Ruthenium(II)-catalyzed selective monoarylation in water and sequential functionalisations of C–H bonds†
Percia B. Arockiam, Cédric Fischmeister, Christian Bruneau* and Pierre H. Dixneuf*
Centre of Catalysis and Green Chemistry, OMC-Institut Sciences Chimiques de Rennes, UMR 6226: CNRS-Université de Rennes, Campus de Beaulieu, 35042 Rennes, France. E-mail: christian.bruneau@univ-rennes1.fr; pierre.dixneuf@univ-rennes1.fr
First published on 1st October 2012
Abstract
The ruthenium(II)-phosphine catalyst RuCl2(PPh3)(p-cymene) operating water selectively leads to ortho monoarylation, with arylchlorides and heteroarylhalides, of functional arenes. Further catalytic heteroarylation with Ru(OAc)2(p-cymene) in water produces mixed bifunctional derivatives.
Introduction
The direct catalytic functionalisation of C–H bonds is attracting tremendous interest to develop at low cost C–C bond cross-coupling reactions without the use of a stoichiometric amount of organometallic reagents, thus via a better atom economy and greener process.1 However many influencing factors need to be elucidated and improved such as catalyst efficiency and cost, compatible non-toxic solvents, and multiple controlled regioselectivities. Another rising objective consists in the consecutive regioselective functionalisations of several C–H bonds for both synthetic efficiency and green chemistry contribution. The diarylation with arylhalides and heteroarylhalides of ortho C–H bonds of functional arenes can now be achieved easily,1 especially with cheap ruthenium(II) catalysts in the presence of catalytic amounts of carboxylate.2–4 The sequential monoarylations with two different functional (hetero)arylhalides of two identical C–H bonds have not been achieved yet in spite of their potential to reach new mixed bifunctional polyaryl molecules of interest for molecular materials and as polymer precursors.Selective monoarylations are easily achieved only when one ortho substituent is present or when only one C–H bond is protected by steric hindrance of a substituent at an arene meta position. However a few ruthenium(II)-phosphine catalysts operating in organic solvents have shown the ability to preferentially generate monoarylated arenes with rather good selectivity.4 The first example of direct arylation with arylbromides using the ruthenium(II)-PPh3 catalyst was shown by Oi and Inoue and led predominantly to the ortho monoarylation of 2-phenylpyridine and arylimines in NMP.4a,b Chanjuan Xi et al.4c demonstrated a selective monoarylation of 2-phenylpyridine in the presence of a [RuCl2(PPh2(–C(Ph)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHPh)(arene))] catalyst precursor. The [RuCl2(L)(p-cymene)]2 complex containing a bulky monophosphine L ligand was efficient to monoarylate 2-phenylpyridine and N-phenylpyrazole with arylchlorides in NMP at 120 °C as reported by Doherty et al.4d In parallel the carboxylate-ruthenium(II) catalysts when operating in water were revealed to provide in a rare example a high ratio of mono(hetero)arylation of functional arene C–H bonds.3c Thus we have investigated the positive influence of these two factors: ruthenium(II)-phosphine catalyst and ruthenium catalysis in water in the search for selective monoarylation, thus in a non-toxic solvent and under greener chemistry conditions. Several industrial processes are already operating under aqueous phase organometallic-catalyzed reaction conditions and there are already some technical solutions such as membrane filtration or phase separation for water recovery.5
CHPh)(arene))] catalyst precursor. The [RuCl2(L)(p-cymene)]2 complex containing a bulky monophosphine L ligand was efficient to monoarylate 2-phenylpyridine and N-phenylpyrazole with arylchlorides in NMP at 120 °C as reported by Doherty et al.4d In parallel the carboxylate-ruthenium(II) catalysts when operating in water were revealed to provide in a rare example a high ratio of mono(hetero)arylation of functional arene C–H bonds.3c Thus we have investigated the positive influence of these two factors: ruthenium(II)-phosphine catalyst and ruthenium catalysis in water in the search for selective monoarylation, thus in a non-toxic solvent and under greener chemistry conditions. Several industrial processes are already operating under aqueous phase organometallic-catalyzed reaction conditions and there are already some technical solutions such as membrane filtration or phase separation for water recovery.5
We now report that Ru(II)-PPh3 catalyst RuCl2(PPh3)(p-cymene) operating in water, without a carboxylate promoter, constitutes a good catalytic system for selective monoarylation of functional arenes and that the sequential catalytic functionalisations in water of two ortho C–H bonds of functional arenes can be applied to reach unsymmetrical ortho diarylated arenes and one example of mixed ortho arylated and alkenylated arenes.
Results and discussion
The activity of an arylation catalyst was often revealed by the ratio of the diarylation of (hetero)arenes as mono and diarylations had almost similar rates.3 Thus conditions were searched to differentiate these two reaction rates. We first investigated the reaction of a slight excess of 2-phenylpyridine (0.6 mmol) with chlorobenzene (0.5 mmol) in the presence of various catalysts in water at 100 °C and in the presence of K2CO3 as a base, as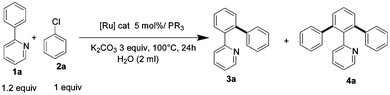 | (1) |
| Entry | Catalyst | Time (h)/Convb (%) | 3a/4ac |
|---|---|---|---|
| a Reaction conditions: 0.6 mmol of 2-phenylpyridine, 5 mol% of [Ru], 3 equiv. of K2CO3, 10 μL of tetradecane (internal standard) for GC, 0.5 mmol of chlorobenzene in 2 mL of water, 24 h, 100 °C.b Conversion determined by gas chromatography based on arylhalide.c 3a/4a ratio determined by gas chromatography.d Reaction at 80 °C.e Reaction performed in NMP as a solvent.f Reaction at 60 °C. | |||
| 1 | [RuCl2(p-cymene)]2 | 24 h – 97 | 78/22 |
| 2 | Ru(OPiv)2(p-cymene) | 24 h – 98 | 78/22 |
| 3 | Ru(OPiv)2(p-cymene) + 2 equiv. PPh3 | 24 h – 100 | 82/12 |
| 4 | Ru(OAc)2(p-cymene) | 24 h – 97 | 80/20 |
| 5 | Ru(OAc)2(p-cymene) + 2 equiv. PPh3 | 24 h – 89 | 94/6 |
| 6 | RuCl2(PPh3)(p-cymene) | 24 h – 100 | 89/11 |
| 7 | RuCl2(P(CH2Ph)3)(p-cymene) | 24 h – 98 | 85/15 |
| 8 | RuCl2(PCy3)(p-cymene) | 24 h – 94 | 89/11 |
| 9 | RuCl2(Pi-Pr3)(p-cymene) | 24 h – 93 | 85/15 |
| 10 | [RuCl2(p-cymene)]2 + (P(o-Me-C6H4)3) | 24 h – 96 | 80/20 |
| 11d | RuCl2(PPh3)(p-cymene) | 3 h – 81 | 96/4 |
| 5 h – 92 | 95/5 | ||
| 5 h – 45e | 98/2 | ||
| 12f | RuCl2(PPh3)(p-cymene) | 16 h – 70 | 98/2 |
The reactions produced only 3a and 4a derivatives without side products such as homocoupling products. With [RuCl2(p-cymene)]2 as the catalyst precursor in the absence of a carboxylate ligand a conversion of 97% was reached in 24 h with a 3a/4a ratio of 78/22 (Table 1, entry 1). The reaction performed in the presence of Ru(OPiv)2(p-cymene) and Ru(OAc)2(p-cymene), which were previously shown to be efficient catalysts for diarylation in water,3c,7 led to 97% conversion, with a moderate 3a/4a ratio (Table 1, entries 2, 4). Addition of 2 equiv. of PPh3 to the ruthenium carboxylate catalysts showed only a slight improvement in the selectivity towards 3a (Table 1, entries 3, 5 vs. 2, 4).
Better conversion and selectivity towards the monoarylated product were obtained after 24 h with 5 mol% of RuCl2(PPh3)(p-cymene) (Table 1, entry 6). The replacement of PPh3 by bulkier and more electron donating phosphines such as P(CH2Ph)3, PCy3, Pi-Pr3 or P(o-Me-C6H4)3 did not improve the selectivity of the formation of 3a (Table 1, entries 7–10). Neither steric nor electronic property of the phosphine ligand had a significant positive effect on the selective formation of 3a with respect to PPh3 in water. Noteworthily, the selective formation of 3a could be achieved at 80 °C in 5 h, and even at 60 °C in 16 h (Table 1, entries 11–12). The reaction performed in NMP at 80 °C for 5 h provided only 45% of conversion (entry 11). This reveals that water is a more efficient solvent at lower temperature than NMP for monoarylation.
We performed a kinetic study, in order to better observe the variations of conversion and selectivity as a function of the reaction time with 5 mol% of the RuCl2(PPh3)(p-cymene) catalyst in water. After 1 h, the conversion was only 71% with 99% of selectivity towards the monoarylated product. When the reaction was carried out for 24 h, the selectivity of 3a decreased from 99% to 89%, which showed that the diarylated product was formed during the course of the reaction at the expense of the monoarylated product. We observed that the best formation of monoarylated 2-phenylpyridine 3a was obtained in 3 h (Table S1†). The reaction was performed with various catalysts in 3 h at 100 °C in order to evaluate the influence of various phosphorous and arene ligands coordinated to ruthenium (Tables S2 and S3†). Hence, after the screening of the reaction of 2-phenylpyridine with chlorobenzene it could be concluded that the best conditions to reach the monoarylated product 3a were to use the RuCl2(PPh3)(p-cymene) catalyst: (i) at 80 °C, with 5 mol% of the catalyst with a reaction time of 5 h, or (ii) at 100 °C, with 5 mol% of the catalyst with a reaction time of 3 h.
Selective monoarylation of 2-phenylpyridine with (hetero)aryl halides in water
The selective monoarylation with various (hetero)arylhalides was investigated using 5 mol% of the RuCl2(PPh3)(p-cymene) catalyst in water at 80 °C (eqn (2), Table 2).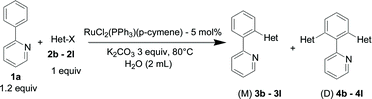 | (2) |
| Entry | Het-X | T (h) | Convb (%) | M/Dc | Yieldd (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 0.6 mmol of 2-phenylpyridine, 5 mol% of RuCl2(PPh3)(p-cymene), 3 equiv. of K2CO3, 10 μL of tetradecane (internal standard) for GC, 0.5 mmol of Het-X in 2 mL of water, 80 °C.b Conversion determined by gas chromatography based on (hetero)arylhalide.c M/D ratio determined by gas chromatography.d Isolated yield. | |||||
| 1 |  | 5 | 98 | 94/6 |  |
| 2 | 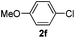 | 8 | 88 | 96/4 | 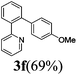 |
| 10 | 89 | 96/4 | |||
| 3 | 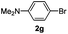 | 13 | 90 | 90/10 | 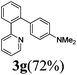 |
| 14 | 92 | 90/10 | |||
| 4 | 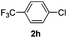 | 6 | 78 | 96/4 | 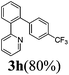 |
| 13 | 99 | 90/10 | |||
| 5 | 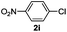 | 6 | 10 | — | |
| 6 | 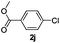 | 3 | 96 | 96/4 | 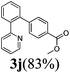 |
| 4 | 97 | 95/5 | |||
| 7 | 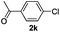 | 6 | 90 | 90/10 | 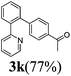 |
| 10 | 96 | 88/12 | |||
| 8 | 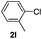 | 8 | 50 | 88/12 | 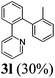 |
| 24 | 60 | 80/20 | |||
| 9 | 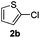 | 3 | 100 | 86/14 | 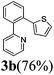 |
| 4 | 100 | 85/15 | |||
| 10 | 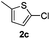 | 3 | 100 | 89/11 | 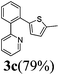 |
| 4 | 100 | 87/13 | |||
| 11 | 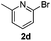 | 3 | 81 | 81/19 | 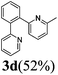 |
| 4 | 83 | 80/20 | |||
| 6 | 100 | 73/22 | |||
The reaction of 2-phenylpyridine with various electron donating and electron withdrawing heteroarenes was performed. The monophenylated 2-phenylpyridine 3a was obtained by the reaction with phenylchloride in 5 h with 76% isolated yield (Table 2, entry 1). The reaction with arylhalides containing the para substituted electron donating groups, –OMe, –NMe2, gave 69% and 72% yield, respectively in 8–13 h (Table 2, entries 2–3). With the electron withdrawing para CF3-substituted phenylchloride 99% of conversion was obtained in 13 h leading to 80% of monoarylated product 3h (Table 2, entry 4). With 4-methyl chlorobenzoate 2j and 4-chloroacetophenone 2k the reactions were completed in shorter reaction times (3–10 h), providing 83% and 77% of monoarylated products 3j and 3k, respectively (Table 2, entries 6–7). With o-methylchlorobenzene 2l only 30% of product 3l was isolated after 24 h (Table 2, entry 8).
The reaction with heterocyclic halides such as 2-chlorothiophene, 5-methyl-2-chlorothiophene, 6-bromo-2-methylpyridine was performed successfully with short reaction times (Table 2, entries 9–11). With 2b and 2c, the reaction went to completion within 3 h affording 3b and 3c in 76–79% isolated yields, whereas with 2d, only 81% conversion of 2-phenylpyridine was obtained in 3 h, but the complete conversion was obtained in 6 h leading only to 52% of isolated 3d (Table 2, entry 11).
Selective monoarylation of N-phenylpyrazole with (hetero)aryl halides
The reaction of N-phenylpyrazole 1b with 4-chloromethoxybenzene and 4-methyl chlorobenzoate was performed in the presence of RuCl2(PPh3)(p-cymene) and 3 equiv. of K2CO3 in water at 80 °C, affording the monoarylated products 5f and 5j in a moderate yield of 52–53% (eqn (3), Table 3, entries 1a–2a). Bidentate ligands 5c and 5d were isolated in moderate yields from the reaction of N-phenylpyrazole with 5-methyl-2-chlorothiophene and 6-bromo-2-methylpyridine (Table 3, entries 3a–4a). It is noteworthy that the efficient catalytic system allowed us to carry out the reaction in water at 65 °C in reasonable reaction times (14–24 h), with good conversion and selectivity (Table 3, entries 1b–4b). | (3) |
| Entry | Het-X | T (h) | Convb (%) | M/Dc | Yd (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 0.6 mmol of phenylpyrazole, 5 mol% of RuCl2(PPh3)(p-cymene), 3 equiv. of K2CO3, 10 μL of tetradecane (internal standard) for GC, 0.5 mmol of Het-X in 2 mL of water, 80 °C.b Conversion determined by gas chromatography.c M/D ratio determined by gas chromatography.d Isolated yield. | |||||
| 1a | 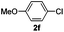 | 7 | 97 | 90/10 | 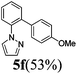 |
| 1b | 14 (65 °C) | 62 | 93/7 | ||
| 2a | 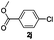 | 5 | 96 | 89/11 | 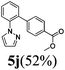 |
| 2b | 20 (65 °C) | 98 | 90/10 | ||
| 3a |  | 5 | 80 | 78/22 | 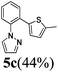 |
| 3b | 7 | 84 | 75/25 | ||
| 20 (65 °C) | 98 | 74/26 | |||
| 4a | 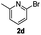 | 8 | 78 | 84/16 | 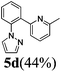 |
| 4b | 24 (65 °C) | 91 | 75/25 | ||
Similar conditions for selective catalytic monoarylation were used to produce new monoarylated products in water from functional arenes containing already one ortho substituent. As there is no risk to get parasite ortho diarylation the phosphine can be replaced by the carboxylate ligand. Thus the [RuCl2(p-cymene)]2–4 KOPiv catalytic system was applied for the monoarylation of the ortho C–H bond of 2-o-tolylpyridine 1c. Thus the ortho (hetero)arylated derivatives 4e (71%), 4f (60%), 4g (63%), 4h (58%) were obtained by direct arylation with chlorobenzene, and the heterocyclic halides 2b, 2c and 2d (Scheme 1).
![Selective monoarylation of 2-o-tolylpyridine with (hetero)arylhalides in water. (a) Reaction conditions: 0.5 mmol of 2-o-tolylpyridine, 5 mol% of [RuCl2(p-cymene)]2, 20 mol% of KOPiv, 3 equiv. of K2CO3, 10 μL of tetradecane (internal standard) for GC, 1.25 mmol of aryl(hetero)halide in 2 mL of water. (b) Conversion determined by gas chromatography. (c) Isolated yield.](/image/article/2013/GC/c2gc36222h/c2gc36222h-s1.gif) | ||
| Scheme 1 Selective monoarylation of 2-o-tolylpyridine with (hetero)arylhalides in water. (a) Reaction conditions: 0.5 mmol of 2-o-tolylpyridine, 5 mol% of [RuCl2(p-cymene)]2, 20 mol% of KOPiv, 3 equiv. of K2CO3, 10 μL of tetradecane (internal standard) for GC, 1.25 mmol of aryl(hetero)halide in 2 mL of water. (b) Conversion determined by gas chromatography. (c) Isolated yield. | ||
Successive monoarylations in water
One major aim of the selective monoarylation reaction is to utilize the remaining available ortho C–H bond, as a functional group, for further functionalisation in order to prepare unsymmetrical bifunctional arenes.The monoheteroarylated derivative 3j was first obtained from the monoarylation of phenylpyridine with 4-chlorobenzoate in water in the presence of RuCl2(PPh3)(p-cymene) (Table 2). The reaction of 3j with 2-chlorothiophene was then performed in the presence of RuCl2(PPh3)(p-cymene) in water for 20 h to give only 70% of conversion. Then the same reaction in the presence of Ru(OAc)2(p-cymene) or Ru(OPiv)2(p-cymene)7 was carried out at 120 °C and provided the mixed diarylated product with 99% conversion in 20 h and afforded 77% isolated yield of 7 (Scheme 2). For comparison, when the same reaction with 3j was performed in NMP as a solvent, only 20% conversion was obtained after 20 h. The advantage of using water as a solvent medium was obvious as water not only acts as a solvent but also enhances the rate of the reaction for the formation of unsymmetrical or mixed diarylated products.
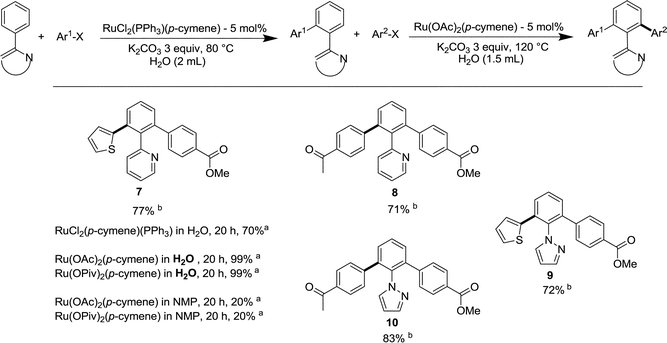 | ||
| Scheme 2 Successive arylations in water and preparation of difunctional unsymmetrical diarylated arenes. Reaction conditions: 0.25 mmol of monoarylated heteroarenes, 5 mol% of Ru(OAc)2(p-cymene), 3 equiv. of K2CO3, 10 μL of tetradecane (internal standard) for GC, 0.5 mmol of Het-X in 1.5 mL of water. (a) Conversion determined by gas chromatography. (b) Isolated yield. | ||
The transformation in water of 3j with 4-chloroacetophenone provided the mixed ketone carboxylate product 8 isolated in 71% yield after 24 h. Similarly, the monoarylated pyrazole 5j was reacted in water with 2-chlorothiophene and 4-chloroacetophenone. After 24 h the unsymmetrical diarylated products 9 and 10 were isolated in 72 and 83% yields.
Successive alkenylation of monoarylated heteroarene
The ruthenium(II)-catalyzed alkenylation of monoarylated heteroarenes was then investigated. The reaction of monoarylated 2-phenylpyridine and phenylpyrazole derivatives 3j and 5j with n-butyl acrylate was attempted in the presence of a [RuCl2(p-cymene)]2/AgSbF6 catalyst8 with Cu(OAc)2·H2O as an oxidant which was shown to be an efficient catalytic system for the alkenylation of arenes containing weakly coordinating directing groups.8 Unfortunately, this catalytic system did not provide any conversion of 3j and 5j.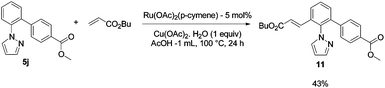 | (4) |
However it was recently established that arenes containing a nitrogen directing group were successfully alkenylated with a carboxylate-ruthenium(II) catalyst but in acetic acid.9 The alkenylation of monoarylated product 5j with n-butyl acrylate was attempted using these conditions in the presence of Ru(OAc)2(p-cymene) as a catalyst with Cu(OAc)2·H2O as an oxidant in AcOH at 100 °C during 24 h. The difunctional mixed aryl alkenylheteroarene 11 was thus successfully isolated in 43% yield (eqn (4)).
Conclusion
The above results show the selective ortho monoarylation of functional arenes directed by N-containing heterocyclic directing groups. It is selectively obtained using the RuCl2(PPh3)(p-cymene) catalyst operating in water, for which both phosphine PPh3 linked to ruthenium(II) and water medium are cooperative to the monoarylation catalyst activity. The profit of this selective mono(hetero)arylation is shown in the sequential preparations of bifunctional polyaryl derivatives. One example of synthesis of mixed ortho aryl alkenyl benzene is shown and mixed ortho diarylated arenes are produced using the Ru(O2CMe)2(p-cymene) catalyst in water for the second monoarylation step.Acknowledgements
The authors are grateful to the CNRS, the French Ministry for Research, the Institut Universitaire de France (P. H. D.), and the ANR program 09-Blanc-0101-01 for support and for a PhD grant to P. B. A.References
- For review on C–H bond functionalisation see: (a) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS; (b) C. L. Sun, B. J. Li and Z. J. Shi, Chem. Rev., 2011, 111, 1293 CrossRef CAS; (c) Y. Nakao, Synthesis, 2011, 3209 CrossRef; (d) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS; (e) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS; (f) S. Messaoudi, J. D. Brion and M. Alami, Eur. J. Org. Chem., 2010, 6495 CrossRef CAS; (g) A. Gunay and K. H. Theopold, Chem. Rev., 2010, 110, 1060 CrossRef CAS; (h) G. P. McGlacken and L. M. Bateman, Chem. Soc. Rev., 2009, 38, 2447 RSC; (i) A. A. Kulkarni and O. Daugulis, Synthesis, 2009, 4087 CAS; (j) R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS; (k) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS; (l) I. V. Seregin and V. Gevorgyan, Chem. Soc. Rev., 2007, 36, 1173 RSC; (m) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS; (n) A. R. Dick and M. S. Sanford, Tetrahedron, 2006, 62, 2439 CrossRef CAS; (o) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731 CrossRef CAS; (p) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507 CrossRef CAS; (q) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633 CrossRef CAS; (r) A. Sen, Acc. Chem. Res., 1998, 31, 550 CrossRef CAS.
- (a) L. Ackermann, P. Novak, R. Vicente and N. Hofmann, Angew. Chem., Int. Ed., 2009, 48, 6045 CrossRef CAS; (b) L. Ackermann and P. Novak, Org. Lett., 2009, 11, 4966 CrossRef CAS; (c) L. Ackermann, P. Novak, R. Vicente, V. Pirovano and H. K. Potukuchi, Synthesis, 2010, 2245 CrossRef CAS; (d) L. Ackermann, R. Vicente, H. K. Potukuchi and V. Pirovano, Org. Lett., 2010, 12, 5032 CrossRef CAS; (e) L. Ackermann and A. V. Lygin, Org. Lett., 2011, 13, 3332 CrossRef CAS; (f) L. Ackermann, N. Hofmann and R. Vicente, Org. Lett., 2011, 13, 1875 CrossRef CAS; (g) L. Ackermann, J. Pospech and H. K. Potukuchi, Org. Lett., 2012, 14, 2146 Search PubMed.
- (a) P. Arockiam, V. Poirier, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2009, 11, 1871 RSC; (b) F. Pozgan and P. H. Dixneuf, Adv. Synth. Catal., 2009, 351, 1737 CrossRef CAS; (c) P. B. Arockiam, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Angew. Chem., Int. Ed., 2010, 49, 6629 CrossRef CAS; (d) W. B. Li, P. B. Arockiam, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2011, 13, 2315 RSC; (e) B. Li, C. B. Bheeter, C. Darcel and P. H. Dixneuf, ACS Catal., 2011, 1, 1221 Search PubMed; (f) B. Li, K. Devaraj, C. Darcel and P. H. Dixneuf, Tetrahedron, 2012, 68, 5179 CrossRef CAS.
- (a) S. Oi, S. Fukita, N. Hirata, N. Watanuki, S. Miyano and Y. Inoue, Org. Lett., 2001, 3, 2579 CrossRef CAS; (b) S. Oi, Y. Ogino, S. Fukita and Y. Inoue, Org. Lett., 2002, 4, 1783 CrossRef CAS; (c) B. R. Yu, X. Y. Yan, S. Wang, N. Tang and C. J. Xi, Organometallics, 2010, 29, 3222 Search PubMed; (d) S. Doherty, J. G. Knight, C. R. Addyman, C. H. Smyth, N. A. B. Ward and R. W. Harrington, Organometallics, 2011, 30, 6010 CrossRef CAS.
- (a) A. Behr, “Technical solutions”, in “Aqueous-phase Organometallic Catalysis – Concepts and Applications”, ed. B. Cornils and W. A. Herrmann, Wiley, Weinheim, 1998, pp. 163–180 Search PubMed; (b) H. Bahrmann and B. Cornils, “Membrane techniques”, in “Aqueous-phase Organometallic Catalysis – Concepts and Applications”, ed. B. Cornils and W. A. Herrmann, Wiley, Weinheim, 1998, pp. 190–192 Search PubMed.
- I. Özdemir, S. Demir, B. Çetinkaya, C. Gourlaouen, F. Maseras, C. Bruneau and P. H. Dixneuf, J. Am. Chem. Soc., 2008, 130, 1156 CrossRef.
- E. Ferrer-Flegeau, C. Bruneau, P. H. Dixneuf and A. Jutand, J. Am. Chem. Soc., 2011, 133, 10161 CrossRef CAS.
- (a) K. Padala and M. Jeganmohan, Org. Lett., 2011, 13, 6144 CrossRef; (b) K. Padala and M. Jeganmohan, Org. Lett., 2012, 14, 1134 CrossRef; (c) B. Li, J. Ma, N. Wang, H. Feng, S. Xu and B. Wang, Org. Lett., 2012, 14, 736 CrossRef CAS; (d) Y. Hashimoto, T. Ortloff, K. Hirano, T. Satoh, C. Bolm and M. Miura, Chem. Lett., 2012, 41, 151 CrossRef CAS; (e) L. Ackermann, L. Wang, R. Wolfram and A. V. Lygin, Org. Lett., 2012, 14, 728 CrossRef CAS; (f) R. K. Chinnagolla and M. Jeganmohan, Chem. Commun., 2012, 48, 2030 RSC.
- P. B. Arockiam, C. Fischmeister, C. Bruneau and P. H. Dixneuf, Green Chem., 2011, 13, 3075 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic and analytical data. See DOI: 10.1039/c2gc36222h |
| This journal is © The Royal Society of Chemistry 2013 |