyst bed. In a first set of experiments, recirculation runs were performed under previously optimized conditions for single-pass operation using 140 °C, 25 bar oxygen pressure and n-heptane–dioxane 2:1 as a solvent. Unfortunately, recirculation of the product solution accelerated catalyst deactivation, with the reaction already terminating between 30–50% conversion (Table 5, also see Fig. S11 in the ESI†).
Deactivation of the catalyst was obviously accompanied by the formation of significant amounts of benzyl formate (see Fig. S11†). We hypothesized that deposition of carbonaceous species by fouling29 would be conceivable since there is no plausible mechanism for the formylation of benzyl alcohol without the participation of C1 molecule fragments. In the absence of dioxane as a co-solvent, the formation of benzyl formate was not observed during recirculation runs. Nevertheless, the catalyst changes its selectivity towards alkylation, using a non-coordinating solvent, generating a mixture of dibenzyl ether, (benzyl)phenylmethanol isomers and a variety of other unknown byproducts (see Fig. S11 in the ESI†).
Gratifyingly, benzyl formate formation and ether formation were avoided by increasing the amount of TEMPO from 5 to 10 mol%, retaining the initial catalyst activity and allowed us to reach full conversion (95% calibrated HPLC yield) of benzyl alcohol selectively to benzaldehyde (Table 6). The required circulation time in the reactor was ∼1 h at 120 °C, 35 bar oxygen and a flow rate of 0.22 mL min−1 (measured at the outlet) on a 0.5 mmol scale. Although in this case a very low-loaded heterogeneous iron catalyst has been used, the corresponding TON of 14 (Table 6, entry 3) is remarkable since it matches the performance of already published batch results using a high loading homogeneous iron protocol (8 mol% FeCl3/TEMPO/silica gel), from which a TON of 12 for the selective conversion of benzyl alcohol to benzaldehyde was derived.18g
While the use of TEMPO may appear acceptable to study the concept of heterogeneous iron oxide catalysis, for a convenient product isolation a switch from TEMPO to its water-soluble analog 4-OH-TEMPO (4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl, TEMPOL) can be envisaged which would allow separation (e.g., by extraction) during workup.30 Recycling of n-heptane or its “greener” analogues (isooctane, methylcyclohexane) may be achieved by simple distillation from the formed aldehyde product.
Conclusions
A first proof-of-principle study on the use of a heterogeneous iron oxide nanocatalyst for the aerobic oxidation of an (activated) primary alcohol has been reported. The use of an immobilized nanoparticle catalyst in combination with continuous flow technology (“flow-nanocatalysis”) as a process intensification tool promoted the selective oxidation of benzyl alcohol to benzaldehyde as simple model transformation for the oxidation of activated primary alcohols. Applying a low-volume capillary reactor in this study allowed a particularly safe handling of otherwise notoriously dangerous reaction conditions involving high oxygen pressures at high temperatures. No metal leaching was detectable under the examined reaction conditions in the aerobic oxidation with iron oxide/TEMPO in non-polar solvents, supporting assumptions on the heterogeneity of the reaction mechanism. Careful tuning of both process-relevant aspects (backpressure and vapor pressure management) and reaction conditions using TEMPO as a co-catalyst was a key to achieve conversions up to 42% in a single pass of the catalyst cartridge under high selectivity. Continuous recirculation was used to achieve full conversion of benzyl alcohol to benzaldehyde, simulating a hypothetical, significantly longer catalyst bed.Given further advances in the development of heterogeneous iron catalysts, we believe that iron nanocatalysts have potential to become a niche player for selected oxidation processes as non-toxic and cost-effective replacement of already known noble-metal and heavy-metal based catalyst systems. Further investigations on the hitherto unknown mechanistic aspects of iron oxide nanocatalysis in aerobic oxidations as well as further work on the substrate scope of this reaction are currently ongoing in our laboratories.
Experimental section
General remarks
GC/MS monitoring was based on electron impact ionization (70 eV) using an HP/5MS column (30 m × 0.250 mm × 0.025 mm). After 1 min at 50 °C the temperature was increased in 25 °C min−1 steps up to 300 °C and kept at 300 °C for 4 min. The carrier gas was helium and the flow rate 1.0 mL min−1 in constant-flow mode. ICPMS analyses were made on an Agilent 7500ce inductively coupled plasma mass-spectrometer. After the element screening in the digest of the Fe/Al-SBA15 catalyst material the elements listed in Table 3 were quantitatively determined with appropriate ICP standard solutions. Analytical HPLC analysis was carried out on a C18 reversed-phase (RP) analytical column (150 × 4.6 mm, particle size 5 μm) at 37 °C using a mobile phase A (water–acetonitrile 90:10 (v/v) + 0.1% TFA) and B (MeCN + 0.1% TFA) at a flow rate of 1.5 mL min−1. The following gradient was applied: linear increase from solution 30% B to 100% B in 9 min, hold at 100% solution B for 2 min. The identity of starting materials and products was confirmed by referencing against samples of the pure compounds and corresponding GC/MS spectra. Absolute quantities (yields) of starting materials and products were calculated from HPLC areas at 215 nm using 0.25–1 equiv. of o-xylene as an internal standard, added prior to the reactions. An internal standard calibration was provided by measuring mixtures of known composition containing the analytes and o-xylene at 215 nm in the HPLC. The inertness of o-xylene under the used reaction conditions was constantly monitored via the checksum of the calibrated analytes, and by GC/MS. The TON in the recycle experiments was calculated by dividing the amount of aldehyde formed (mmol) with the amount of iron catalysts in the fixed bed reactor (mmol) used in the reaction. All chemicals and solvents were obtained from known commercial suppliers and were used without any further purification. Iron oxide catalysts and supports were packed into catalyst cartridges at the facility of Thales Nanotechnology, Hungary. All continuous flow experiments were performed using a stainless steel microreactor with a packed-bed catalyst (H-Cube Pro with Gas-Module, or the X-Cube), similar in construction (both Thales Nanotechnology Inc.).24,27
General experimental procedure for continuous flow screenings
All screenings with the exception of the data presented in Table 1 were done in the H-Cube Pro (Thales Nanotechnology Inc.) continuous flow reactor with a gas module. For a typical experiment, 10–15 mL of a 0.1 M benzyl alcohol solution with 0–5 mol% of TEMPO (2,2,6,6-tetramethylpiperidine-1-oxyl) or NHPI (N-hydroxyphthalimide) as a co-catalyst was prepared. Prior to the experiment, 0.25–1 equiv. of o-xylene was added as an internal standard. Oxygen gas was supplied at 75–140 bar pressure by an external gas-bottle connected via a 1-stage pressure reducer to the gas-module for providing a variable gas-flow (0–98 mL min−1) at variable pressures. Since the mesoporous silica cartridges in some cases developed very high backpressures (up to 100 bar) at room temperature, a special procedure was used to prevent safety termination by the software due to high pressure peaks (>140 bar) during equilibration of the instrument's control circle for gas addition. A fresh catalyst cartridge (70 × 4 mm i.d.; ∼200 mg catalyst) was installed, and then heated to the target temperature between 100 and 140 °C (without switching to oxygen gas) at a set flow rate of 0.3 mL min−1 (0.22 mL min−1 measured at the outlet) with the working solvent. Using the “stop + keep” option, the temperature of the cartridge was kept constant, followed by the start of the screening run using a gas-module setting of 5–98 mL min−1 for the oxygen flow and a gas-pressure of 12–100 bar, as adjustable by the liquid backpressure valve. Depending on screening conditions, the HPLC pump was set to a flow rate of 0.3–0.5 mL min−1. After switching to the reactant solution and discarding the dead-volume of around 1.5 mL of the reactor, the outlet was switched to product collection and 1 mL fractions were collected. 100 μL of each sample was diluted to 1 mL with dioxane and subjected to HPLC analysis at 215 nm using an internal standard calibration. Selected fractions were further subjected to GC/MS analysis and ICPMS for elemental trace analysis (see Table S11†).General experimental procedure for recirculation
Recirculation runs were conducted using the reactor-startup procedure already described for the H-Cube Pro with a gas-module and using a constant pumping speed of 0.3 mL min−1 (0.22 mL min−1 as measured at the reactor outlet). A 10 mL filter-syringe with a removed plunger which acted as a buffer-vessel for gas–liquid separation and access for sample-withdrawal was connected upside-down to the inline-switching valve of the microreactor. The effluent of the reactor at the outlet switching valve was recirculated to the filter-syringe via PTFE tubing. For the full conversion of benzyl alcohol to benzaldehyde, a volume of 5–10 mL of a 0.1 M benzyl alcohol solution with 5 mol% TEMPO in n-heptane–dioxane 2:1 or pure n-heptane with 10 mol% TEMPO was prepared. This reaction mixture was circulated for 1–7 h at 120–140 °C and 25–35 bar oxygen pressure at a gas-flow rate of 5 mL min−1. During recirculation, 10 μL samples were taken from the buffering vessel every 1–3 h to monitor the conversion. In parallel, a make-up solvent was added periodically to compensate the evaporation-losses (0.5–1 mL h−1) caused by the vigorous gas-stream. The optimization trials were partly performed without an internal standard (to prevent side reactions by alkylation of o-xylene), whereas under optimized conditions, the aerobic oxidation was performed with 0.25 equiv. of o-xylene as an internal standard.
Acknowledgements
This work was supported by a grant from the Christian Doppler Research Society (CDG). Rafael Luque gratefully acknowledges the Spanish MICINN for financial support via the concession of a RyC contract (ref: RYC-2009-04199) and funding under project CTQ2011-28954-C02-02. Consejeria de Ciencia e Innovacion, Junta de Andalucia is also gratefully acknowledged for funding project P10-FQM-6711. Both institutions gratefully acknowledge support from MICINN under an Integrated Action Austria-Spain (ES 09/2010; AT2009-0031).References
- (a) R. A. Sheldon and J. K. Kochi, Metal-Catalyzed Oxidations of Organic Compounds, Academic Press, New York, 1981 Search PubMed; (b) Modern Oxidation Methods, ed. J. E. Bäckvall, Wiley-VCH, Weinheim, 2004 Search PubMed; (c) M. Hudlicky, Oxidations in Organic Chemistry, ACS, Washington, DC, 1990 Search PubMed.
- For a recent review on transition-metal catalyzed aerobic oxidations of alcohols see: C. Parmeggiani and F. Cardona, Green Chem., 2012, 14, 547–564 RSC.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155–4156 CrossRef CAS.
- A. J. Mancuso, S. L. Huang and D. J. Swern, J. Org. Chem., 1978, 12, 2480–2482 CrossRef.
- S. V. Ley, J. W. Norman, P. Griffith and S. P. Marsden, Synthesis, 1994, 639–666 CrossRef.
- R. Ben-Daniel, P. Alsters and R. Neumann, J. Org. Chem., 2001, 66, 8650–8653 CrossRef CAS.
- D. Geißlmeir, W. G. Jary and H. Falk, Monatsh. Chem., 2005, 136, 1591–1599 CrossRef.
- (a) Guideline on the specification limits for residues of metal catalysts from the European Agency for the Evaluation of Medicinal Products (EMEA), 2007 (http://www.emea.europa.eu). According to the guideline, only iron residues are considered entirely unproblematic. The limit for the total amount of platinoid-group metals is 5 mg kg−1 (for dosages of up to 10 g per day). Compliance to this limit is usually assured by post-reaction scavenging/purification (see ref. 9a–c); ; (b) For an article treating the role of gold as an allergic sensitizer: M. Bruze and K. E. Andersen, Contact Dermatitis, 1999, 40, 1–5 CrossRef.
- For transition metal separation in pharmaceutical synthesis, see: (a) C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889–900 CrossRef CAS; (b) C. J. Welch, J. Albaneze-Walker, W. R. Leonard, M. Biba, J. DaSilva, D. Henderson, B. Laing, D. J. Mathre, S. Spencer, X. Bu and T. Wang, Org. Process Res. Dev., 2005, 9, 198–205 CrossRef CAS; (c) J. T. Bien, G. C. Lane and M. R. Oberholzer, Top. Organmet. Chem., 2004, 6, 263–283 CAS.
- H.-H. Sun, Q. Hua, F.-F. Guo, Z.-Y. Wang and W.-X. Huang, Adv. Synth. Catal., 2012, 354, 569–573 CrossRef CAS.
- (a) F. Shi, M. K. Tse, M.-M. Pohl, A. Brückner, S. Zhang and M. Beller, Angew. Chem., Int. Ed., 2007, 46, 8866–8868 CrossRef CAS; (b) B. Join, K. Möller, C. Ziebart, K. Schröder, D. Gördes, K. Thurow, A. Spannenberg, K. Junge and M. Beller, Adv. Synth. Catal., 2011, 353, 3023–3030 CrossRef CAS.
- (a) C. Gonzalez-Arellano, J. M. Campelo, D. J. Macquarrie, J. M. Marinas, A. A. Romero and R. Luque, ChemSusChem, 2008, 1, 746–750 CrossRef CAS; (b) A. M. Balu, A. Pineda, K. Yoshida, J. M. Campelo, P. L. Gai and R. Luque, Chem. Commun., 2010, 46, 7825–7827 RSC.
- (a) Z. Hou, N. Theyssen, A. Brinkmann and W. Leitner, Angew. Chem., Int. Ed., 2005, 44, 1346–1349 CrossRef CAS; (b) K. Kaizuka, K.-Y. Lee, H. Miyamura and S. Kobayashi, J. Flow Chem., 2012, 1, 1–4 CrossRef.
- N. Wang, T. Matsumoto, M. Ueno, H. Miyamura and S. Kobayashi, Angew. Chem., Int. Ed., 2009, 121, 4838–4840 CrossRef.
- (a) E. Cao, W. B. Motherwell and A. Gavriilidis, Chem. Eng. Technol., 2006, 29, 1372–1375 CrossRef CAS; (b) N. Zotova, K. Hellgardt, G. H. Kelsall, A. S. Jessiman and K. K. Hii, Green Chem., 2010, 12, 2157–2163 RSC; (c) D. V. Bavykin, A. A. Lapkin, S. T. Kolaczkowski and P. K. Plucinski, Appl. Catal., A, 2005, 288, 175–184 CrossRef CAS; (d) A. Bleloch, B. F. G. Johnson, S. V. Ley, A. J. Price, D. S. Shephard and A. W. Thomas, Chem. Commun., 1999, 1907–1190 RSC.
- (a) F. Rajabi, N. Karimi, M. Reza Saidi, A. Primo, R. S. Varma and R. Luque, Adv. Synth. Catal., 2012, 354, 1707–1711 CrossRef CAS; (b) A. Pineda, A. M. Balu, J. M. Campelo, A. A. Romero, D. Carmona, F. Balas, J. Santamaria and R. Luque, ChemSusChem, 2011, 4, 1561–1565 CrossRef CAS; (c) F. Rajabi, S. Naresian, A. Primo and R. Luque, Adv. Synth. Catal., 2011, 353, 2060–2066 CrossRef CAS.
- (a) A. M. Balu, C. S. K. Lin, H. Liu, C. Vargas, Y. Li and R. Luque, Appl. Catal., A, 2013, 455, 261–266 CrossRef CAS; (b) F. Rajabi, A. Pineda, S. Nahresian, A. M. Balu, A. A. Romero and R. Luque, Green Chem., 2013 10.1039/c3gc40110c; (c) A. I. Carrillo, E. Serrano, R. Luque and J. Garcia-Martinez, Appl. Catal., A, 2013, 483, 383–390 CrossRef.
- (a) S. E. Martin and D. F. Suarez, Tetrahedron Lett., 2002, 43, 4475–4479 CrossRef CAS; (b) N. Wang, R. Liu, J. Chen and X. Liang, Chem. Commun., 2005, 5322–5324 RSC; (c) W. Yin, C. Chu, Q. Lu, J. Tao, X. Liang and R. Liu, Adv. Synth. Catal., 2010, 352, 113–118 CrossRef CAS; (d) X. Wang and X. Liang, Chin. J. Catal., 2008, 29, 935–939 CrossRef CAS; (e) S. Ma, J. Liu, S. Li, B. Chen, J. Cheng, J. Kuang, Y. Liu, B. Wan, Y. Wang, J. Ye, Q. Yu, W. Yuan and S. Yu, Adv. Synth. Catal., 2011, 353, 1005–1017 CrossRef CAS; (f) C.-X. Miao, J.-Q. Wang, B. Yu, W. G. Cheng, J. Sun, S. Chanfreau, L.-N. He and S.-J. Zhang, Chem. Commun., 2011, 47, 2697–2699 RSC; (g) L. Wang, J. Li, Y. Lv, G. Zhaoa and S. Gao, Appl. Organomet. Chem., 2012, 26, 37–43 CrossRef CAS.
- For recent reviews on microwave-assisted organic chemistry, see: (a) S. Caddick and R. Fitzmaurice, Tetrahedron, 2009, 65, 3325–3355 CrossRef CAS; (b) C. O. Kappe and D. Dallinger, Mol. Diversity, 2009, 13, 71–193 CrossRef CAS.
- For general methodologies to prepare supported iron-nanoparticles see: (a) J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2, 18–45 CrossRef CAS; (b) Z. Ma and S. Dai, Nano Res., 2011, 4, 3–32 CrossRef CAS; (c) B. Karimi, H. Behzadnia, E. Farhangi, E. Jafari and A. Zamani, Curr. Org. Synth., 2010, 7, 543–567 CrossRef CAS.
- A. M. Balu, D. Dallinger, D. Obermayer, J. M. Campelo, A. A. Romero, D. Carmona, F. Balas, K. Yohida, P. L. Gai, C. Vargas, C. O. Kappe and R. Luque, Green Chem., 2012, 14, 393–402 RSC.
- For a selection of gas–liquid–solid microreactor applications see: (a) Micro Reaction Technology in Organic Synthesis, ed. C. Wiles, P. Watts, CRC Press, Boca Raton, 2011, pp. 60–75 Search PubMed; (b) Micro Process Engineering, Vol. 2: Devices, Reactions and Applications, ed. V. Hessel, A. Renken, J. C. Schouten, J. Yoshida, Wiley-VCH, Weinheim, 2009, pp. 131–182 Search PubMed; (c) M. Irfan, T. N. Glasnov and C. O. Kappe, ChemSusChem, 2011, 4, 300–316 CrossRef CAS; (d) J. Kobayashi, Y. Mori, K. Okamoto, R. Akiyama, M. Ueno, T. Kitamori and S. Kobayashi, Science, 2004, 304, 1305–1308 CrossRef CAS; (e) X. Liu, B. Ünal and K. F. Jensen, Catal. Sci. Technol., 2012, 2, 2134–2138 RSC; (f) See also ref. 13–15.
- For a selection of reviews and articles discussing safety aspects of oxidations and hydrogenations in microreactors see: (a) V. Hessel, Chem. Eng. Technol., 2009, 32, 1655–1681 CrossRef CAS; (b) A. de Mello and R. Wootton, Lab Chip, 2002, 2, 7N13N Search PubMed; (c) T. Inoue, M. A. Schmidt and K. F. Jensen, Ind. Eng. Chem. Res., 2007, 46, 1153–1160 CrossRef CAS; (d) M. Hamano, K. D. Nagy and K. F. Jensen, Chem. Commun., 2012, 2086–2088 RSC.
- For more details on the H-Cube Pro reactor (Thales Nanotechnology Inc.), see: http://www.thalesnano.com.
- A. Pineda, A. M. Balu, J. M. Campelo, R. Luque, A. A. Romero and J. C. Serrano-Ruiz, Catal. Today, 2012, 187, 65–69 CrossRef CAS.
- (a) CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, 90th edn, 2010, pp. F38–F40 Search PubMed; (b) Tabellenbuch Chemie, ed. R. Kaltofen, Verlag Harry Deutsch, Frankfurt am Main, 13th edn, 1998, p. 233 Search PubMed.
- The X-Cube from Thales Nanotechnology is identical to the H-Cube Pro in its basic construction and capabilities, but cannot be used in combination with a mass-flow-controller to vary the gas liquid ratio. For a description of the X-Cube see ref. 15b.
- For a review on nitroxyl radical mediated organic oxidations see: R. E. Sheldon and I. W. C. Arends, Adv. Synth. Catal., 2004, 346, 1051–1071 CrossRef CAS.
- For mechanisms and definitions of catalyst deactivation processes see: C. H. Bartholomew, Appl. Catal., 2001, 212, 17–60 CrossRef CAS.
- R. Ciriminna and M. Pagliaro, Org. Process Res. Dev., 2010, 14, 245–251 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3gc40307f |
| This journal is © The Royal Society of Chemistry 2013 |