Integrated conversion of hemicellulose and cellulose from lignocellulosic biomass†
David Martin
Alonso
a,
Stephanie G.
Wettstein
ab,
Max A.
Mellmer
ab,
Elif I.
Gurbuz
ab and
James A.
Dumesic
*ab
aDepartment of Chemical and Biological Engineering, University of Wisconsin-Madison, 1415 Engineering Drive, Madison, Wisconsin 53706, USA. E-mail: dumesic@engr.wisc.edu
bDOE Great Lakes Bioenergy Research Center, University of Wisconsin-Madison, Madison, Wisconsin 53706, USA
First published on 1st November 2012
Abstract
Using gamma-valerolactone (GVL) as solvent, the cellulosic fraction of lignocellulosic biomass can be converted into levulinic acid (LA), while at the same conditions the hemicellulose fraction can be converted into furfural. This process allows for the conversion of hemicellulose and cellulose simultaneously in a single reactor, thus eliminating pre-treatment steps to fractionate biomass and simplifying product separation.
Broader contextReplacing petroleum with renewable sources, such as lignocellulosic biomass, requires that current processes be simplified by reducing the number of steps necessary to achieve the final product. This paper reports a process to convert simultaneously the hemicellulose and cellulose fractions of lignocellulosic biomass, thereby eliminating the need for pretreatment steps to fractionate biomass. This process uses gamma-valerolactone (GVL) as a solvent, thereby simplifying separation steps, because GVL is one of the reaction products. Based on the yields achieved, this process is comparable in the production of fuels with the production of ethanol by fermentation. Additionally, the GVL solubilizes the degradation products typically formed during biomass deconstruction. Thus, the strategy can be implemented using continuous flow reactors without problems associated with the deposition and accumulation of solid residues. The possibility of producing a single product, GVL, from the hemicellulose and cellulose fractions of lignocellulosic biomass, the elimination of pretreatment steps, and the simplification of separation steps should improve the economics for production of chemicals and fuels from biomass. |
Large-scale production of biofuels requires utilization of lignocellulosic biomass feedstocks that do not compete with food sources (e.g., corn stover).1 A key challenge for biomass processing is that the physical and chemical properties of hemicellulose and cellulose (the main lignocellulose components) are significantly different.2 Therefore, typical processing strategies include a pretreatment step3 in which the C5 sugars (hemicellulose) are removed from the C6 sugars (cellulose), allowing the sugars to be processed by separate routes.
Horvath, a pioneer in gamma-valerolactone (GVL) valorization,4–7 identified GVL as a green solvent for various applications. We report here that by using GVL as the solvent, it is possible to achieve the integrated catalytic conversion of hemicellulose and cellulose to furfural and levulinic acid (LA), respectively, followed by conversion to GVL using corn stover as the feed. As depicted in Fig. 1, an open reactor system (i.e., valves A and B open) can be employed to produce furfural from hemicellulose. Since furfural (b.p. 441 K) has a lower boiling point than GVL (b.p. 481 K), it is continuously removed by distillation. The cellulose is converted to LA, passing through the intermediate formation of hydroxymethylfurfural.8,9 In this approach, the continuous removal of furfural from the reactive liquid minimizes furfural degradation and leads to high yields of furfural (e.g., 80%) from direct conversion of hemicellulose in corn stover. The less reactive cellulose fraction is converted with high yield to LA (e.g., 60%) by increasing the residence time in the reactor. The furfural, LA, and GVL can be used as chemical intermediates6,10,11 or can be converted to fuel components such as methylfuran,10,12 diesel,13 and butene oligomers.14 Alternatively, valves A and B (Fig. 1) can be closed to create a closed batch reactor that simultaneously converts hemicellulose and cellulose to furfural and LA, respectively, and then, without separation, these intermediates can be upgraded catalytically to GVL. Furfural can be converted to furfuryl alcohol in the GVL solvent, followed by conversion over an acid catalyst to produce LA,15 such that high yields of LA are achieved from both the hemicellulose and cellulose fractions of lignocellulosic biomass, completed by reduction of LA to GVL over a RuSn catalyst.16,17 The lignin and degradation products formed during the biomass deconstruction reaction are soluble in the GVL solvent; however, they can be precipitated upon addition of water and be removed by filtration (see Fig. 1).
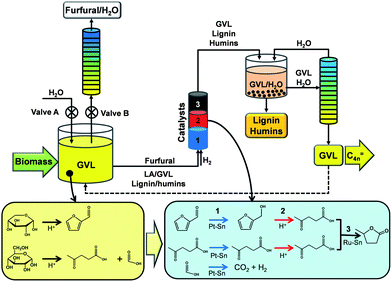 | ||
Fig. 1 Schematic representation of the integrated conversion of hemicellulose and cellulose portions of lignocellulosic biomass (e.g., corn stover) to furfural and GVL, using a portion of the GVL as a solvent and the remainder for conversion to butene oligomers (C4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ) as hydrocarbon. ) as hydrocarbon. | ||
Using GVL as solvent offers important advantages. (1) It eliminates or simplifies separation steps since GVL is a product of the process. (2) At the reaction conditions, the GVL effectively solubilizes the biomass, thereby eliminating the formation of solid deposits that typically lead to reactor clogging and solids handling problems.18,19 (3) Importantly, GVL decreases the rate of furfural degradation and increases the rate of cellulose conversion. Thus, furfural can be evaporated from the less volatile GVL solvent at elevated temperatures (e.g., 410 K) without undergoing significant degradation. (4) The GVL solvent broadens the optimal conditions for the separate processing of hemicellulose and cellulose, such that these conditions overlap, which allows hemicellulose and cellulose to be processed at high yields under the same conditions. (5) Finally, GVL is completely miscible with water, allowing wet biomass to be used in the process.
Using the open reaction system shown in Fig. 1, it is possible to remove the furfural from the reactive mixture of LA, GVL, and mineral acid (e.g., sulfuric acid (SA)) used for biomass deconstruction. Operating the open reactor (see ESI†) at 433 K and using 0.05 M SA as the catalyst (Table 1), results in a high furfural yield (79%) from the hemicellulose fraction of the corn stover, with 99% recovery of furfural. However, the yield of LA from the cellulose remains low (21%), even after further reaction in a closed batch reactor for 16 h (27%) (see ESI Table S1†) which indicates that the cellulose fraction is degraded during the reaction. Since GVL solubilizes the solids completely, it is not possible to calculate the cellulose conversion by typical methods, and further reaction to produce LA was thus used as reference to determine cellulose conversion. Low acid concentrations and high temperatures are optimal for feedstocks rich in C5 sugars that contain small amounts of C6 sugars, such as pre-hydrolysis liquors (PHL) from dissolving pulp applications or papermaking waste streams due to the high furfural yields obtained.
Decreasing the temperature decreases the amount of furfural recovered from the reactor (77%; Table 1, entry 2), but increases the stability of the cellulose fraction. Even when low yields of LA (15%) are achieved at 413 K, higher yields result after further reaction in a closed reactor at 443 K (Table S1†). Increasing the SA concentration decreases the furfural yield (70%), but increases the LA yield (42%; Table 1, entry 3) without needing further processing in the closed reactor. These conditions are optimal for feedstocks containing significant amounts of both C5 and C6 sugars (e.g., corn stover), where high yields from both types of sugars are required.
At 393 K with 0.1 M SA, the overall furfural yield (75%; Table 1, entry 4) increases, but the amount of furfural removed from the reactor (43%) decreases due to the lower temperature. The conversion of cellulose remains low under these reaction conditions, such that high LA yields (68%) can be achieved by increasing the temperature to 443 K during subsequent processing in a closed reactor. This approach is appropriate for feedstocks with a low content of C5 sugars and high content of C6 sugars, such as loblolly pine.
If the final goal is to produce fuels from both the hemicellulose and cellulose fractions of the biomass, then the processing strategy of Fig. 1 can be employed with valves A and B closed. In this case, the furfural from hemicellulose is not separated and is processed together with the LA from cellulose. The hemicellulose fraction of corn stover is easier to hydrolyzed than the cellulose fraction, and thus, as seen in Table 2, furfural can be produced from hemicellulose at high yields, 63% and 87%, using dilute solutions of mineral acids (0.02 M SA; entry 1) or using a solid acid catalyst (mordenite; entry 2), respectively.
| Entry | Feed | Sulfuric acid (M) | Solvent | Time (h) | Furfural yield (%) | Formic acid yield (%) | Levulinic acid yield (%) | |
|---|---|---|---|---|---|---|---|---|
| GVL (wt%) | H2O (wt%) | |||||||
| a Mordenite catalyst: 0.9 g catalyst per 1 g of corn stover. | ||||||||
| 1 | 6.6 wt% corn stover | 0.02 | 90 | 10 | 3 | 63 | 0 | 5 |
| 2 | 0a | 90 | 10 | 2 | 87 | 0 | 11 | |
| 3 | 0.1 | 80 | 20 | 1.5 | 56 | 79 | 61 | |
| 4 | 0.2 | 80 | 20 | 19 | 0 | 20 | 66 | |
| 5 | 5 wt% AHP corn stover | 0.025 | 90 | 10 | 0.5 | 96 | 11 | 10 |
| 6 | 0.025 | 90 | 10 | 4 | 73 | 54 | 51 | |
| 7 | 0.1 | 80 | 20 | 1 | 67 | 77 | 58 | |
| 8 | 0.1 | 0 | 100 | 1 | 53 | 20 | 8 | |
| 9 | 0.1 | 0 | 100 | 2 | 40 | 32 | 18 | |
Short reaction times and low acid concentrations prevent furfural degradation reactions and leave the cellulose mostly unconverted, leading to low LA yields (less than 11%). We note that some of the furfural produced may arise from C6 sugars, as reported previously.20 To obtain higher yields of LA (61%), the acid concentration must be increased (entry 3), which leads to low amounts of furfural degradation (56% yield). A further increase in the acid concentration and reaction time (entry 4) results in the highest LA yield (66%), but also leads to complete degradation of furfural. At optimum conditions, the presence of GVL increases the stability of the furfural and the reaction rate of cellulose conversion; therefore, high yields of furfural and LA can be achieved from corn stover using low SA concentrations (0.1 M). However, using low acid concentrations is problematic, because the lignin present in the biomass consumes a fraction of the SA during the reaction. Due to the low initial concentration of acid, the amount of acid lost is significant, and the LA yield decreases from 57% to 27% when 3 batches of corn stover are added to the reaction mixture (Table S2†). Typically, a higher SA concentration is used in the process (0.5 M),21,22 and therefore, this effect may have not been realized.
Although removing the lignin is not necessary for the process, the effect lignin had on the acid concentration during reaction was studied using corn stover that had been subjected to an alkaline hydrogen peroxide pretreatment (AHP), which partially removes the lignin.23,24 Using AHP corn stover, the yield of LA increases slightly to 69% in the first batch, and remains high (overall 53%) after the third batch of stover. Since the SA is retained in the reactor, and not consumed by the lignin, the use of the AHP corn stover allows lower acid concentrations and shorter reaction times to be employed (Table 2, entry 5), resulting in furfural yields up to 96%, with low conversion of cellulose. Increasing the reaction time at low acid concentration (entry 6) increases the LA yield (51%), but decreases the furfural yield (73%) due to degradation reactions. Importantly, the reaction conditions can be optimized to achieve high yields of both furfural and LA (entry 7), which is not possible when only water is used as a solvent (entries 8 and 9). Comparing entries 3 and 7, the presence of lignin only slightly modifies the optimum reaction conditions, and similar yields of LA and furfural can be achieved with regular and AHP corn stover.
At this point in the process, the hemicellulose has been converted to furfural and the cellulose to LA. To continue the processing strategy using the closed system (Fig. 1), it is necessary to achieve catalytic conversion of furfural to LA in the GVL solvent in the presence of the LA produced from cellulose. This conversion can be accomplished using bimetallic and acid catalysts. The first catalytic bed uses a Pt3Sn/SiO2 catalyst to hydrogenate furfural to furfuryl alcohol at 373 K with high yields (90%; commercial feed) without converting the GVL solvent or LA (see ESI†). This behavior is consistent with previous literature that used PtSn-based catalysts for liquid-phase furfural hydrogenation.25 After the Pt3Sn/SiO2 catalyst bed, the furfuryl alcohol passes over an acid catalyst (Amberlyst 70) that converts it to LA with yields of 70%, using the water already present in the solution (20 wt%) (Table S3†). An overall yield of 60% was achieved for the production of LA from furfural (commercial feed), even after 50 h time-on-stream over the dual-catalyst bed (Fig. S3†). When SA was added to the feed to simulate the SA remaining from the biomass deconstruction step, the furfuryl alcohol yield over Pt3Sn/SiO2 decreased to 70%, and the catalyst deactivated continuously until it showed no activity after 50 h time-on-stream (Fig. S4†). Thus, it is necessary to remove or neutralize the residual SA present in the GVL solution prior to catalytic conversion over the dual-catalyst bed (Fig. S5†). Since the GVL solvent allows for using lower concentrations of SA, the amount of SA that needs to be neutralized is an order of magnitude lower than other typical biomass deconstruction processes.26 The neutralization amount could be lower due to the presence of lignin in the feedstock.
The dual-catalyst bed of Pt3Sn/SiO2 and Amberlyst 70 successfully converted a liquid feed produced from corn stover deconstruction (80 wt% GVL/20 wt% water and 0.1 M SA overall concentration), followed by neutralization of SA, in a flow reactor. AHP-pretreated corn stover was used to determine the amount of SA which needs to be neutralized (see ESI†). Fig. 2 shows that the yield of furfuryl alcohol over Pt3Sn/SiO2 remains high (90%) for over 60 h time-on-stream for this biomass-derived feed (100% conversion), and the yield of LA over Amberlyst 70 remained at 70% for over 60 h time-on-stream (100% conversion). As noted above, LA can then be hydrogenated to GVL with high yields using a RuSn catalyst, without hydrogenation of the GVL,16,27 thereby completing the process of Fig. 1.
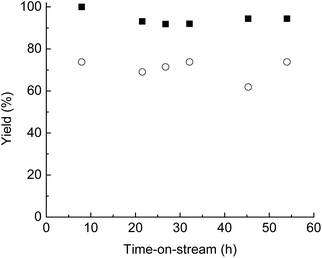 | ||
| Fig. 2 Furfuryl alcohol yield over Pt3Sn/SiO2 (■) and LA yield over Amberlyst 70 (○) versus time-on-stream at 35 bar H2. The conversions of furfural over Pt3Sn/SiO2 (373 K and 0.13 h−1) and furfuryl alcohol over Amberlyst 70 (398 K and 0.04 h−1) were both 100%. The feed to the dual-catalyst bed was prepared by deconstruction of AHP corn stover in 80 wt% GVL/20 wt% water using 0.1 M SA overall concentration. | ||
An advantage of this processing approach is that GVL solubilizes lignin and other degradation products typically produced during acid-catalyzed biomass reactions using water as solvent.9,28 These solid residues (i.e., humins) are responsible for plugging problems when flow reactors are used for biomass processing and can accumulate on the walls of batch reactors. Using water as a solvent to process corn stover led to the formation of 0.32 kg of solids per kg of corn stover (Fig. 3A) (0.23 kg solids per kg of AHP corn stover; Fig. 3C). In contrast, only 0.05 kg of solids per kg of corn stover (Fig. 3B) (0.02 kg solids per kg of AHP corn stover; Fig. 3D) were formed when GVL is used as the solvent. The bottom images in Fig. 3A and B show the significant difference in the thickness of the water-based retentate and the GVL-based retentate, respectively. Solubilizing the lignin and degradation products also allows for the possibility of upgrading these species to other products, such as the production of aromatic compounds from lignin.29 Additionally, the GVL solvent allows these species to be removed from the biomass deconstruction reactor as solubilized species in the GVL solution. In our experiments using heterogeneous catalysts in a flow reactor to upgrade the furfural and LA to GVL, we did not observe any reactor clogging problems or changes in the drop pressure across the reactor, indicating that the lignin and humins remain soluble at the reaction conditions and are not retained in the reactor. These results are in agreement with our previous results, where humins from cellulose deconstruction in GVL were not deposited in a flow reactor loaded with a Ru–Sn/C catalyst.16 Indeed, similar yields were obtained with simulated and corn stover feeds, indicating that the accumulation of lignin and humins on the catalyst surface is minimal.
 | ||
| Fig. 3 Images after monophasic reaction of the retentate after filtration and before oven-drying (top) and the liquid solution before filtering (middle, under 50× optical microscopy). (A) Corn stover in H2O/SA (bottom, side view of retentate), (B) corn stover in GVL/SA (bottom, side view of retentate), (C) AHP corn stover in H2O/SA, (D) AHP corn stover in GVL/SA, and (E) AHP corn stover in GVL/SA after addition of water. | ||
The lignin and degradation species can be recovered from the GVL solution by precipitation upon addition of water, as observed in Fig. 3E. This method for separation of lignin and humins from GVL can be implemented in our overall processing approach, as depicted in Fig. 1.
It has been reported that 39 gal of ethanol can be produced per ton of dry corn stover by fermentation of the cellulosic fraction.30 This amount can be increased to 59 gal of ethanol if the hemicellulose fraction is fermented as well. According to the yields in the present paper, up to 28 gal of high energy density liquid fuel (equivalent to 42 gal of ethanol in energy content) can be produced from the cellulose fraction of the corn stover (60% of the theoretical maximum). This amount can be increased to 37 gal of high energy density liquid fuel (equivalent to 57 gal of ethanol in energy content) if the hemicellulose fraction is also converted into liquid fuels (Table S4†). Alternatively, the hemicellulose can be converted to furfural that is distilled from the reactor during the process, such that 101 kg of furfural can be produced (62% of theoretical maximum), while still producing 25 gal of liquid fuels (equivalent to 38 gal of ethanol) per ton of corn stover without extra steps to separate hemicellulose and cellulose.
Conclusions
Simultaneous conversion of hemicellulose to furfural and cellulose to LA at high yield is possible using GVL as solvent. The furfural can be separated by distillation during the reaction or can be kept in the reactor and subsequently processed to produce furfuryl alcohol and LA. The LA can be upgraded to other platform molecules, such as GVL, or used as precursor to produce liquid fuels. This process not only benefits from the simultaneous production of fuels and chemicals by utilization of both hemicellulose and cellulose, but it also benefits from the elimination of pretreatment and extraction/separation steps, which can account for up to 30% of the total capital cost necessary to implement a biofuels production plant.26 Finally the GVL solubilizes the lignin and typical degradation products, eliminating solids accumulation problems in flow reactors and solids accumulation/handling issues in batch reactors.Acknowledgements
This work was funded in part by the DOE Great Lakes Bioenergy Research Center (DOE Office of Science BER DE-FC02-07ER64494). In addition, this work was supported through funding from the Defense Advanced Research Projects Agency (Surf-cat: Catalysts for Production of JP-8 range molecules from Lignocellulosic Biomass). The views, opinions, and/or findings contained in this article are those of the author and should not be interpreted as representing the official views or policies, either expressed or implied, of the Defense Advanced Research Projects Agency or the Department of Defense. The authors would like to thank Prof. David Hodge from Michigan State University for providing the AHP corn stover and thank Hui Chin Wong and Jher Hau Yeap for performing experiments reported in this paper.Notes and references
- D. Tilman, R. Socolow, J. A. Foley, J. Hill, E. Larson, L. Lynd, S. Pacala, J. Reilly, T. Searchinger, C. Somerville and R. Williams, Science, 2009, 325, 270–271 CrossRef CAS.
- C. E. Wyman, B. E. Dale, R. T. Elander, M. Holtzapple, M. R. Ladisch and Y. Y. Lee, Bioresour. Technol., 2005, 96, 2026–2032 CrossRef CAS.
- N. Mosier, C. Wyman, B. Dale, R. Elander, Y. Y. Lee, M. Holtzapple and M. Ladisch, Bioresour. Technol., 2005, 96, 673–686 CrossRef CAS.
- I. T. Horvath, Green Chem., 2008, 10, 1024–1028 RSC.
- I. T. Horvath, H. Mehdi, V. Fabos, L. Boda and L. T. Mika, Green Chem., 2008, 10, 238–242 RSC.
- H. Mehdi, V. Fabos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horvath, Top. Catal., 2008, 48, 49–54 CrossRef CAS.
- D. Fegyverneki, L. Orha, G. Lang and I. T. Horvath, Tetrahedron, 2010, 66, 1078–1081 CrossRef CAS.
- B. Girisuta, L. Janssen and H. J. Heeres, Chem. Eng. Res. Des., 2006, 84, 339–349 CrossRef CAS.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Green Chem., 2006, 8, 701–709 RSC.
- J. P. Lange, E. van der Heide, J. van Buijtenen and R. Price, ChemSusChem, 2012, 5, 150–166 CrossRef CAS.
- J. J. Bozell, L. Moens, D. C. Elliott, Y. Wang, G. G. Neuenscwander, S. W. Fitzpatrick, R. J. Bilski and J. L. Jarnefeld, Resour., Conserv. Recycl., 2000, 28, 227–239 CrossRef.
- A. Corma, O. de la Torre, M. Renz and N. Villandier, Angew. Chem., Int. Ed., 2011, 50, 2375–2378 CAS.
- D. M. Alonso, J. Q. Bond, J. C. Serrano-Ruiz and J. A. Dumesic, Green Chem., 2010, 12, 992–999 RSC.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS.
- J. P. Lange, W. D. van de Graaf and R. J. Haan, ChemSusChem, 2009, 2, 437–441 CrossRef CAS.
- S. G. Wettstein, D. M. Alonso, Y. Chong and J. A. Dumesic, Energy Environ. Sci., 2012, 5, 8199–8203 CAS.
- S. G. Wettstein, J. Q. Bond, D. M. Alonso, H. N. Pham, A. K. Datye and J. A. Dumesic, Appl. Catal., B, 2012, 117–118, 321–329 CrossRef CAS.
- I. Iliuta and F. Larachi, AIChE J., 2004, 50, 2541–2551 CrossRef CAS.
- T. Noel, J. R. Naber, R. L. Hartman, J. P. McMullen, K. F. Jensen and S. L. Buchwald, Chem. Sci., 2011, 2, 287–290 RSC.
- A. Ohnishi, K. Kato and E. Takagi, Polym. J., 1975, 7, 431–437 CrossRef CAS.
- S. W. Fitzpatrick, US Pat., US 5608105, 1997.
- J. C. Serrano-Ruiz, D. J. Braden, R. M. West and J. A. Dumesic, Appl. Catal., B, 2010, 100, 184–189 CrossRef CAS.
- G. Banerjee, S. Car, T. Liu, D. L. Williams, S. L. Meza, J. D. Walton and D. B. Hodge, Biotechnol. Bioeng., 2012, 109, 922–931 CrossRef CAS.
- G. Banerjee, S. Car, J. S. Scott-Craig, D. B. Hodge and J. D. Walton, Biotechnol. Biofuels, 2011, 4 DOI:10.1186/1754-6834-4-16.
- A. B. Merlo, V. Vetere, J. F. Ruggera and M. L. Casella, Catal. Commun., 2009, 10, 1665–1669 CrossRef CAS.
- D. J. Braden, C. A. Henao, J. Heltzel, C. T. Maravelias and J. A. Dumesic, Green Chem., 2011, 13, 1755–1765 RSC.
- D. M. Alonso, S. G. Wettstein, J. Q. Bond, T. W. Root and J. A. Dumesic, ChemSusChem, 2011, 4, 1078–1081 CrossRef CAS.
- S. K. R. Patil and C. R. F. Lund, Energy Fuels, 2011, 25, 4745–4755 CrossRef CAS.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS.
- A. Demirbas, Energy Sources, 2005, 27, 327–337 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2ee23617f |
| This journal is © The Royal Society of Chemistry 2013 |