Fractionation of ethylcellulose oleogels during setting
Andrew J.
Gravelle
,
Shai
Barbut
and
Alejandro G.
Marangoni
*
Department of Food Science, University of Guelph, Guelph, ON, Canada N1G2W1. E-mail: amarango@uoguelph.ca; Fax: +1 519 824 6631; Tel: +1 519 824 4120 ext. 54340
First published on 9th November 2012
Abstract
The use of ethylcellulose (EC) polymers as a means to structure edible oils for fat replacement is beginning to show great promise and the use of these ‘oleogels’ has recently been shown to be feasible in food products. These gels are very versatile, as the mechanical properties can be tailored by altering either the fatty acid profile of the oil component, or the viscosity or concentration of the polymer component. Here we report the observation that certain formulation of EC oleogels tend to separate into two distinct phases; a soft interior core surrounded by a firm exterior sheath. It was found that the extent of this effect depends on EC viscosity, and can also be induced through the addition of certain surfactants, such as sorbitan monostearate and sorbitan monooleate, though not by glycerol monooleate. Although the two visually distinct regions were shown to be chemically indistinct, the cooling rate during gel setting was found to play a large role; rapid setting of the gels reduces the fractionation effect, while slow cooling produced a completely homogeneous structure. In addition, by reheating only the soft region of the gel, a firm and soft fractionated gel could again be produced. Finally, it was observed that oleogels prepared with castor oil or mineral oil have the ability to remove or induce the gel separation, respectively. Taken together, these results indicate chemical interactions may incite the separation into two distinct phases, but the process also seems to be driven by the cooling conditions during gel setting. These findings lend insight into the EC-oleogel gelation process and should provide a stepping stone for future research into the manufacturing of these products.
1 Introduction
With the continual rise in obesity rates and cardiovascular diseases in both developed and developing countries,1,2 and the mounting evidence that excessive consumption of trans- and saturated fatty acids can increase the risk of metabolic syndrome,3 there has been an increasing push to develop a suitable alternative for these structured fats. In the World Health Organization's (WHO) Global Strategy on diet, physical activity and health, it is advised that the consumption of saturated and trans-fats be limited and dietary fats come mainly from unsaturated fatty acids.4 In accordance, many governing bodies are developing strategies to reduce consumption of saturated and trans-fats. For example, the most recent dietary guidelines developed by the United States and Canadian governments5,6 reflect the need to adjust dietary fat intake as outlined by the WHO. The European Food Safety Authority, under request from the European Commission, has also recently published its scientific opinion on dietary reference values for fats,7 recommending the dietary intake of saturated and trans-fatty acids should be minimized.In response, the food industry has been attempting to reduce the level of saturated and trans-fats in food products. The most straight-forward approach would be to simply replace common fat sources rich in these fatty acids, such as butter, palm oil, hydrogenated oils, lard, and tallow with those high in unsaturated fatty acids, like vegetable oils. However, the physical aspects of structured fats provide both textural properties, such as mouthfeel and creaminess, as well as mechanical properties including product firmness and the snap associated with chocolate. However, these desirable qualities are the result of the ability of triacylglycerides to form a colloidal fat crystal network within the food matrix.8,9 Therefore, direct replacement of fats which maintain a solid structure at room temperature with liquid oils can produce adverse effects in complex food systems.10,11 As a result, the study of organogels for potential uses as novel structuring agents in food systems has surged in popularity in recent years.12,13 The use of organogelators to structure edible oils has also become a popular strategy to provide these liquids with the structure necessary to mimic their solid counterparts without compromising the organoleptic properties of the final product.
A working definition of an organogelator can be adapted from the description of a ‘gel’ as proposed by Hermans;14 a molecule which is capable of forming a coherent colloid dispersed system, producing a continuous network which imparts solid-like properties to an organic liquid, typically at relatively low concentrations. A variety of organogelators and combinations of molecules capable of forming organogels have been identified as having potential uses for structuring oils for applications in food systems.13,15–17 For example, mono- di- and triacylglycerides, n-alkanes, waxes such as candelilla wax, carnauba wax, and rice bran wax, lecithin–sorbitan tristearate mixtures, sorbitan monostearate, and fatty acids, fatty alcohols, and mixtures thereof have all been shown to have the ability to form organogels. Certain organogelators provide structure to the liquid phase by forming self-assembled fibrillar networks (SAFiNs). Some of these include ceramides, 12-hydroxistearic acid, ricinelaidic acid, γ-oryzanol, a variety of phytosterols, and peptide chains.
Another promising organogelator for use in food applications is ethylcellulose (EC), a modified version of the naturally abundant cellulose polymer. Although cellulose itself is hydrophilic, commercially available versions of EC contain approximately 48–49.5% ethoxyl content,18 which translates to ethoxyl substitutions at roughly 2.5 of the 3 available hydroxyl sites per glucose monomer.19 This level of substitution also results in a minimum in the thermoplastic transition temperature of the polymer and renders it insoluble in water, but soluble in a variety of organic solvents.18,19 EC is well known for its excellent film forming abilities and is commonly used in industry for encapsulation applications.18 The polymer is “generally regarded as safe” (GRAS) by the U.S. Food and Drug Administration for indirect food usage (21 CFR Section: 182.90) and has been approved by the European Union for food applications.20 Commercial versions of EC are available in a variety of different viscosities, which are determined at 25 °C using a 5% EC solution dissolved in a mixture of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 toluene–ethanol. Higher viscosity versions of the polymer will have higher average molecular weights (MWs), however the functional range for applications in oleogels seem to fall in the range of 10–100 mPa s or centiPoise (cP).21
:20 toluene–ethanol. Higher viscosity versions of the polymer will have higher average molecular weights (MWs), however the functional range for applications in oleogels seem to fall in the range of 10–100 mPa s or centiPoise (cP).21
Recent work on vegetable oil-based EC organogels has focused on how their mechanical properties are affected by both the type of oil being gelled, and how the fatty acid profile of different oils may play a role in determining the macroscopic properties of the resulting gels. Laredo et al.22 first demonstrated the substantial differences of 45 cP EC oleogels prepared with canola, soybean, and flax oils which are predominantly (>50%) made up of oleic, linoleic, and α-linolenic acids, respectively. This work showed that an increasing level of unsaturation in the fatty acids leads to an increase in the flexibility of the carbon chain, thus increasing the oil's compactibility, which results in a higher density of the oil phase. This work was also the first to demonstrate at the molecular level, the mechanism by which these gels are supported; the formation of hydrogen bonds between the un-substituted hydroxyl groups found in the carbohydrate backbone of the EC polymers. In addition, the authors demonstrated that hydrogen bonding did not take place between the polymer network and the entrapped oil, thus indicating the oil is a passive filler within the gel matrix and does not affect the mechanical properties of the gel through chemical interactions. The authors therefore concluded that the substantial differences in mechanical properties of gels made from the three different oils were due only to the differences in oil density.
A follow-up study by Zetzl et al.21 also considered the effect of oil type, as well as how independently altering the polymer concentration and viscosity affects the mechanical properties of the oleogel. Their findings were in agreement with the conclusion drawn from the aforementioned work when comparing the three oils independently. It was also demonstrated that as the viscosity of the polymer increases over the range of 10 to 45 to 100 cP, so too does the mechanical strength of the gel. This is a somewhat intuitive result because, as mentioned above, an increase in viscosity of EC should correspond to an increase in the average MW of the polymer chains. This, in turn, should provide more opportunities for the formation of physical junctions, producing a more integrated, stabilized network. The authors also demonstrated that increasing the volume fraction of 45 cP EC exponentially increases the strength of the oleogel. Finally, this work was the first to demonstrate the feasibility of using EC oleogels as an alternative fat source in a food system. The oleogels were successfully incorporated into an all-beef frankfurter as a substitute for beef fat, while maintaining comparable textural properties to that of the control product containing beef fat as the sole fat source.
In the present work we report some interesting observations pertaining to edible oil-based oleogels prepared with low- moderate- and higher-range viscosity versions of EC. We address how the gel structure can be drastically affected in several ways, including the addition of some common food-grade surfactants, altering the level of polar molecules in the oil phase, or simply changing the cooling rate as the gel sets. Through this work we hope to gain a better understanding of EC–-solvent interactions during the formation of the gel network, which dictate the structural and mechanical properties of the final product.
2 Materials and methods
2.1 Materials
Soybean oil was purchased from Nealanders International Inc. (Missassauga, ON), heavy mineral oil was obtained from Fisher Scientific (Ottawa, ON), and castor oil was purchased from a commercial source. The surfactants glycerol monooleate (GMO) and sorbitan monostearate (SMS) were purchased from HallStar (Bedford Park, IL, USA) and Danisco A/S (Scarborough, ON) respectively. Sorbitan monooleate (SMO) and sorbitan tristearate (STS) were purchased from Sigma-Aldrich Canada Ltd. (Oakville, ON). As noted above, the functional range of EC for oleogel applications is approximately 10–100 cP, however difficulties often arise when using the 100 cP version, such as increased heating time, oil smoking, and excessive bubble formation within the sample. For this reason the three versions of EC selected for this study were limited to the 10–45 cP range. ETHOCEL™ std. 10, 20, and 45, (EC10, EC20, and EC45 respectively), have corresponding viscosities of 10, 20, and 45 cP. All versions were obtained from Dow Wolff Cellulosics through Colorcon Inc. (St. Laurent, QC).2.2 Methods
Upon completion of the heating process, the molten gel was split into 6 aliquots for mechanical testing. Five 30 ml samples were prepared for back extrusion in 50 ml polypropylene centrifuge tubes (Fisher Scientific). The final aliquot of approximately 45 ml was poured into an open-ended cylindrical glass tube (height: 14.5 cm, diameter: 1.9 cm) lined with aluminum foil and plugged at the base with a #3 rubber stopper (Fisher Scientific). This apparatus (displayed in Fig. 1), is a modified version of that adopted by Zetzl et al.21 which allowed for easier removal of the set gel from the glass tube, thus minimizing the potential for damage to the gel structure. This modification has also allowed for the successful recovery of firmer gels without causing them damage, thus increasing the potential use of texture profile analysis (TPA)24 as a technique to evaluate the mechanical properties of these oleogels. In the current work, gels prepared in these tubes were used for visual determination of gel fractionation. Samples prepared for lipid–EC separation via lipid extraction were instead poured into several of the glass tubes described above. In all cases, after pouring the molten gels were allowed to cool to 20 °C in an incubator unit for a minimum of one hour and then stored overnight in a 5 °C refrigerator.
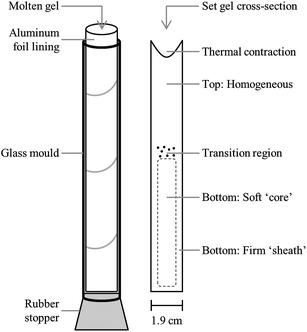 | ||
| Fig. 1 Schematic diagram of (left) an oleogel cooling vessel lined with aluminum foil and plugged with a rubber stopper (right) cross-section of an oleogel taken out of the vessel after cooling, which undergoes fractionation. The different regions which exhibit different physical properties within the gel have been labelled. | ||
 | ||
| Fig. 2 Comparison of oleogels prepared using a variety of formulations and cooling methods. All oleogels were prepared using soybean oil and 11 wt% EC (viscosity indicated by superscript), with the exception of the gel containing mineral oil, which was prepared with 14 wt% EC. Surfactants, where noted, were included at 3.67 wt%. Castor oil and mineral oil were included as a wt% replacement of the oil component. All samples were taken from the bottom few cm of the oleogels. | ||
Viscosity measurements were carried out using an AR 2000 controlled-stress rheometer (TA Instruments, New Castle, DE, USA) fitted with a cone and plate geometry. The geometry selected for this study was a 6 cm diameter stainless steel cone with a 2° angle and a truncation gap of 52 μm. Temperature was maintained at 25 °C using a Peltier plate. To minimize the potential for solvent evaporation, samples were kept at 25 °C prior to measurement, so no equilibration time was used. A solvent trap was also placed over the geometry during testing. Viscosity measurements were recorded in the range of 10–100 s−1, in 10 s−1 intervals. Due to variability at the lowest shear rates, the 10 and 20 s−1 values were discarded and the average of the remaining 8 data points was used as the reported sample viscosity. Measurements could be reproducibly repeated, indicating no significant effect of solvent evaporation.
:methanol solution (2:1, v/v; Fisher Scientific). Samples were vortexed for 1 min, flushed with nitrogen gas (Boc gases, Guelph, ON) and incubated at 4 °C overnight. On the following day, samples were centrifuged at 1000 rpm for 10 min at 21 °C to separate phases. The lower chloroform layer was extracted and transferred to a fresh test tube and dried down with a gentle stream of nitrogen gas. The lipid was saponified in 0.5 M potassium hydroxide in methanol and heated for 1 h at 100 °C. Phospholipids were converted to fatty acid methyl esters with the addition of 14% boron trifluoride (Sigma Aldrich)/methanol and incubation at 100 °C for 1 h.
FAME were quantified on an Agilent 7890A gas chromatograph equipped with an FID and separated on an Agilent J&W fused-silica capillary column (DB-FFAP; 15 m, 0.1 μm film thickness, 0.1 mm inside diameter; Agilent, Pal Alto, CA, USA). Samples were injected in split 1:200 mode. The injector and detector ports were set at 250 °C. FAME were eluted using a temperature program set initially at 150 °C and held for 0.25 min, increased at 35 °C min−1 and held at 170 °C for 3 min, increased at 9 °C min−1, and held at 225 °C for 0.5 min, increased at 80 °C min−1 and finally, held at 245 °C for 2.2 min to complete the run. Total run time was 12.88 min. The carrier gas was hydrogen, set to a 30 ml min−1 constant flow rate.
3 Results and discussion
3.1 Gel fractionation and effect of surfactants
It has been observed that there are several gel formulations which reproducibly result in two distinct phases forming in the oleogels. These two regions are always made up of a firmer exterior ‘sheath’ surrounding a softer interior ‘core’ region. A selection of such gels prepared with soybean oil is presented in Fig. 2, however the same effect has also been noted when gels are prepared with canola oil. Although this fractionation phenomenon does not occur for all formulations, one discernible pattern has emerged; for oil–EC and oil–EC–surfactant combinations in which fractionation does occur, there seems to be some critical concentration of EC and/or surfactant above which fractionation is no longer observed. However, in our experience, this concentration seems to depend on viscosity of EC used, type of surfactant, and type of oil. Therefore, due to the wide variety of surfactants, oil types, and combinations thereof, such critical concentrations were not investigated further for this study.Notwithstanding, several notable observations have been made regarding the fractionation phenomenon. Firstly, in many cases when fractionation is observed, the extent of separation seems to be enhanced as the viscosity of EC used in the formulation is increased. The gels containing SMS presented in Fig. 2 display minimal separation when prepared with EC10, while those prepared with EC20 and EC45 not only have an increasing degree of fractionation, but the difference in mechanical strength between these two regions also becomes more distinct; i.e. the sheath becomes increasingly firm, while the core remains very soft. Second, the presence of a surfactant also plays a role in the gel separation. For example, soybean oil gels prepared with 11% EC45 do not exhibit any fractionation, but when either SMS or SMO are added to the gel system, severe fractionation is observed (see Fig. 2). Interestingly, this effect is not induced upon addition of GMO, which suggests the surfactant head-group must play some role in mediating the structure of the gel as it sets. As noted above, the EC gel network is supported by intermolecular hydrogen bonding which form junctions between neighbouring polymer strands. It would therefore be expected that the effect these surfactants have on the resulting gel should be the caused by some interaction between the hydroxyl groups of the polymer and the surfactant. Considering that both sorbitan and glycerol have multiple available hydroxyl groups, their differing effects on the EC oleogels suggests the structure of these molecules must somehow alter the interaction. Interestingly, when SMS is replaced with STS, fractionation is greatly reduced. This could possibly be due to STS having a greater affinity for the lipid phase which may reduce the ability of the bulkier sorbitan head-group to interact, and possibly interfere with, polymer–polymer junction zones.
3.2 Effect of altering oil polarity and cooling rate on oleogel properties
The addition of the above mentioned surfactants also generally increases the mechanical strength of the oleogel. This not only supports the idea that the hydroxyls in the head group directly interact with the EC matrix, but also suggests the mechanical properties of the gel could be controlled by altering the polarity of the oil component. To investigate this, a portion of the oil component was substituted with either castor oil or mineral oil. Castor oil is largely made up of ricinoleic acid, which contains an additional hydroxyl group in the fatty acid tail. Conversely, mineral oil is made up exclusively of alkanes and therefore has no ability to form hydrogen bonds. It should also be noted that EC is not soluble in mineral oil, and when attempting to produce a gel using exclusively mineral oil as the liquid phase, EC can be taken through the glass transition temperature, however it will then aggregate and form a solid precipitant. When attempting to use a mixture containing a 50:50 vegetable oil and mineral oil EC does not precipitate during heating, however a large portion of the mineral oil is expelled from the gel during cooling. When lower concentrations of mineral oil are used, a gel can be formed without any of the components coming out of solution. Therefore, the replacement of vegetable oil with either castor oil or mineral oil was limited to 10 wt%. The back extrusion results are displayed in Fig. 3 and the corresponding force at maximum penetration values are presented in Table 1. Accompanying these are values for formulations containing SMS which are known to fractionate, as well as these same formulations in which 10 wt% of the oil component is replaced with castor oil.
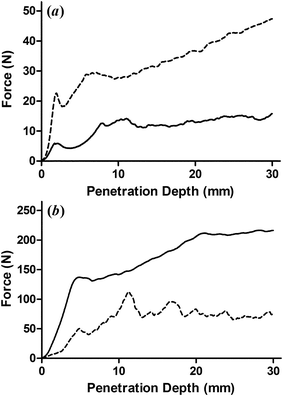 | ||
| Fig. 3 Back extrusion results for soybean oil-based EC20 oleogels for which 10% of the oil component was replaced with either (a) castor oil or (b) heavy mineral oil (dashed lines). For reference, these are compared to the same formulation having exclusively soybean oil as the oil component (solid line). Curves displayed are an average of three replicates. | ||
| Oleogel formulation | Force at maximum penetration (N) | Effect of oil replacement |
|---|---|---|
| 11% EC20 | 15.8 ± 0.8 | — |
| 11% EC20, oil 10% castor | 47.4 ± 0.8 | Increased 3.0× |
| 11% EC20, 1.83% SMS | 46.4 ± 4.0 | — |
| 11% EC20, 1.83% SMS, oil 10% castor | 104.8 ± 4.9 | Increased 2.3× |
| 11% EC20, 3.67% SMS | 103.8 ± 9.2 | — |
| 11% EC20, 3.67% SMS, oil 10% castor | 147.2 ± 14.2 | Increased 1.4× |
| 14% EC20 | 216.1 ± 11.4 | — |
| 14% EC20, oil 10% heavy mineral | 73.9 ± 6.4 | Decreased 2.9× |
These results clearly demonstrate that the composition of the oil plays a large role in determining the mechanical properties of the resulting oleogel. As noted above, it has previously been reported that the oil component of EC oleogels acts as a passive filler and differences in mechanical properties of gels having different fatty acid profiles has been attributed to differences in their densities.22 However, castor oil and heavy mineral oil have nearly identical densities, so the increase in mechanical strength seen in the gels containing 10 wt% castor oil, and decrease in those prepared with 10 wt% mineral oil demonstrates that the oil component must interact with the EC network in some way. Interestingly, in the absence of a surfactant, the addition of castor oil increased the gel strength by three-fold while the addition of mineral oil decreased the strength of the gel nearly three-fold. However, as the molar percentage of SMS is increased, the increase in gel strength upon addition of castor oil is reduced.
Another interesting observation to note is how the introduction of these two oils to the lipid phase can affect the resulting gel structure. One example is seen in formulations containing 11% EC20 or 11% EC45 and some level of SMS which normally exhibit fractionation; however with the addition of castor oil, this phenomenon no longer occurs and the gel is entirely homogeneous. Conversely, the 14% EC20 gel which otherwise does not fractionate, has been seen to exhibit severe separation upon partial replacement of the oil component with mineral oil (see Fig. 2). These observations support the idea that the composition of the solute can induce the separation of the gel into two distinct phases.
The distinct physical difference between the fractionated regions of the gels suggests that there is some type of molecular arrangement occurring while the gels are allowed to set. It would therefore seem reasonable to assume that by altering the amount of time the molecules are able to rearrange should have an effect on the degree of fractionation. By cooling the gel more rapidly, the EC will not have as much time to rearrange/segregate, while slowing the cooling rate would provide more time for this process to continue, and possibly exaggerating the degree of fractionation. To explore this hypothesis, a single batch of soybean oleogel was prepared using a formulation known to fractionate (11% EC45 and 3.67% SMS). Upon completion of the heating process, the molten gel was split into three separate glass moulds and each was cooled at a different rate. The first gel was cooled rapidly by plunging the entire mould into a propylene glycol solution kept at −18 °C, and appeared to be solidified after approximately 2 minutes. The second gel was allowed to set in a 20 °C incubator, as outlined in Section 2.2.1. Finally, the third gel was cooled slowly by using a pre-heated mould and placing the vessel containing the molten solution back in the oven in which the gel was prepared, and subsequently turning the oven off to allow the entire system to cool to room temperature. This gel appeared to still be in a liquid state after approximately 30 min, but solid after 45 min of cooling.
After cold storage, the gels were removed from their moulds and cut into 1 cm thick pucks for comparison. The quench-cooled gel displayed less fractionation than that seen in the air-cooled sample, but the effect was not fully removed. Surprisingly, and possibly somewhat counter-intuitively, the slowly cooled gel showed absolutely no fractionation. The same effect was also seen in a slowly cooled gel when SMS was substituted with SMO (see Fig. 2). This result demonstrates that the cooling rate can play a major role in the formation of the gel network. One possible explanation for the lack of segregation seen in the slow-cooled gels is the removal of the large temperature gradient between the centre of the sample and the walls of the mould.
3.3 Lipid extraction and EC viscosity from fractionated gel samples
There are several possible explanations for the formation of the core and sheath within the gel. As the cooling gradient works its way towards the centre of the gel, so too will the formation of the self-supporting gel structure. One possibility is that as the gel is setting, the EC which remains in solution may become entangled in or drawn towards the growing ‘gel-front’ which may result in a physical depletion of EC at the centre of the gel. Alternatively, it might be expected that the thermal energy required to keep higher MW polymers in a ‘flexible’ state would exceed that of the shorter polymer strands. It has also been noted that higher viscosity versions of EC require either longer heating times or higher temperatures to achieve full dissolution of the polymer. It may therefore be possible for the larger MW polymers to re-solidify and begin to form intermolecular contacts at temperatures higher than those of a lower MW. As the outermost portion of the gel cools first, this region may contain a higher level of larger MW polymer strands, which could consequently reduce the amount of higher MW polymers available to form contacts in the interior of the gel. In other words, the firmer sheath may have a higher concentration of longer polymer chains, while the softer core would contain a higher concentration of shorter chains which are not able to form as many physical contacts as their larger counterparts.To determine if gel fractionation results from a difference in EC concentration in the two regions, lipid extraction was performed on two soybean oleogel formulations; 11 wt% EC45 and 3.67 wt% of either SMS or SMO. Quantification of the lipid content in each distinct region allowed for an indirect measure of the EC concentration in each of the soft core, the firm sheath, and the homogeneous top portion of the gels. The results from the Soxhlet lipid extraction are shown in Table 2. These results demonstrate that there is no significant difference between the wt% EC solids in the soft interior and the firm exterior of the fractionated region in the gel. This suggests that the two distinct phases are not caused by a physical depletion of the EC at the interior of the fractionated region. It may however suggest that, as discussed above, there is a preference for the higher MW polymer strands to assemble at the exterior of the vessel. To determine if such a phenomenon does occur, the viscosities of the EC recovered from the two regions of the gel after lipid extraction were determined. For reference, the viscosity of EC10 and EC45 samples taken from stock were also evaluated. The results (presented in Table 2) indicate that there are also no differences in the viscosities, and thus the average MW of the EC in each region.
| Sample | EC content (wt%) | Extracted EC viscosity (cP) |
|---|---|---|
| EC10 | — | 10.66 ± 0.99 |
| EC45 | — | 48.73 ± 1.37 |
| Soft core (SMS) | 12.25 ± 0.28 | 41.83 ± 1.69 |
| Firm sheath (SMS) | 11.44 ± 0.63 | 39.85 ± 0.83 |
| Top region (SMS) | 11.84 ± 0.15 | 41.96 ± 0.85 |
| Soft core (SMO) | 11.75 ± 0.39 | 40.16 ± 1.97 |
| Firm sheath (SMO) | 11.75 ± 0.11 | 37.70 ± 0.14 |
| Top region (SMO) | 11.64 ± 0.25 | 42.40 ± 1.36 |
Fatty acid analysis was performed on the samples used for lipid extraction. Analysis was performed on the soft and firm regions separately to determine if the separation may have resulted in an accumulation of the surfactant to either the inner or outer portion of the gel during setting. The results from gas chromatography displayed in Table 3 demonstrate that there is not a significant difference in the amount of stearic or oleic acid present in the two regions of the gels containing SMS and SMO respectively, thus indicating the amount of surfactant in the two regions is equivalent. It is also apparent that there is no preference for any particular type of fatty acid to migrate to either region of the gel during setting.
| Sample | 16:0 |
18:0 |
18:1 |
18:2 |
18:3 |
|---|---|---|---|---|---|
| Soft core (SMS) | 12.27 ± 0.02 | 5.56 ± 0.02 | 19.21 ± 0.04 | 52.07 ± 0.06 | 8.65 ± 0.03 |
| Firm sheath (SMS) | 12.12 ± 0.05 | 5.38 ± 0.02 | 19.13 ± 0.07 | 52.24 ± 0.08 | 8.68 ± 0.03 |
| Top region (SMS) | 12.07 ± 0.03 | 5.47 ± 0.05 | 19.36 ± 0.05 | 52.28 ± 0.09 | 8.67 ± 0.03 |
| Soft core (SMO) | 11.11 ± 0.64 | 4.30 ± 0.61 | 21.16 ± 1.10 | 52.76 ± 0.04 | 8.73 ± 0.05 |
| Firm sheath (SMO) | 10.77 ± 0.03 | 3.96 ± 0.01 | 21.86 ± 0.09 | 52.76 ± 0.23 | 8.75 ± 0.04 |
| Top region (SMO) | 10.72 ± 0.02 | 3.98 ± 0.07 | 21.87 ± 0.09 | 52.65 ± 0.09 | 8.74 ± 0.03 |
These results indicate that there is neither a depletion of EC in the interior of the gel, nor is there a segregation of either higher and lower MW polymers, or of the surfactant. That is, despite the two regions having distinctly different physical properties, all evidence suggests that they are chemically indistinct. It would therefore seem that the observed difference must be due to molecular interactions and/or structural arrangement within the gel. For example, the fractionation could potentially be the result of a preferential molecular arrangement. EC is well known for its film-forming properties,18 so it may be that a certain level of preferential molecular arrangement allows a film-like structure to form at the exterior of the gel, potentially as a result of the rapid cooling of the molten gel as the solution comes into contact with the vessel walls. This ‘film’ may then act as a nucleator for molecular arrangement, however this arrangement may not persist as the formation of the gel continues towards the interior of the vessel, leaving the interior region disordered. It is also possible that as the gel cools, the polymers in the interior of the vessel could begin to take on less extended conformations, which would limit their ability to form intermolecular contacts, and thus their contribution to the gel network.
3.4 Effect of re-melting the soft gel interior
If the formation of the soft and hard regions in the fractionated gels is indeed induced solely by the cooling conditions, one would expect that the contents of the soft region should have just as much potential to form a stiffer network than that of the firm region. To determine if this is the case, two batches of soybean oleogels containing 11% EC45 and 3.67% SMO were prepared and each were split into five glass vessels (as depicted in Fig. 1). After storage, the liquidy interior of each of the ten gels was removed and pooled, producing a ∼100 g solution with a consistency comparable to that of refrigerated soybean oil; somewhat viscous, yet still free-flowing. This mixture was then re-heated using the gel preparation procedure described in Section 2.2.1 and then again poured into glass vessels for cooling and storage. Upon completion of the re-melting process, the resulting gels were essentially identical in structure to the original gels from which the liquidy centres were removed. That is, although the re-melted gels were made up of the fractions which could seemingly not take on a self-supporting structure, re-plasticizing the EC within the mixture again allowed for the formation of a firm sheath and viscous, liquid centre. This observation not only strongly supports the presented findings which indicate the two regions are chemically equivalent, but also clearly demonstrates that the formation of the gel network is highly dependent on kinetic and/or thermodynamic processes occurring during the setting of the gel.4 Conclusion
In the current work, the fractionation phenomenon of EC oleogels during setting has been reported for the first time and had been identified as an inhomogeneity in the mechanical properties of the oleogels, as opposed to a true fractionation of the chemical components within. The extent of this effect was determined to be dependent on both polymer viscosity, as well as the presence of surfactants. In particular, SMS and SMO have the ability to induce fractionation in some formulations while GMO does not, indicating the head group plays a major role in determining the physical properties of the oleogel during setting. In addition, sample homogeneity can be achieved by reducing the cooling rate, and thus the temperature gradient within the sample during gel formation. Finally, it has been demonstrated that the two physically distinct regions are chemically equivalent, and that the softer region can also be re-melted to form a newly fractionated gel, again containing both firm and soft regions. These observations indicate the formation of two distinct regions is highly dependent on the temperature gradient within the gel during setting. However, whether this phenomenon is the result of a kinetically or thermodynamically driven process, or if it is due to the EC polymer taking on a less extended conformation as the gel cools, has yet to be elucidated.Previous studies on EC oleogels have demonstrated that the fatty acid composition of the oil phase plays a large role in determining the mechanical strength of the gel21,22 as a result of differences in oil density.22 Contrary to this finding, we have shown that altering the hydroxyl content without changing the density of the oil still has a drastic effect on both the physical and mechanical properties of the resulting gel. Introducing hydroxyl groups into the fatty acid tail by partial substitution with castor oil significantly increases the gel firmness and can also reduce, and even eliminate fractionation. In contrast, by reducing the amount of hydroxyls (in the glycerol head groups) by partial replacement with heavy mineral oil, one can both induce fractionation and substantially reduce the rigidity of the gel structure. This work adds to the growing knowledgebase on vegetable oil-based EC oleogels and also sheds light on the gelation process and what factors need be considered for sample homogeneity. This is particularly true for higher viscosity versions of EC, as they generally require lower concentrations of the polymer to form self-supporting gels and therefore represent the most straight-forward method for cost-reduction in these oleogel systems.
5 Abbreviations
| Canola oil | CO |
| Ethylcellulose | EC |
| Fatty acid methyl ester | FAME |
| Glycerol monooleate | GMO |
| Molecular weight | MW |
| Sorbitan monooleate | SMO |
| Sorbitan monostearate | SMS |
| Sorbitan tristearate | STS |
Acknowledgements
We would like to thank Dr. Lyn Hillyer of the Department of Human Health and Nutritional Sciences at the University of Guelph for performing the Fatty acid methyl ester (FAME) analysis for determination of lipid content. We would also like to acknowledge the Ontario Ministry of Agriculture, Food and Rural Affairs for providing funding for this research.References
- World Health Organization, The World Health Report, 2002: Reducing Risks, Promoting Healthy Life, World Health Organization, Geneva, 2002 Search PubMed.
- V. L. Roger, A. S. Go, D. M. Lloyd-Jones, E. J. Benjamin, J. D. Berry, W. B. Borden, D. M. Bravata, S. Dai, E. S. Ford, C. S. Fox, H. J. Fullerton, C. Gillespie, S. M. Hailpern, J. A. Heit, V. J. Howard, B. M. Kissela, S. J. Kittner, D. T. Lackland, J. H. Lichtman, L. D. Lisabeth, D. M. Makuc, G. M. Marcus, A. Marelli, D. B. Matchar, C. S. Moy, D. Mozaffarian, M. E. Mussolino, G. Nichol, N. P. Paynter, E. Z. Soliman, P. D. Sorlie, N. Sotoodehnia, T. N. Turan, S. S. Virani, N. D. Wong, D. Woo and M. B. Turner, American heart association statistics committee and stroke statistics subcommittee, Circulation, 2012, 125, e2–e220 CrossRef.
- R. P. Mensink, P. L. Zock, A. D. M. Kester and M. B. Katan, Am. J. Clin. Nutr., 2003, 77, 1146–1155 CAS.
- World Health Organization, Global Strategy on Diet, Physical Activity and Health, World Health Organization, Geneva, Switzerland, 2004 Search PubMed.
- United States Department of Agriculture and United States Department of Health and Human Services, Dietary Guidelines for Americans, U.S. Government Printing Office, Washington, D.C., 7th edn, 2010, http://www.health.gov/dietaryguidelines/2010.asp Search PubMed.
- Health Canada, Fats: The Good the Bad and the Ugly, Health Canada, Ottawa, ON, CA, 2012, http://www.hc-sc.gc.ca/hl-vs/alt_formats/pdf/iyh-vsv/med/fats-gras-eng.pdf Search PubMed.
- European Food Safety Authority, Scientific panel on dietetic products, nutrition and allergies (NDA), EFSA J., 2010, 8, 1461 Search PubMed.
- D. M. Tang and A. G. Marangoni, Adv. Colloid Interface Sci., 2006, 128, 257–265 CrossRef.
- A. G. Marangoni, N. Acevedo, F. Maleky, E. Co, F. Peyronel, G. Mazzanti, B. Quinn and D. Pink, Soft Matter, 2012, 8, 1275–1300 RSC.
- M. K. Youssef and S. Barbut, Meat Sci., 2009, 82, 228–233 CrossRef CAS.
- F. Maleky and A. Marangoni, Soft Matter, 2011, 7, 6012–6024 RSC.
- M. Pernetti, K. F. van Malssen, E. Flöter and A. Bot, Curr. Opin. Colloid Interface Sci., 2007, 12, 221–231 CrossRef CAS.
- Edible Oleogels: Structure and Health Implications, ed. A. G. Marangoni and N. Garti, AOCS Press, Urbana, IL, USA, 2011 Search PubMed.
- P. H. Hermans, in Colloid Science, ed. H. R. Kruyt, Elsevier, Amsterdam, 1949, vol. 2, pp. 483–651 Search PubMed.
- M. A. Rogers, Food Res. Int., 2009, 42, 747–753 CrossRef CAS.
- E. D. Co and A. G. Marangoni, J. Am. Oil Chem. Soc., 2012, 89, 749–780 CrossRef.
- N. E. Hughes, A. G. Marangoni, A. J. Wright, M. A. Rogers and J. W. E. Rush, Trends Food Sci. Technol., 2009, 20, 470–480 CrossRef CAS.
- Dow Cellulosics, ETHOCELTM: Ethylcellulose Polymers Technical Handbook, The Dow Chemical Company, 2005, http://www.dow.com/dowwolff/en/pdf/192-00818.pdf Search PubMed.
- W. Koch, Ind. Eng. Chem., 1937, 29, 687–690 CrossRef CAS.
- European Food and Safety Authority, Scientific panel on food additives, processing aids and materials in contact with food, EFSA J., 2004, 2, 1–6 Search PubMed.
- A. K. Zetzl, A. G. Marangoni and S. Barbut, Food Funct., 2012, 3, 327–337 CAS.
- T. Laredo, S. Barbut and A. G. Marangoni, Soft Matter, 2011, 7, 2734–2743 RSC.
- A. J. Gravelle, S. Barbut and A. G. Marangoni, Food Res. Int., 2012, 48, 578–583 CrossRef CAS.
- M. C. Bourne, Food Tech., 1978, 32, 62–66 Search PubMed.
- F. A. Osorio and J. F. Steffe, Rheol. Acta, 1991, 30, 549–558 CrossRef CAS.
- D. Folch, M. Lees and G. H. S. Stanley, J. Appl. Chem., 1957, 226, 497–509 Search PubMed.
- J. C. Touchstone, J. C. Chen and K. M. Beaver, Lipids, 1980, 15, 61–62 CrossRef CAS.
- W. W. Christie, Lipid Analysis, Pergamon Press, Oxford, England, 2nd edn, 1982 Search PubMed.
- W. W. Christie, J. Lipid Res., 1982, 23, 1072–1075 CAS.
| This journal is © The Royal Society of Chemistry 2013 |