Influence of the dye molecular structure on the TiO2 conduction band in dye-sensitized solar cells: disentangling charge transfer and electrostatic effects†
Enrico
Ronca
ab,
Mariachiara
Pastore
a,
Leonardo
Belpassi
a,
Francesco
Tarantelli
ab and
Filippo
De Angelis
*a
aComputational Laboratory for Hybrid/Organic Photovoltaics (CLHYO), Istituto CNR di Scienze e Tecnologie Molecolari, via Elce di Sotto 8, I-06123, Perugia, Italy. E-mail: filippo@thch.unipg.it
bDipartimento di Chimica, Università degli Studi di Perugia, via Elce di Sotto 8, I-06123, Perugia, Italy
First published on 19th October 2012
Abstract
We report a thorough theoretical and computational investigation of the effect of dye adsorption on the TiO2 conduction band energy in dye-sensitized solar cells that is aimed at assessing the origin of the shifts induced by surface adsorbed species in the position of the TiO2 conduction band. We thus investigate a series of working dye sensitizers and prototypical surface adsorbers and apply an innovative approach to disentangle electrostatic and charge-transfer effects occurring at the crucial dye–TiO2 interface. We clearly demonstrate that an extensive charge rearrangement accompanies the dye–TiO2 interaction, which amounts to transfer of up to 0.3–0.4 electrons from the dyes bound in a dissociative mode to the semiconductor. Molecular monodentate adsorption leads to a much smaller CT. We also find that the amount of CT is modulated by the dye donor groups, with the coumarin dyes showing a stronger CT. A subtle modulation of the semiconductor conduction band edge energy is found by varying the nature of the dye, in line with the experimental data from the literature obtained by capacitance and open circuit voltage measurements. We then decompose the total conduction band shift into contributions directly related to the sensitizer properties, considering the effect of the electric field generated by the dye on the semiconductor conduction band. This effect, which amounts to ca. 40% of the total shift, shows a linear correlation with the TiO2 conduction band shifts. A direct correlation between the dye dipole and the observed conduction band shift is retrieved only for dyes of similar structure and dimensions. We finally found a near-exact proportionality between the amount of charge transfer and the residual contribution to the conduction band shift, which may be as large as 60% of the total shift. The present findings constitute the basis for obtaining a deeper understanding of the crucial interactions taking place at the dye–semiconductor interface, and establish new design rules for dyes with improved DSC functionality.
Broader contextThe effect of surface-adsorbed species on the TiO2 conduction band energy is a highly debated issue in the field of dye-sensitized solar cells. The possible modulation of the position of the TiO2 conduction band appears to be a viable way to obtain higher cell open circuit voltages, and thus higher solar energy conversion efficiency. By applying first principles computational modeling, we investigate a series of working dyes and co-adsorbers to disentangle electrostatic and charge-transfer effects occurring at the dye–TiO2 interface. We clearly demonstrate that an extensive charge rearrangement accompanies the dye–TiO2 interaction. A subtle modulation of the TiO2 conduction band is found by varying the nature of the dye, in line with available experimental data. Such conduction band shifts are decomposed into contributions directly related to the sensitizer properties. A linear correlation is found between the dye electrostatic potential and the conduction band shift, which is proportional to the dye dipole for dyes of similar structure and dimensions. We also found a near-exact proportionality between the charge transfer and the residual contribution to the conduction band shift. The present findings constitute the basis for a deeper understanding of dye–sensitized semiconductors, with possible implications for the functioning of a wide range of optoelectronic devices. |
1. Introduction
Dye-sensitized solar cells (DSCs)1–4 have attracted significant attention as low-cost alternatives to conventional photovoltaic devices for the conversion of sunlight into electricity, with a highest certified efficiency of 11.4%.5 In DSCs, a dye sensitizer, anchored to the surface of a mesoporous oxide layer (usually TiO2), absorbs the solar radiation and transfers a photoexcited electron to the wide band gap semiconductor electrode consisting of nanometer-sized particles, while the concomitant hole is transferred to a redox shuttle in solution or to a solid state hole conductor, see Fig. 1.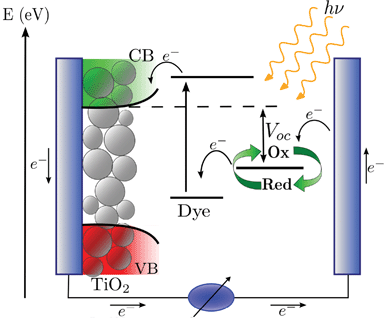 | ||
| Fig. 1 Schematic representation of the charge flow and of the energy levels of a dye-sensitized solar cell. | ||
To obtain further progress and a wider DSC uptake, higher conversion efficiencies need to be achieved. The three ingredients of a DSC, namely the dye, the semiconductor oxide and the redox shuttle, can be individually or simultaneously optimized for higher efficiencies. Out of the three DSC components, variations in the dye chemical nature and structure have been widely investigated.
Traditionally, the most commonly used dyes in DSCs are transition metal polypyridyl complexes,3 usually containing Ru(II) centers.2–5 Recently, Zn(II)-porphyrins have reached even higher efficiency when coupled to Co(II)/Co(III) redox shuttles, exceeding 12%.6 Fully organic dyes have also attracted considerable interest,3,7–12 in light of the anticipated superior synthetic flexibility, scalability and reduced environmental impact. The most efficient organic sensitizers have typically a D–π–A structure, with the donor group (D) being an electron-rich unit, linked through a conjugated linker (π) to the electron acceptor group (A), which is directly bound to the semiconductor surface, usually through a carboxylic or cyanoacrylic function.13–15 DSCs employing these dyes have obtained efficiencies close to 10%.13–16
The solar energy-to-electricity conversion efficiency (η) of a DSC is determined by the short circuit photocurrent density (Jsc), the open circuit voltage (Voc), the fill factor (ff) of the cell, and the intensity of the incident light (Is), namely:
| η = JscVocff/Is | (1) |
The photocurrent density is directly related to the charge generation and collection efficiency, which in turn depend upon the dye's light-harvesting and the kinetics of electron injection, dye regeneration and charge transport within the cell. Thus, the charge generation contribution to Jsc depends directly on the dye. The DSC open circuit voltage represents the difference between the quasi-Fermi level of the semiconductor under illumination and the redox potential of the electrolyte, see Fig. 1, the latter being nearly constant under operation conditions due to the high concentration of redox species.17
The quasi-Fermi level (EF,n) of the semiconductor depends “statically” on the semiconductor conduction band (CB) energy and “dynamically” on the charge density (n) accumulated in the semiconductor,12 according to the following equation:
| EF,n = ECB + kBT ln[n/Nc] | (2) |
Organic dyes usually show good light-harvesting properties, due to their high extinction coefficients, so the limitations in the efficiency with these materials can be mainly attributed to recombination processes of the injected electrons with the oxidized dye or electrolyte and/or to the formation of dye-aggregates on the titania surface,18–26 which provide light filtering and a means of excited state deactivation. While dye aggregation can be controlled to some extent by the use of co-adsorbents,27 the effect of the dye structure on the recombination process is more complicated and several studies have been devoted to understanding this aspect.11,19,20,28–36
From a different perspective, the energetics of the TiO2 conduction band are known to depend on several factors, such as the local pH,37–39 the concentration of potential determining ions (e.g. Li+),39,40 and, possibly in relation to acid–base equilibria, also on the nature of the electrolyte solvent.40,41 The role of surface adsorbed molecules, including the dye, in determining the TiO2 CB energetics is much less clear.17,42–49 Previous works on ruthenium dyes have shown a correlation between the dye protonation state and the DSC performance,45,46,49 with dyes carrying a higher number of protons leading to higher Jsc but lower Voc. An interesting correlation between the dipole moment of co-adsorbing species, mainly substituted benzoic acids, and the corresponding DSC Voc was observed by Rühle et al.,44 who pointed out a linear relationship between the dye coverage (N), the dipole (μ) component normal to the surface (θ is the molecule tilting angle) and the potential shift (ΔV) at the interface affecting the TiO2 CB energy:
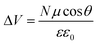 | (3) |
For ruthenium dyes, we reported a correlation between the dye adsorption mode and the corresponding DSC Voc, with the homoleptic N719 dye, adsorbing through three carboxylic groups, constantly showing higher cell voltages than comparably efficient heteroleptic dyes.46 Later work on solid-state DSC has clearly shown a greater than 100 mV TiO2 conduction band shift between a heteroleptic ruthenium dye and an organic dye, which was interpreted in terms of a dipole-induced TiO2 CB shift of different sign.50 Such shifts are generally more difficult to be observed in DSC based on a liquid electrolyte,20 in which the high ion strength and the effect of thermal motion may hinder the role of interface dipoles. Nevertheless, Kusama et al. reported a combined experimental and theoretical study which showed a clear correlation between the dipole moment of electrolyte additives and their DSC Voc.51
When a dye binds to a semiconductor surface, two effects might be at work: (a) the aforementioned electrostatic (EL) effect, due to the dye dipole moment; and (b) the effect of the charge transfer (CT) between the dye and the semiconductor, which may accompany the dye–semiconductor bond formation. To our knowledge, the latter effect has never been investigated in the DSCs framework, although the interfacial ground state CT could easily lead to semiconductor CB shifts. CT occurrence and extent upon formation of chemical bonds is controversial and often difficult to assess. We can successfully investigate it using the Charge Displacement (CD) analysis, introduced by some of us.52 This approach is based on a partial integration of the electron density difference between a bound system and its separated fragments, and its application led to significant progress in the comprehension of the chemical bond in various systems,52–57 including organometallic complexes.58
Motivated by the high interest in providing a clear understanding of surface adsorbed species-induced CB shifts, thus tuning the DSCs open circuit voltage, in this paper we computationally investigate the adsorption of several prototypical organic dyes and co-adsorbents on TiO2 models, disentangling the effects of EL and CT contributions to the TiO2 CB energetics. We find substantial differences in both EL and CT effects as a function of the dye structure and adsorption mode, and we correlate them with available experimental information. Our results provide a unified view of dye–semiconductor interactions, which reconcile apparent discrepancies between previous data and contribute to designing new and more efficient solar cell sensitizers.
2. Models and methods
We investigate a wide series of organic dyes with a push–pull (D–π–A) structure (see Fig. 2), effectively employed in DSC devices. In this set of compounds we include dyes with the same donor and π spacer, but with different anchoring units, such as L0 (ref. 3,7 and 47) and rh-L0,47 that differ only in the substitution of the conjugated cyanoacrylic group (in the former) with a non-conjugated rhodanine-3-acetic unit (in the latter). We have also analyzed sensitizers with the same donor and anchoring groups but with π-spacers of different length, such as the two coumarin dyes NKX-2587 and NKX-2697 that only differ in the number of thiophenes contained in the central part of the sensitizer (one in NKX-2587 and three in NKX-2697).3,18,59 Along the same lines, we investigated L1 (ref. 7) and D5 (ref. 47, 60 and 61) dyes, that differ in the nature of the π-spacer. In addition, we also investigated a set of small organic molecules commonly used as prototypes for the simulation of co-adsorbents on TiO2. This series includes benzoic acid,44 BA, and acetic acid, AA, with some of their functionalized derivatives (nitro-benzoic acid,44 NO2-BA, 4-aminobenzoic acid, NH2-BA, and trifluoroacetic acid, AAF3) and the prototype formic acid, FA.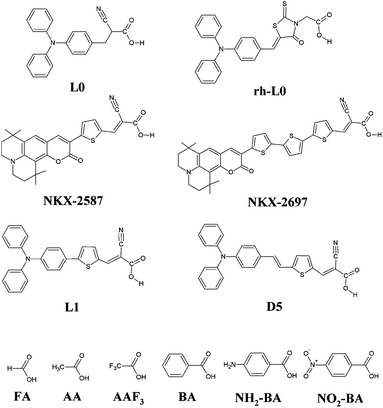 | ||
| Fig. 2 Molecular structures of the L0, rh-L0, NKX-2587, NKX-2697, L1 and D5 dyes, and of benzoic acid (BA), 4-aminobenzoic acid (NH2-BA), nitrobenzoic acid (NO2-BA), acetic acid (AA), trifluoroacetic acid (AAF3) and formic acid (FA). | ||
The compounds described above have been anchored on the titania surface by the carboxylic group in both dissociative bridged bidentate (BB) and molecular monodentate (M) adsorption modes, Fig. 3.47,62 In the BB adsorption mode, the sensitizer's proton is transferred to one of the TiO2 oxygens, and the dye's carboxylic group tends to interact with two surface titanium atoms, while in the M adsorption mode the dye carboxylic –OH group interacts by hydrogen bonding with a surface oxygen which is not directly bound to the Ti atom anchoring the dye.47
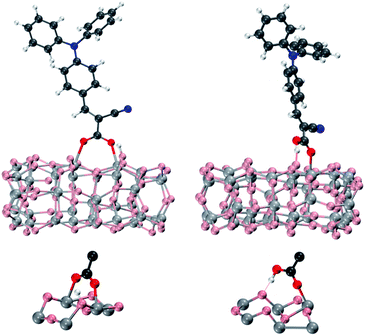 | ||
| Fig. 3 Top: optimized geometries of the L0 dye adsorbed onto the (TiO2)38 cluster in bridged bidentate (BB, left) and monodentate (M, right) anchoring geometries. Bottom: details of the anchoring region for BB and M adsorption modes. | ||
To model the TiO2 surface, we used two clusters, (TiO2)38 (ref. 63–65) and (TiO2)82,66 both obtained by appropriately “cutting” an anatase slab exposing the majority (101) surface.67 The considered clusters represent a reasonable tradeoff between accuracy and computational convenience and nicely reproduce the main electronic characteristics of TiO2 nanoparticles. Indeed, the lowest transition of the (TiO2)38 cluster calculated using time-dependent density functional theory (TDDFT) is 3.20 eV,68 in good agreement with the experimental bandgaps typical of TiO2 nanoparticles of a few nanometers in size (3.2–3.3 eV).69,70 The TiO2 conduction band edge was calculated at ca. −4 eV vs. vacuum, in good agreement with experimental values.71 The larger (TiO2)82 cluster shows a similar conduction band structure, within 0.1 eV, to the corresponding periodic model.72 We thus use the (TiO2)38 cluster as our “workhorse”, resorting to the larger (TiO2)82 cluster to check the accuracy of our results. We also notice that even though the use of relatively small clusters might introduce some artifacts in the description of the dye–semiconductor interaction, we are mainly interested here in relative variations along a series of dyes, which should be much less affected. The dye-adsorbed geometries were optimized in the gas phase using density functional theory (DFT) with the ADF program package73 employing the PBE exchange correlation functional74 with a TZVP (DZVP) basis set for Ti (H, C, N, O, S) atoms.
A key approach in our analysis is the study of the electron density changes taking place upon formation of the dye–TiO2 adducts. The electron density change (Δρ) is defined as the density difference between the interacting complex and the isolated, non-interacting, partners placed at the same position they have in the bonded adduct. For the BB-anchored systems, we chose as fragments the deprotonated dye (negatively charged) and the protonated TiO2 (positively charged), while for the M-bound species we considered the neutral dye–TiO2 fragments. Based on our previous results,75 we located for all the investigated systems the dissociated proton on the surface oxygen atom closest to the deprotonated carboxylic group. To analyze the electron density rearrangement, we define the charge displacement (CD) along the z direction as:
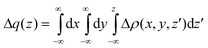 | (4) |
The electronic densities were obtained in all cases by performing single point calculations at the DFT/B3LYP level of theory using the SVP basis set76,77 on the ADF optimized geometries. Solvation effects (acetonitrile) have been taken into account by means of the C-PCM78,79 solvation model as implemented in Gaussian 09 (G09).80 The chosen combination of methods ensures efficient geometry optimizations, which are necessary for the rather large systems studied here, while retaining the accuracy typical of hybrid functionals, e.g. in terms of TiO2 band gap, for the description of the electronic structure of the dye–semiconductor assemblies. We also checked the basis set effect by calculating the CD curve of Fig. 4 with a larger 6-31G* basis, finding negligible differences, ESI.† In order to extract information regarding the energetic position of the CB edge of the semiconductor, we evaluated the Partial Density of States (PDOS) profiles on TiO2 calculated at the B3LYP/SVP level of theory in acetonitrile solution considering a window of 40 virtual orbitals. The resulting PDOS have been plotted using a Gaussian function with a standard deviation σ = 0.20 eV. We then performed a linear fitting of the low energies profile in a range from 20 to 80% of the maximum height, see ESI.† The intercept of this line with the energy axis was taken as the CB edge. The chosen 20–80% interval was employed since it allows us to include the possible deformation of the DOS curves on the low energy side. We checked that the relative trends are unaffected by the choice of the interval.
 | ||
| Fig. 4 Isodensity contour plot and charge displacement curve for L0 adsorbed onto TiO2 in a BB configuration. Yellow surfaces identify regions in which the electron density decreases whereas zones of density accumulation are marked by dark blue surfaces. The density value at the surfaces is ±0.0005 e au−3. | ||
3. Results and discussion
3.1 Charge displacement analysis
We start our analysis by performing a study of the interaction between the sensitizer and TiO2. In Fig. 4 we show the isodensity contour plot of the electron density difference and the CD curve, calculated by eqn (4) for the prototypical L0 dye adsorbed onto TiO2 in the BB configuration. The density deformation plot shows an evident rearrangement of both dye and semiconductor's electronic structure as a consequence of the dye–surface interaction. In particular, we can see significant charge depletion lobes in the areas around the carboxylic group, the nitrogen atom of the nitrile group and the phenyl bound to the cyanoacrylic moiety. Charge accumulation lobes can be seen instead on the central carbon of the cyanoacrylic anchoring group of the sensitizer, in proximity of the proton detached from the dye and adsorbed on the TiO2 surface and, in particular, in regions of the semiconductor cluster. Turning to the CD curve, the pronounced rearrangement of the electron density in the sensitizer is evident from the shape of the curve, which slowly rises in the region of the triphenylamine donor group, to increase more rapidly in proximity of the cyanoacrylic linker. Note that the curve is largely positive across the entire adduct, implying a continuous charge transfer from the dye to the TiO2. The maximum of the CD curve in the inter-fragment region is 0.36 electrons, see Table 1. This is clearly a pronounced CT, which reflects the strong interaction between the oppositely charged sensitizer and semiconductor, as demonstrated also by the large binding energy, see below.| Dye | ΔCBTOT | μ z | V EL | V DIP | ΔCBEL | ΔCBCT | CT |
|---|---|---|---|---|---|---|---|
| Dissociative bridged bidentate adsorption | |||||||
| NKX-2587 | 0.29 | 18.5 | −2.76 | — | 0.09 | 0.20 | 0.40 |
| D5 | 0.28 | 8.9 | −2.58 | — | 0.09 | 0.18 | 0.37 |
| L1 | 0.28 | 6.8 | −2.57 | — | 0.09 | 0.18 | 0.36 |
| L0 | 0.27 | 6.4 | −2.59 | — | 0.10 | 0.17 | 0.36 |
| rh-L0 | 0.26 | 3.9 | −2.67 | — | 0.11 | 0.16 | 0.34 |
| NKX-2697 | 0.25 | 21.1 | −2.64 | — | 0.09 | 0.16 | 0.36 |
| NH2-BA | 0.31 | 5.3 | −2.71 | — | 0.13 | 0.18 | 0.39 |
| BA | 0.28 | 1.5 | −2.59 | — | 0.10 | 0.18 | 0.36 |
| NO2-BA | 0.27 | −3.8 | −2.41 | — | 0.10 | 0.17 | 0.34 |
| AA | 0.30 | 0.8 | −2.65 | — | 0.13 | 0.17 | 0.36 |
| AAF3 | 0.25 | −1.8 | −2.49 | — | 0.11 | 0.15 | 0.30 |
| Undissociated monodentate adsorption | |||||||
| NKX-2697 | 0.07 | 18.5 | −0.20 | −0.17 | −0.01 | 0.06 | 0.14 |
| NKX-2587 | 0.07 | 18.9 | −0.30 | −0.29 | −0.01 | 0.06 | 0.14 |
| L0 | 0.04 | 8.0 | −0.20 | −0.15 | −0.01 | 0.05 | 0.10 |
| NH2-BA | 0.04 | 5.3 | −0.20 | −0.20 | −0.01 | 0.06 | 0.13 |
| BA | 0.02 | 1.6 | −0.07 | −0.05 | −0.02 | 0.05 | 0.11 |
| NO2-BA | 0.01 | −3.9 | 0.09 | 0.16 | −0.03 | 0.04 | 0.10 |
| AA | 0.02 | 1.0 | −0.06 | −0.05 | −0.02 | 0.04 | 0.10 |
| AAF3 | −0.02 | −1.9 | 0.10 | 0.11 | −0.04 | 0.02 | 0.06 |
| FA | 0.02 | 0.2 | 0.01 | 0.07 | −0.02 | 0.04 | 0.09 |
The CD curves for the entire series of molecules are presented in the ESI.† It suffices here to notice that a stronger maximum CT (0.40 electrons), Table 1, and a higher binding energy for the dissociative BB adsorption mode (2.25 eV when accounting for basis set superposition error, BSSE) are calculated for the NKX-2587 coumarin dye, suggesting that the coumarin unit is a more efficient donor group than the TPA one, as previously found by Wang and Liu.81 As a check of our methodology, we also calculated the binding energy for undissociated M formic acid adsorption to TiO2, finding a BSSE-corrected value of 1.02 eV, very close to the 0.92 value reported by Vittadini et al.82 The large difference between BB and M binding energy data is related to the different choice of the fragments, whereby for BB the negative dye interacts with the positively charged protonated TiO2, also leading to a larger CT. It is also interesting to notice that a comparable amount of CT is calculated from the CD curves and from the partial dye charges obtained for the interacting dye–semiconductor assemblies, see ESI.† Finally, to check the convergence of our results with respect to the size of the TiO2 substrate, we repeated the CD analysis for NKX-2587 adsorbed on the larger (TiO2)82 cluster and found a CT of 0.39 electrons, which confirms the adequacy of the approach employed.
3.2 Dye effect on the TiO2 conduction band
In the previous section we have shown that a substantial charge rearrangement takes place upon dye adsorption onto the semiconductor surface. This charge redistribution can cause significant modifications on the TiO2 electronic structure, with likely consequences on the CB energetics. As discussed in the introduction, however, a pure electrostatic (EL) contribution, due primarily to the dye dipole, is also able to alter the TiO2 CB energy.17 It is thus very desirable to try to disentangle CT and EL effects. For this purpose, we formulate here a simple model enabling us to associate the calculated CB shift to the varying degree of EL and, in turn, CT contributions. We assume that the total TiO2 CB shift, ΔCBTOT, can be expressed as the sum of various contributions:| ΔCBTOT = ΔCBsolv + ΔCBions + ΔCBEL + ΔCBCT | (5) |
The total calculated CB shifts due to the presence of the various investigated systems are reported in Table 1 (see Fig. 6 below for the corresponding PDOS profiles in the L0 case, and ESI†). As can be noticed in Table 1, the different systems anchored in the same BB geometry show quite similar perturbations of the TiO2 CB edge, varying from 0.24 to 0.31 eV. In all cases the observed differences lead to a CB energy upshift, as previously reported for organic dyes.50 As mentioned above, the calculated shifts do not take into account the effect of ionic screening, and the solvent effects are approximated by a continuum model, so they should be considered only for comparison purposes.
Despite the small differences among the investigated systems, some interesting information can be extracted from these data and compared to available experimental data. For example, comparing the results obtained for the two coumarin dyes, we can see that (as pointed out above also for CT) the shorter NKX-2587 sensitizer shows a more significant CB energy upshift than that produced by the longer NKX-2697 dye. The difference, amounting to 40 meV, is in exact agreement with the experimental results obtained through capacitance measurements by Miyashita et al. in ref. 20. Furthermore, Liu et al.11 reported experimental investigations about oligothiophene dyes and observed, as in our case, a decline of the open circuit voltage by increasing the number of thiophene units in the dye π-spacer. This can be partly related to the CB energy downshift that we observe here on going from NKX-2587 to NKX-2697. A trend can also be outlined for the series of substituted benzoic acids. In this case, the CB shift tends to increase with the donor character of the substituent, in agreement with the results obtained by open circuit voltage measurements by Rühle et al. in ref. 44.
It is also very interesting to note that a significantly different perturbation is observed when anchoring the dye in the monodentate versus bidentate geometry, the former showing a CB edge shift significantly lower (in the range 10–70 meV) than the latter (∼200 to 300 meV). This highlights the importance of the dye anchoring mode for tuning the TiO2 CB energy. We shall further discuss and clarify this point later.
An additional useful comparison can be made by looking at the total CB shift calculated for the NKX-2587 dye when adsorbed on (TiO2)38 or (TiO2)82, the latter exposing a surface roughly three times larger. A reduced total CB shift is calculated for the larger (TiO2)n cluster (0.23 vs. 0.28 eV, for n = 82 and 38, respectively), which is consistent with the observation that the dye-induced shift of the TiO2 CB is a function of the dye coverage (see eqn (3)),44 a fact that should always be taken into account to make meaningful comparisons. In this regard, it is important to highlight that by scaling the ΔCBTOT relative to the benzoic acids in the BB anchoring geometry for the coverage density (∼1018 molecules per m2 in our case, to be compared to the 2 × 1017 molecules per m2 value of Rühle et al. in ref. 44), we obtain ΔCBTOT values in the range 50–60 mV (57 mV for BA, 61 mV for NH2-BA and 54 mV for NO2-BA), in line with the results of ref. 44. Our results also suggest that the investigated dyes are less likely to be adsorbed on TiO2 in a monodentate geometry, because the shifts calculated for molecules in this adsorption mode are one order of magnitude smaller than those observed experimentally.
3.3 Electrostatic effects
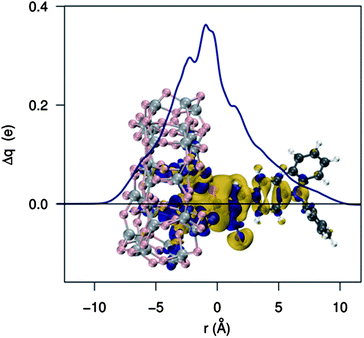
Let us now begin the analysis of electrostatic effects (EL) by assessing the effect of an applied dipole on the TiO2 electronic structure. Following our previous work,46 we computed the TiO2 DOS in the presence of an artificial dipole applied in the direction z normal to the surface plane. The results are shown in Fig. 5 where we show how the CB edge energy changes by varying the distance of a 20 D dipole from the semiconductor surface. The plot displays the CB position versus the dipole distance from the surface, which falls off as the inverse square distance: it clearly appears that, as previously pointed out,44,48 dipoles pointing away from the semiconductor (positive dipoles) produce CB energy upshifts; the opposite holds for negative dipoles. Moving the dipole away from the surface (i.e., as the potential decreases), the magnitude of the CB shift tends to decrease, reaching the value of the bare TiO2 CB in the absence of the field (∼−3.6 eV at the present level of theory). An accurate quadratic fit is found throughout, which corresponds to a linear dependence of the shift on the dipole magnitude for a given dipole distance.44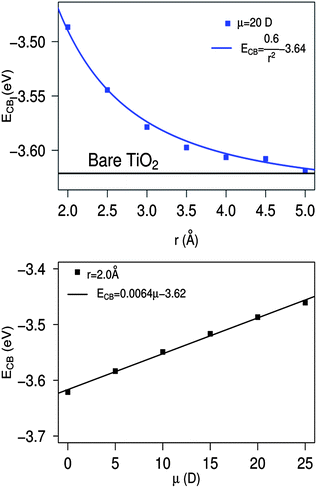 | ||
| Fig. 5 Upper panel: TiO2 conduction band energy as a function of the distance due to a nearby electric dipole. A 20 D dipole, pointing toward the TiO2 surface, was employed. Lower panel: effect of the dipole strength on the TiO2 conduction band energy (the distance of the dipole center from the semiconductor is 2 Å). | ||
To estimate now the extent of the EL effects due to the surface adsorbed species and to decouple them from the CT effects, we simulate the electrostatic field due to the dye on the TiO2 CB by using a simple point-charge model, Fig. 6. We obtain a set of atomic charges by fitting the dye electrostatic potential through the Merz–Singh–Kollan method.83,84 In these calculations the isolated dyes are calculated in solution at their optimized geometry when adsorbed on TiO2. We then simply calculate the electronic structure of the (TiO2)38 cluster in the presence of the calculated charges at the optimized interacting geometry, Fig. 6. Note that this simple approach, which corresponds to the classic long range definition of the electrostatic potential, has the advantage of being independent of the choice of a charge center, and it can be used also for charged molecules (such as the dyes in the BB anchoring mode) whose multipoles depend on the reference system origin. At the same time the employed approach does not account for dynamic polarization of the conjugated molecules in the presence of the TiO2 nanoparticles.
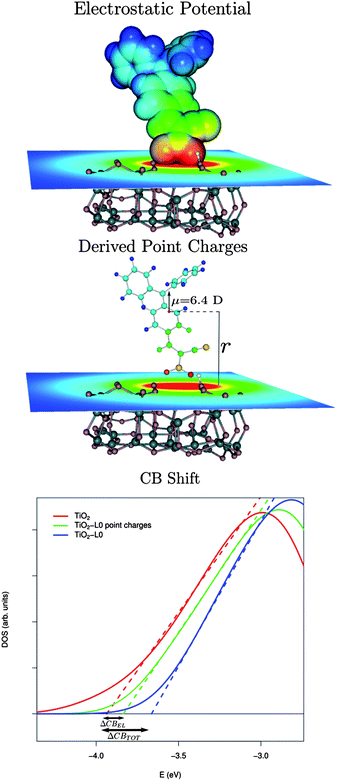 | ||
| Fig. 6 Top-middle panels: schematic representation of the model used to assess the CB shift due to the electrostatic potential generated by the sensitizer for the L0 case. Bottom: calculated conduction band PDOS for the bare TiO2 (red solid line) for TiO2 in the presence of the L0 point charges (green solid line) and for the fully interacting L0–TiO2 system (blue solid line). The dashed lines intercepts with the energy axis correspond to the calculated CB edges. | ||
The calculated electrostatic CB edge shifts, ΔCBEL, evaluated in this model are reported in Table 1 for all the investigated systems. As can be observed, these shifts vary in the range 0.08–0.13 eV, thus amounting to 30–40% of the total calculated shifts (ΔCBTOT). It is worth noticing that the monodentate-anchored systems give rise to smaller electrostatic perturbations (10–40 meV), due to the fact that in this configuration the dye is neutral.
Comparing the CB shifts due to the presence of the dye point charges to the corresponding neutral dye dipole moment component perpendicular to the semiconductor surface (Table 1), we notice there is not a clear correlation between the two quantities. This behavior is evident by comparing NH2-BA and NO2-BA in the BB adsorption mode. Despite the fact that NO2-BA has a dipole opposite to NH2-BA, it shows a comparable CB electrostatic shift of the same sign with respect to that of the unsubstituted BA. The same behavior can be observed for the AA and AAF3 systems. Another clear example can be seen by comparing the dyes in the bidentate anchoring geometry with their analogs in the monodentate one. Surprisingly small differences in the dipoles (e.g. less than 3 D for L0) give rise to a consistent CB shift variation (e.g. ∼0.11 eV for L0) whereas the much larger difference of ∼11 D between L0 and NKX-2587 (in the BB geometry) produces a change on ΔCBEL of only 0.01 eV.
These apparent inconsistencies are lifted if we recognize that the CB shift should be considered in relation to the electrostatic potential generated by the dye molecule rather than with its standalone dipole moment. Thus, we evaluate the average dye electrostatic potential on the TiO2 surface by using the following relationship:
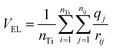 | (6) |
The relationship between the TiO2 CB shift and the electrostatic potential of eqn (6) is reported for the investigated dye series in Fig. 7. Looking at the main panel, we can immediately notice an approximately linear relationship between the electrostatic potential and the CB shift for the dyes adsorbed on TiO2 in a monodentate way. The accuracy of the fit, characterized by R2 = 0.86, is definitely good, considering the simplicity of the model and the heterogeneity of the investigated systems. Including also the dyes anchored on TiO2 in the BB geometry, we can see that the linear trend persists and actually the fit quality improves (R2 = 0.97). It is of course eye-catching that the BB dyes generate a significantly higher potential on the TiO2 surface with consequent larger shifts. This behavior can be simply explained considering that, as pointed out earlier, the species adsorbed on the semiconductor in BB geometry are charged. Our results suggest then that to obtain conduction band shifts comparable with those measured experimentally, we need to consider charged molecules adsorbed on TiO2. In comparison with the M-adsorbed dyes, the BB systems show a larger deviation from the linear trend. This can be explained by taking into account the irregularities in the low energy DOS profile due to the presence of the proton adsorbed on the surface and the larger perturbation generated by a charged species on the semiconductor.
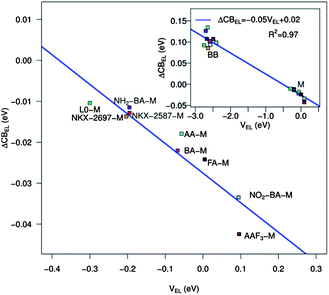 | ||
| Fig. 7 CB shift due to the electric field generated by the adsorbed dye molecule as a function of the electrostatic potential (eqn (6)) generated by the dye point charges for all the dyes in their monodentate adsorption mode. Inset: same but including the charged dyes in the bidentate adsorption mode. | ||
The above results confirm the direct proportionality between the electrostatic component of the CB shift and the electrostatic potential generated by the sensitizer. Now it would be very interesting to investigate, for the systems in the monodentate geometry (neutral species), the relationship connecting the TiO2 electrostatic CB shift with the dipole moment of the isolated dye. In order to do this, we evaluated the average dipolar electrostatic potential (VDIP) generated by each molecule on the TiO2 surface by the following relationship:
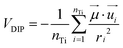 | (7) |
We would like to finally comment on the seeming inconsistency that the lines in Fig. 7 do not cross zero for V = 0 (the point roughly corresponding to the FA adsorbate). We can possibly attribute this small systematic negative CB shift to the effect of the protonated carboxylic group. To quantify this aspect, we resort to the prototypical formic acid case, which produces an almost zero electrostatic potential to the surface, Table 1, yet it shows a small negative TiO2 CB shift. We trace this effect back to the interaction between the dye-bound proton and the TiO2 surface, which slightly downshifts the TiO2 CB energy. An alternative plausible explanation is that due to the delocalized nature of the electron density, some residual perturbation on the semiconductor is present even when VEL = 0. Nevertheless, shifting the data in Fig. 7 by the ΔCBEL value of FA restores the expected crossing at V = 0.
3.4 Charge transfer effects
Having satisfactorily described and explained the electrostatic ΔCB component, we now turn our attention to the fact that, as mentioned above, this contribution is found to be only a fraction of the total shift. In particular, we would like to establish whether the residual shift component can confidently be attributed to CT effects. To verify this supposition, we first investigate if a meaningful relationship can be established between the residual shift (ΔCBTOT − ΔCBEL) and the CT amount predicted by the charge displacement analysis along the series of dye–TiO2 systems. This is displayed in Fig. 8, which evidences beyond doubt a surprisingly accurate linear correlation between the two computed quantities (R2 = 1.00).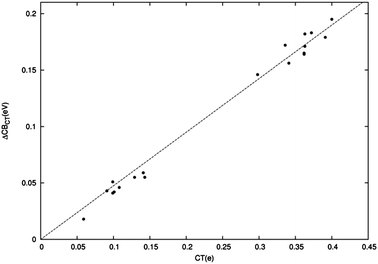 | ||
| Fig. 8 Difference in eV between the total CB energy shift and its electrostatic component plotted versus the amount of CT (electrons). The top right part of the graph refers to BB-anchored dyes, the bottom left part of the graph to M-anchored dye. | ||
As usual, the complexes at hand cluster in two distinct groups, one corresponding to the neutral M-bound dyes, at smaller shifts and CT values, and the other to the charged BB-anchored systems. The linear correlation appears to fit both groups equally accurately. This finding clearly strengthens our claim that the additional CB shift component must be attributed to CT, besides confirming the adequacy of the charge displacement analysis as a tool to measure CT and supporting our estimate of the electrostatic contribution to the band shift. We can however attempt to further validate our conclusions by independently investigating how charge transfer affects the position of the TiO2 CB edge. This can be very simply done by evaluating the TiO2 DOS when electrons are added or subtracted. We have thus computed the CB shift with respect to the neutral TiO2 for TiO2+, TiO2− and TiO22−, see ESI,† as a function of the cluster charge. Again, a very accurate linear correlation is found (R2 = 0.97) with the points corresponding to the investigated actual systems (the same as Fig. 8) lying very close to, although always above, the best-fit line found, firmly supporting the emerging picture.
On the basis of the above results, we believe it may be safely concluded that, in the dye–TiO2 interaction, CT effects induce a CB shift which for BB anchored systems is much larger than for the M-bound ones, and this, as well as the electrostatic effect, contributes to their larger upward CB shift. It thus turns out that the CT component of the CB shift may in fact be the dominant one (60% of the total), with a relative variability comparable to that exhibited by the electrostatic component.
4. Conclusions
We have set up and applied to a series of working dye sensitizers and prototypical surface adsorbers an innovative approach to disentangle electrostatic and charge-transfer effects occurring at the crucial dye–TiO2 interface, which lies at the heart of dye-sensitized solar cells (DSCs). Our focus is on the effect of the dye on the TiO2 conduction band energy in an effort to provide fundamental insight useful to obtain higher DSC open circuit output voltages and to lift apparent discrepancies among data obtained in different conditions.We clearly demonstrated that an extensive charge rearrangement accompanies the dye–TiO2 interaction which, using the well-established charge-displacement analysis, we estimated to generate a net charge transfer of up to 0.3–0.4 electrons from the dyes to the semiconductor. Different dyes adsorbed in the same dissociative bridged bidentate adsorption mode show little variations in the amount of CT, while the molecular monodentate adsorption geometry leads to a much smaller CT. We showed that the amount of CT is modulated by the dye donor groups, with the coumarin dyes showing a stronger CT, for the same acceptor and π-spacer groups.
We evaluated the TiO2 partial density of states in the presence of a sensitizer for a wide series of organic molecules, finding a subtle modulation of the semiconductor conduction band edge energy. Our results are perfectly consistent with the experimental data from the literature obtained by capacitance and open circuit voltage measurements.
To give a physical interpretation of these observations, and to assist the design of new dyes potentially able to shift the TiO2 conduction band in a predictable way, we decomposed the total conduction band shift into contributions directly related to the sensitizer properties. First, we evaluated the effect of the electric field generated by the dye molecules on the semiconductor conduction band. We found that the electrostatic potential generated by the dye charge distribution correlates linearly and very accurately with the observed TiO2 conduction band shifts. This implies that a direct correlation between the dye dipole and the observed conduction band shift effectively exists only for dyes of similar structure and dimensions. By varying the dimensions of the employed TiO2 model, we also verified that such electrostatic shift scales with the surface coverage.
The estimated electrostatic contribution to the conduction band shift amounts to about 40% of the total calculated shifts. Considering the pronounced ground state dye → semiconductor charge transfer, we investigated the supposition that the remaining contribution to the semiconductor conduction band shift is directly due to charge transfer effects. We established that there is indeed near-exact proportionality between the amount of charge transfer calculated by the charge displacement analysis and the residual contribution to the conduction band shift. We thus found that the CT induced CB shift may be as large as 60% of the total shift. The results of our work suggest that in order to obtain higher Voc, a possible recipe is the use of organic dyes with rather short π-spacers but strong donor groups, to improve the electrostatic effect on the TiO2 surface. The CT component could be tuned again by using stronger donor units and by exploiting new anchoring units able to transfer a larger amount of charge upon interacting with the semiconductor.
In our view, the present findings provide useful insights and a solid basis for obtaining a deeper understanding of the crucial interactions taking place at the dye–semiconductor interface, and establish a novel computational tool which can be used to design new dyes with improved functionality in DSCs.
Acknowledgements
We thank FP7-ENERGY 2010, Project “ESCORT” for financial support. ER thanks Fondazione Istituto Italiano di Tecnologia, Platform Computation, Project SEED 2009 “HELYOS” for financial support.Notes and references
- B. O'regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835–16847 CrossRef CAS.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS.
- Michael Graetzel Festschrift, a Tribute for this 60th Birthday: Dye Sensitized Solar Cells, ed. M. K. Nazeeruddin, Elsevier, Amsterdam, 2004, vol. 248 Search PubMed.
- H. Liyuan, I. Ashraful, C. Han, M. Chandrasekharam, C. Barreddi, Z. Shufang, Y. Xudong and Y. Masatoshi, Energy Environ. Sci., 2012, 5, 6057–6060 Search PubMed.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS.
- D. P. Hagberg, T. Marinado, K. M. Karlsson, K. Nonomura, P. Qin, G. Boschloo, T. Brinck, A. Hagfeldt and L. Sun, J. Org. Chem., 2007, 72, 9550–9556 CrossRef CAS.
- M. Grätzel, Acc. Chem. Res., 2009, 42, 1788–1798 CrossRef.
- A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474–2499 CrossRef CAS.
- M. Pastore, S. Fantacci and F. De Angelis, J. Phys. Chem. C, 2010, 114, 22742–22750 CAS.
- J. Liu, R. Li, X. Si, D. Zhou, Y. Shi, Y. Wang, X. Jing and P. Wang, Energy Environ. Sci., 2010, 3, 1924–1928 CAS.
- J. Liu, D. Zhou, M. Xu, X. Jing and P. Wang, Energy Environ. Sci., 2011, 4, 3545–3551 CAS.
- S. Ito, S. M. Zakeeruddin, R. Humphry-Baker, P. Liska, R. Charvet, P. Comte, M. K. Nazeeruddin, P. Péchy, M. Takata, H. Miura, S. Uchida and M. Grätzel, Adv. Mater., 2006, 18, 1202–1205 CrossRef CAS.
- S. Ito, H. Miura, S. Uchida, M. Takata, K. Sumioka, P. Liska, P. Comte, P. Pechy and M. Gratzel, Chem. Commun., 2008, 5194–5196 RSC.
- G. Zhang, H. Bala, Y. Cheng, D. Shi, X. Lv, Q. Yu and P. Wang, Chem. Commun., 2009, 2198–2200 RSC.
- J.-H. Yum, E. Baranoff, F. Kessler, T. Moehl, S. Ahmad, T. Bessho, A. Marchioro, E. Ghadiri, J.-E. Moser, C. Yi, M. K. Nazeeruddin and M. Grätzel, Nat. Commun., 2011, 3, 631 CrossRef.
- S. Rühle and D. Cahen, J. Phys. Chem. B, 2004, 108, 17946–17951 CrossRef.
- M. Pastore, E. Mosconi and F. De Angelis, J. Phys. Chem. C, 2012, 116, 5965–5973 CAS.
- J. Wiberg, T. Marinado, D. P. Hagberg, L. Sun, A. Hagfeldt and B. Albinsson, J. Phys. Chem. C, 2009, 113, 3881–3886 CAS.
- M. Miyashita, K. Sunahara, T. Nishikawa, Y. Uemura, N. Koumura, K. Hara, A. Mori, T. Abe, E. Suzuki and S. Mori, J. Am. Chem. Soc., 2008, 130, 17874–17881 CrossRef CAS.
- M. Pastore and F. D. Angelis, ACS Nano, 2009, 4, 556–562 CrossRef.
- S. Tatay, S. A. Haque, B. O'regan, J. R. Durrant, W. J. H. Verhees, J. M. Kroon, A. Vidal-Ferran, P. Gavina and E. Palomares, J. Mater. Chem., 2007, 17, 3037–3044 RSC.
- K. Sayama, S. Tsukagoshi, K. Hara, Y. Ohga, A. Shinpou, Y. Abe, S. Suga and H. Arakawa, J. Phys. Chem. B, 2002, 106, 1363–1371 CrossRef CAS.
- B. Burfeindt, T. Hannappel, W. Storck and F. Willig, J. Phys. Chem., 1996, 100, 16463–16465 CrossRef CAS.
- D. Liu, R. W. Fessenden, G. L. Hug and P. V. Kamat, J. Phys. Chem. B, 1997, 101, 2583–2590 CrossRef CAS.
- H. Tian, X. Yang, R. Chen, R. Zhang, A. Hagfeldt and L. Sun, J. Phys. Chem. C, 2008, 112, 11023–11033 CAS.
- A. Kay and M. Graetzel, J. Phys. Chem., 1993, 97, 6272–6277 CrossRef CAS.
- M. Planells, L. Pelleja, J. N. Clifford, M. Pastore, F. De Angelis, N. Lopez, S. R. Marder and E. Palomares, Energy Environ. Sci., 2011, 4, 1820–1829 CAS.
- Y. Bai, J. Zhang, D. Zhou, Y. Wang, M. Zhang and P. Wang, J. Am. Chem. Soc., 2011, 133, 11442–11445 CrossRef CAS.
- N. Koumura, Z.-S. Wang, S. Mori, M. Miyashita, E. Suzuki and K. Hara, J. Am. Chem. Soc., 2006, 128, 14256–14257 CrossRef CAS.
- Z.-S. Wang, N. Koumura, Y. Cui, M. Takahashi, H. Sekiguchi, A. Mori, T. Kubo, A. Furube and K. Hara, Chem. Mater., 2008, 20, 3993–4003 CrossRef CAS.
- J.-I. Nishida, T. Masuko, Y. Cui, K. Hara, H. Shibuya, M. Ihara, T. Hosoyama, R. Goto, S. Mori and Y. Yamashita, J. Phys. Chem. C, 2010, 114, 17920–17925 CAS.
- E. M. Barea, C. Zafer, B. Gultekin, B. Aydin, S. Koyuncu, S. Icli, F. F. Santiago and J. Bisquert, J. Phys. Chem. C, 2010, 114, 19840–19848 CAS.
- Y. Bai, J. Zhang, Y. Wang, M. Zhang and P. Wang, Langmuir, 2011, 27, 4749–4755 CrossRef CAS.
- J. Bisquert and I. Mora-Seró, J. Phys. Chem. Lett., 2010, 1, 450–456 CrossRef CAS.
- J. W. Ondersma and T. W. Hamann, J. Am. Chem. Soc., 2011, 133, 8264–8271 CrossRef CAS.
- G. Rothenberger, D. Fitzmaurice and M. Graetzel, J. Phys. Chem., 1992, 96, 5983–5986 CrossRef CAS.
- B. O'regan, M. Grätzel and D. Fitzmaurice, Chem. Phys. Lett., 1991, 183, 89–93 CrossRef CAS.
- G. Boschloo and D. Fitzmaurice, J. Phys. Chem. B, 1999, 103, 7860–7868 CrossRef CAS.
- G. Redmond and D. Fitzmaurice, J. Phys. Chem., 1993, 97, 1426–1430 CrossRef CAS.
- B. Enright, G. Redmond and D. Fitzmaurice, J. Phys. Chem., 1994, 98, 6195–6200 CrossRef CAS.
- S. Ardo and G. J. Meyer, Chem. Soc. Rev., 2009, 38, 115–164 RSC.
- K. Westermark, A. Henningsson, H. Rensmo, S. Södergren, H. Siegbahn and A. Hagfeldt, Chem. Phys., 2002, 285, 157–165 CrossRef CAS.
- S. Rühle, M. Greenshtein, S. G. Chen, A. Merson, H. Pizem, C. S. Sukenik, D. Cahen and A. Zaban, J. Phys. Chem. B, 2005, 109, 18907–18913 CrossRef.
- S. G. Yan and J. T. Hupp, J. Phys. Chem., 1996, 100, 6867–6870 CrossRef CAS.
- F. De Angelis, S. Fantacci, A. Selloni, M. Grätzel and M. K. Nazeeruddin, Nano Lett., 2007, 7, 3189–3195 CrossRef CAS.
- M. Pastore and F. De Angelis, Phys. Chem. Chem. Phys., 2012, 14, 920–928 RSC.
- H. Kusama, H. Orita and H. Sugihara, Langmuir, 2008, 24, 4411–4419 CrossRef CAS.
- Y. Tachibana, S. A. Haque, I. P. Mercer, J. E. Moser, D. R. Klug and J. R. Durrant, J. Phys. Chem. B, 2001, 105, 7424–7431 CrossRef CAS.
- P. Chen, J. H. Yum, F. D. Angelis, E. Mosconi, S. Fantacci, S.-J. Moon, R. H. Baker, J. Ko, M. K. Nazeeruddin and M. Grätzel, Nano Lett., 2009, 9, 2487–2492 CrossRef CAS.
- H. Kusama, H. Orita and H. Sugihara, Langmuir, 2008, 24, 4411–4419 CrossRef CAS.
- L. Belpassi, I. Infante, F. Tarantelli and L. Visscher, J. Am. Chem. Soc., 2008, 130, 1048–1060 CrossRef CAS.
- L. F. Roncaratti, L. Belpassi, D. Cappelletti, F. Pirani and F. Tarantelli, J. Phys. Chem. A, 2009, 113, 15223–15232 CrossRef CAS.
- L. Belpassi, M. L. Reca, F. Tarantelli, L. F. Roncaratti, F. Pirani, D. Cappelletti, A. Faure and Y. Scribano, J. Am. Chem. Soc., 2010, 132, 13046–13058 CrossRef CAS.
- G. Bistoni, L. Belpassi, F. Tarantelli, F. Pirani and D. Cappelletti, J. Phys. Chem. A, 2011, 115, 14657–14666 CrossRef CAS.
- L. Storchi, L. Belpassi, F. Tarantelli, A. Sgamellotti and H. M. Quiney, J. Chem. Theory Comput., 2010, 6, 384–394 CrossRef CAS.
- D. Cappelletti, E. Ronca, L. Belpassi, F. Tarantelli and F. Pirani, Acc. Chem. Res., 2012, 45, 1571–1580 CrossRef CAS.
- N. Salvi, L. Belpassi and F. Tarantelli, Chem.–Eur. J., 2010, 16, 7231–7240 CAS.
- K. Hara, Z.-S. Wang, T. Sato, A. Furube, R. Katoh, H. Sugihara, Y. Dan-Oh, C. Kasada, A. Shinpo and S. Suga, J. Phys. Chem. B, 2005, 109, 15476–15482 CrossRef CAS.
- D. P. Hagberg, J.-H. Yum, H. Lee, F. De Angelis, T. Marinado, K. M. Karlsson, R. Humphry-Baker, L. Sun, A. Hagfeldt, M. Grätzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2008, 130, 6259–6266 CrossRef CAS.
- D. P. Hagberg, T. Edvinsson, T. Marinado, G. Boschloo, A. Hagfeldt and L. Sun, Chem. Commun., 2006, 2245–2247 RSC.
- M. Pastore and F. De Angelis, ACS Nano, 2010, 4, 556–562 CrossRef CAS.
- P. Persson, R. Bergström and S. Lunell, J. Phys. Chem. B, 2000, 104, 10348–10351 CrossRef CAS.
- F. De Angelis, A. Tilocca and A. Selloni, J. Am. Chem. Soc., 2004, 126, 15024–15025 CrossRef CAS.
- M. J. Lundqvist, M. Nilsing, S. Lunell, B. Åkermark and P. Persson, J. Phys. Chem. B, 2006, 110, 20513–20525 CrossRef CAS.
- F. De Angelis, S. Fantacci, A. Selloni, M. K. Nazeeruddin and M. Graätzel, J. Phys. Chem. C, 2010, 114, 6054–6061 CAS.
- A. Vittadini, A. Selloni, F. P. Rotzinger and M. Grätzel, Phys. Rev. Lett., 1998, 81, 2954–2957 CrossRef CAS.
- S. Ito, M. K. Nazeeruddin, P. Liska, P. Comte, R. Charvet, P. Péchy, M. Jirousek, A. Kay, S. M. Zakeeruddin and M. Grätzel, Progr. Photovolt.: Res. Appl., 2006, 14, 589–601 CrossRef CAS.
- Y.-X. Weng, Y.-Q. Wang, J. B. Asbury, H. N. Ghosh and T. Lian, J. Phys. Chem. B, 2000, 104, 93–104 CrossRef CAS.
- M. Khoudiakov, A. R. Parise and B. S. Brunschwig, J. Am. Chem. Soc., 2003, 125, 4637–4642 CrossRef CAS.
- F. De Angelis, S. Fantacci and A. Selloni, Nanotechnology, 2008, 19, 424002 CrossRef.
- F. De Angelis, S. Fantacci, E. Mosconi, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. C, 2011, 115, 8825–8831 CAS.
- G. Te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. Van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- E. Mosconi, A. Selloni and F. De Angelis, J. Phys. Chem., 2012, 16, 5932–5940 Search PubMed.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, in Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- Z.-S. Wang and F. Liu, Front. Chem. China, 2010, 5, 150–161 CrossRef.
- A. Vittadini, A. Selloni, F. P. Rotzinger and M. Grätzel, J. Phys. Chem. B, 2000, 104, 1300–1306 CrossRef CAS.
- U. C. Singh and P. A. Kollman, J. Comput. Chem., 1984, 5, 129–145 CrossRef CAS.
- B. H. Besler, K. M. Merz and P. A. Kollman, J. Comput. Chem., 1990, 11, 431–439 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CD curves, PDOS curves, and additional fits. See DOI: 10.1039/c2ee23170k |
| This journal is © The Royal Society of Chemistry 2013 |