A review of techno-economic models for the retrofitting of conventional pulverised-coal power plants for post-combustion capture (PCC) of CO2
Ming Zhao*, Andrew I. Minett and Andrew T. Harris*
Laboratory for Sustainable Technology, School of Chemical & Biomolecular Engineering, The University of Sydney, NSW 2006, Australia. E-mail: simon.ming.zhao@gmail.com; andrew.harris@sydney.edu.au; Fax: +61-2-9351-2854; Tel: +61-2-9351-2926
First published on 22nd October 2012
Abstract
In this paper, we compare and contrast the four most promising (i.e. commercially viable in the near future) technologies for the post-combustion capture (PCC) of CO2 that can be retrofitted to a conventional pulverised-coal power plant. These are CO2 capture using: (i) chilled ammonia, (ii) alkali-metal carbonates, (iii) membranes and (iv) calcium looping. These four technologies are compared to the benchmark monoethanolamine (MEA) scrubbing process in terms of efficiency penalty and cost indicators. We first review the relevant CO2 capture chemistry and typical process flow schematics, and then discuss energy- and mass-balance considerations. We consider 18 published techno-economic studies on these technologies and highlight the key measures, including net CO2 capture rate, net power output, net plant efficiency, capital costs, cost of electricity and cost of CO2 avoided. Calcium looping technology results in the lowest efficiency penalty (4.6%-points) and cost of PCC (36.3% increase in levelised cost of electricity). In addition, the cost of CO2 avoided by employing calcium looping for PCC can be as low as 29 USD2010 per tCO2. On all three of these criteria, calcium looping performs more than twice as well as the benchmark MEA PCC process.
 Ming Zhao | Ming Zhao obtained his PhD in 2011 from the University of Sydney, Australia. He is currently a research fellow in the Laboratory for Sustainable Technology at the University of Sydney. He is also a major investigator of a large industrial project – “High efficiency post combustion capture of carbon dioxide using solid sorbents” co-funded by ANLEC and Xstrata Coal. His research includes nano-porous materials/composites for catalysis and CO2 capture, and catalyst-assisted, sorption-enhanced H2 production from solid fuels (coal or biomass). He is also interested in catalysed biomass conversion to fuels and high-value chemicals. |
 Andrew I. Minett | Associate Professor Andrew Minett received his PhD in Materials Chemistry from the University of Wollongong (UOW), in 2000. After a fellowship at the Max-Planck-Institut fuer Festkoerperforschung in Stuttgart, he joined the Molecular Electronics and Nanotechnology Group at Trinity College Dublin, as a senior research fellow (2002). In 2005, he was awarded a QEII Research Fellowship to return to Australia, taking up this position at the Intelligent Polymer Research Institute (UOW) and concurrent as a CI in the ARC Centre of Excellence for Electromaterials Science. In March 2011, A/Prof. Minett joined the School of Chemical and Biomolecular Engineering at the University of Sydney where he is co-director of the Laboratory for Sustainable Technology, actively pursuing answers to big-picture issues through a sustained effort in materials science and device engineering. |
 Andrew T. Harris | Prof. Andrew Harris is an Australian Research Council Future Fellow and Professor of Chemical and Biomolecular Engineering at The University of Sydney. He is the co-director of the Laboratory for Sustainable Technology, a multidisciplinary research centre with active research programmes in renewable energy, biomimicry and nanotechnology that span several traditional engineering disciplines. |
Broader contextCarbon capture and storage is one of the hottest research fields of Energy and Environmental Science. The only drive for carbon-capture research is to decrease the cost related to the operation of CO2 capture devices. This work gives a clear view on where we have been so far in the R&D field and how we could move forward towards economically acceptable CO2 capture technologies. We classify post-combustion CO2 capture technologies as being benchmark, near-term commercially viable technologies (reviewed) and “next-generation” options (no in-depth analysis). For the benchmark and selected promising technologies, we not only identify the relevant CO2 capture chemistry and technological characteristics, but also review a wide range of economic models for these technologies highlighting the key measures including net CO2 capture rate, net power output, net plant efficiency, capital costs, cost of electricity and cost of CO2 avoided. A cross-technology comparison shows that a calcium-looping process provides an economic pathway as a near-term alternative to current MEA-based PCC processes. |
1 Introduction
Global efforts to limit emissions from coal-fired power plants, the largest stationary point-source emitters of greenhouse gases to the atmosphere,1,2 include applying CCS (carbon capture and storage) to the process and denying requests to add new coal-fired power plants that lack CO2 controls.3,4 The International Energy Agency (IEA) suggests that, by 2050, global emissions of CO2 from all energy-related technologies be reduced to half their 2007 levels (29 Gt CO2 per annum) in order to stabilise global warming. Approximately 10% of this reduction could be achieved by applying CCS to power plants.5Currently, CCS from power plants can be realised via three general routes: pre-combustion, oxy-fuel combustion and post-combustion.6 Among these, post-combustion capture (PCC) technologies are easiest to retrofit, and maximise the use of existing power-generation infrastructure.7 Furthermore, this end-of-pipe technology is sufficiently flexible to be switched off during periods of peak electricity demand or high market prices. One challenge for this process, however, is the low CO2 concentration (4–14% by volume) present in the flue gas from pulverised-coal power plants. This means that a large volume of gas has to be handled, and that powerful chemical solvents or adsorbents must be used to overcome the low partial pressure of CO2.8
Although most PCC technologies are still at the R&D (Research & Development) stage, amine-based CO2 scrubbing has been commercially demonstrated and thus dominates the current PCC market.9 Nevertheless, due to the high energy penalty and footprint of amine scrubbers,10 substantial technological improvements are needed to lower both the net efficiency penalty and incremental costs (including CO2 capture, compression, and auxiliary energy consumption) of PCC to meet the carbon sequestration program goal, set by U.S. Department of Energy, of capturing 90% of emitted CO2 while increasing the cost of electricity (COE) by <20%.11
Fig. 1 illustrates the potential of the major post-combustion techniques for future commercialisation and cost reduction.6 CaO/CaCO3 looping is among the promising pathways to low-cost sorbents that could reach technological maturity in the near future.12 Other techniques that are near commercialisation include chilled ammonia, dry alkali-metal carbonates, and membrane systems.13–15 On the other hand, ‘next-generation technologies’, such as those based on metal–organic frameworks (MOFs), zeolites, carbons, ionic liquids and enzymes, have been the subjects of fewer studies at the pilot stage, and require significant R&D efforts before they can be used for large-scale production.14 These technologies are not discussed further here.
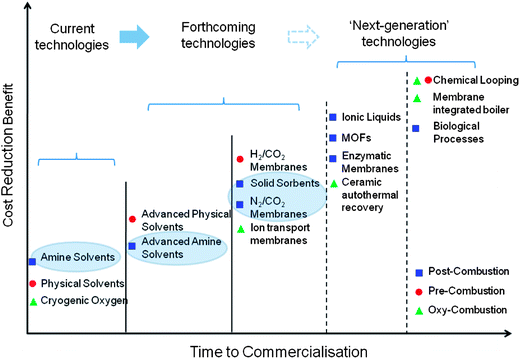 | ||
| Fig. 1 Current and future CCS technologies (adapted from ref. 7). Technologies in ellipses are considered here. | ||
A preliminary techno-economic analysis of emerging technologies is crucial to informing the R&D efforts that are presently investigating a wide range of CO2 capture options.16 Numerous modelling studies have focused on the cost and performance of a particular technology as compared to the well-defined benchmarks of “current technologies”, most often amine scrubbing. However, a comprehensive comparison of techno-economic indicators across the mature PCC technologies (including those forthcoming in the near term) is lacking.
This review deals with the critical issues that determine the efficiency penalty and cost increment of post-combustion CO2 capture. An overview of the technological fundamentals is presented, followed by a summary of published, robust techno-economic studies. The results of these studies are compared in order to draw conclusions about the cost and power plant efficiency penalty associated with each PCC technology.
2 Methodology
2.1 Technologies of interest
Adsorption of CO2 by monoethanolamine (MEA) is the benchmark PCC process, and has been used to remove CO2 from exhaust streams for over 80 years.17 Well-defined engineering-economical calculations are available for this system,1,10,18–22 and therefore the pulverised-coal power station retrofitted with an MEA scrubber is used here as a benchmark to assess the commercial feasibility of four other promising technologies, denoted “future” PCC processes: (i) chilled ammonia, (ii) alkali-metal carbonates, (iii) membranes and (iv) CaO-based sorbents (also known as calcium looping). Chemically, these CO2 capture technologies fall into three categories: chemical absorption, adsorption and membrane separation.2.2 Review of techno-economic studies
The gross size (MW h) of the reference power plant was assumed to be equal to that of the plant with PCC, but the net power output could differ significantly (negatively or positively) between the reference and PCC plants when PCC assemblies extracted or compensated steam cycles compared to the reference plant turbines. Thus, for reports that considered a fixed net plant output, the gross size was adjusted proportionally until it was equal for the reference and retrofitted plants.
All currencies other than USD were converted using the average exchange rate from the year of the cost data,23 and then updated to 2010 using the Chemical Engineering Plant Cost Index (CEPCI);24 conversions between lower (LHV) and higher heating value (HHV) thermal plant efficiencies were based on IEA conversions, and used a 5% difference for coal-fired plants.18
Despite being widely discussed in the literature, capital costs lack a universal metric. In this study, capital costs were considered to include engineering, procurement and construction (EPC) costs and fixed operating costs (FOC), but to exclude the interest accrued during construction and the owner's costs. The levelised cost of electricity (LCOE) is therefore expressed as:
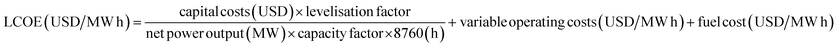 | (1) |
The cost of CO2 avoided (eqn (2)), rather than the cost of CO2 captured or removed, has been considered here. The former is larger than the latter because the energy required to operate the CO2 capture system increases the amount of CO2 emitted per MWe. In evaluating Case 9, in which only the cost of CO2 captured was given,25,26 the cost of CO2 avoided was calculated using eqn (2).
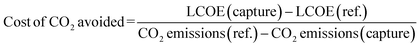 | (2) |
3 Literature review
3.1 Benchmark PCC process: amine scrubbing
In the amine scrubbing process, the chemical absorption of CO2 is based on the reversible reactions of acidic gases (CO2, SO2, etc.) with alkaline absorbents (shown for CO2 in eqn (3)–(5)). Amine absorbents are generally used in aqueous solution and MEA (monoethanolamine), a kind of primary amine (RNH2; for MEA, R = HO(CH2)2–), is the most popular amine used in PCC.| CO2 + H2O ⇌ HCO3− + H+ | (3) |
| RNH2 + H+ ⇌ RNH3+ | (4) |
| RNH2 + CO2 ⇌ RNHCOO− + H+ | (5) |
MEA is the most basic among the amines used for PCC, and presents the highest CO2 carrying capacity by mass among these amines because of its relatively low molecular weight. Typically, a 30 wt% MEA solution is used in PCC, and its reactivity toward CO2 drops as the temperature rises. A typical rich loading is ∼0.45 mol CO2 per mol MEA, at 50 °C; a lean loading can be as little as ∼0.21 mol CO2 per mol MEA, at 130 °C.27 Assuming that ∼50% of the MEA is regenerated, this process can capture more than 90% of the CO2 in the flue gas, producing a regeneration product stream that is >99% CO2.17
Although it removes CO2 effectively, MEA is corrosive and is gradually volatilised; these problems require the use of equipment made from more expensive construction materials. Furthermore, aqueous MEA is very sensitive to SO2, which it binds to form thermally stable salts,17 and also to O2, in the presence of which ammonia and peroxide are generated and can corrode equipment as well as significantly lower the separation performance of MEA.28 Therefore, practically, a very efficient FGD, or an extra gas washer (typically using 2 wt% aqueous Na2CO3 (ref. 29)), is used to hold the scrubber-inlet SO2 concentration to 10–30 mg Nm−3 (3.8–11 ppm)30 and a simultaneous de-oxy catalyst system is introduced to remove most of the oxygen. These extra components in the capture system, along with the additional consumption of chemicals and the handling of the corresponding waste stream, all decrease the economic performance of this process.
Fig. 2 shows a representative process schematic for amine-based PCC, which has been depicted in a number of recent studies.17,21,22,27,29,31–34 Ignoring minor differences among the models, the major energy- and chemical-consuming parts of the configuration, shown in Fig. 2, are: (1) an absorber, running at 30–40 °C and 1 bar (positioned upstream, with blowers and direct contact coolers (DCC)); (2) a stripper, running at 100–120 °C, up to 2 bar, and equipped with pumps, a reboiler, a reclaimer and a CO2 compressor; and (3) intermediate components, including the heat exchanger, mechanical filter, solvent cooler and pumps.
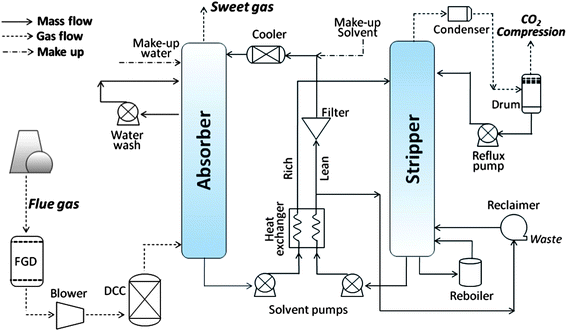 | ||
| Fig. 2 Schematic of the MEA-based PCC process. FGD = flue gas desulfurisation; DCC = Direct Contact Cooler. | ||
Flue gas enters the capture apparatus after passing through the FGD and DCC and exits as sweet gas after making counter-current contact with the MEA solution in the absorber, which is equipped with a water washing unit to return any solvent carried by the gas flow (ensuring <1 ppmv solvent is emitted to atmosphere).35 The rich solvent from the bottom outlet of the absorber is pumped through a heat exchanger and then injected into the top of a stripper that is kept at the temperature required for CO2 release by a counter-current steam flow generated from the reboiler. The lean solvent enters the same heat exchanger to be cooled by the rich solvent flow, while the rich solvent is preheated before reaching the stripper, thereby decreasing the energy penalty of the whole system.
The H2O–CO2 mixture that leaves the stripper is sent to the overhead condenser, which captures most of the water vapour to give a highly pure CO2 stream (>99.5 mol%) that is compressed to 110 bar so that it can to be stored as a liquid.27 The condensed water, meanwhile, is returned as reflux to the counter-current stripper. In some designs, the heat from the condensed steam is recovered in the reboiler.
Another energy-consuming part of the amine-based PCC apparatus is the reclaimer. This is operated, as an intermittent batch process, in parallel to the reboiler in order to control the build-up of heat-stable salts in the lean solvent.36 Nevertheless, a make-up solvent flow is still needed to maintain the expected rate of CO2 capture, and water must be compensated to the lean solvent. The disposal of chemical waste from the reclaimer also adds cost.
Several recent techno-economic models of MEA-based PCC power plants have been published. The key measures from five such studies are summarised in Table 1.
| Literature | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | |
|---|---|---|---|---|---|---|
| 19b | 20c | 21d | 1e | 1e | ||
| a Only the first affiliation for each publication is listed (this is true for all cases in this review). Abbreviations are defined in the Glossary.b The original cost data was in USD2005.c The original cost data was in USD2007.d The original cost data was in Euro2006.e The original cost data was in RMB2010 and AUD2010, respectively.f SCPC = Super Critical Pulverized Coal.g The suffix ‘ref.’ refers to measures for the reference plants without CO2 capture, and ‘cap.’ refers to those for the target plants with PCC.h With respect to the reference case. | ||||||
| Organisationa | CMU | EPRI | TNO | TPRI | CSIRO | |
| Location | USA | USA | EU | China | Australia | |
| Coal type | Bitcoal | Bitcoal | Bitcoal | Bitcoal | Black coal | |
| Steam turbine type | SCPCf | SCPCf | SCPCf | SCPCf | SCPCf | |
| Plant size (gross, MWe) | 575 | 600 | 600 | 600 | 600 | |
| Capacity factor (%) | 75 | 80 | 90 | 90 | 90 | |
| Designed CO2 capture rate (%) | 90 | 85 | 90 | 90 | 90 | |
| CO2 emissions (kg per MW h) | ref.g | 811 | 836 | 772 | 806 | 810 |
| cap.g | 107 | 126 | 112 | 111 | 105 | |
| Net power output (MWe) | ref. g | 528 | 600 | 575 | 570 | 571 |
| cap.g | 493 | 550 | 399 | 412 | 420 | |
| Net plant efficiency (LHV, %) | ref.g | 41.4 | 40.0 | 45.0 | 43.5 | 40.5 |
| cap.g | 31.5 | 29.1 | 31.0 | 31.4 | 29.8 | |
| Efficiency penalty (%-points)h | 9.9 | 10.9 | 14.0 | 12.1 | 10.7 | |
| Capital costs (USD per kWe) | ref.g | 1696 | 2104 | 1420 | 699 | 2522 |
| cap.g | 2759 | 3516 | 2668 | 1296 | 5033 | |
| LCOE (USD per MW h) | ref.g | 62 | 77 | 46 | 44 | 56 |
| cap.g | 104 | 127 | 83 | 65 | 120 | |
| Cost of CO2 avoided (USD per tCO2) | 58 | 71 | 57 | 30 | 91 | |
3.2 Future PCC processes
| (NH4)2CO3(aq) + CO2(g) + H2O(l) ↔ 2NH4HCO3(aq) | (6) |
Although the use of aqueous ammonia is contraindicated by its high volatility and the emission of corrosive and toxic NH3 gas, a promising variation of the wet ammonia process was reported in 2006.38 In it, scrubbing occurs near the condensation point of NH3 to minimise volatilisation and enhance the exothermic CO2 binding process. Fundamental studies regarding the thermodynamics of (NH4)2CO3/NH4HCO3 conversion, the mass and heat balance, and the design of the absorber/stripper are available,41–43 and a medium-scale pilot project has recently been demonstrated by Alstom and EPRI,44 who anticipated commercialising this technology in 2011.45
Fig. 3 shows a typical CAP process schematic that has been adopted in a range of techno-economic assessments.39–41 The main difference between the CAP and MEA-based processes is that the former requires lower operating temperatures in the absorber (<10 °C); this necessitates energy-intensive DCCs (a cooling stream, water at 20 °C, that cools the gas to 30 °C) and chillers (a 0.55 kW per ton refrigeration stream that cools the gas to <10 °C).40 To ensure that the feed gas entering the absorber is sufficiently cleaned and cooled, investigators from Alstom39 used a double-DCC upstream design that differs from the process shown in Fig. 3. Notably, this design was subsequently adopted by some authors for cost estimates,40,41 and the techno-economic data published in all three cases by these authors is summarised in Table 2.
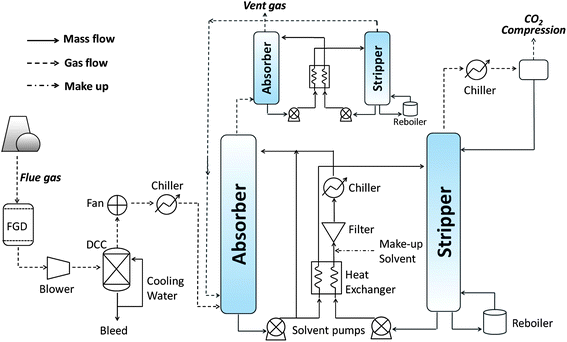 | ||
| Fig. 3 Schematic of chilled-ammonia PCC process. FGD = flue gas desulfurisation; DCC = Direct Contact Cooler. | ||
| Literature | Case 6 | Case 7 | Case 8 | |
|---|---|---|---|---|
| 39a | 40b | 41c | ||
| a This is Case 3 of the original NETL report, in which ALSTOM's CAP process was adopted; the original cost data was in USD2006.b The original cost data was in USD2010.c The original cost data was in Euro2010.d Name of the corresponding author.e This measure was not reported in the original paper.f SCPC = Super Critical Pulverized Coal; USCPC = Ultra Super Critical Pulverised Coal.g The suffix ‘ref.’ refers to the measures for the reference plant without CO2 capture, and ‘cap.’ refers to those of the target plant with PCC.h With respect to the reference case.i Calculated from the original total capital cost (divided by net power output). | ||||
| Organisation | NETL | CMU | Valentid | |
| Location | USA | USA | EU | |
| Coal type | Bitcoal | Bitcoal | —e | |
| Steam turbine type | SCPCd | SCPCd | USCPCf | |
| Plant size (gross, MWe) | 550 | 550 | 758 | |
| Capacity factor (%) | 85 | 85 | —e | |
| Designed CO2 capture rate (%) | 90 | 85 | 85 | |
| CO2 emissions (kg per MW h) | ref.g | 916 | 802 | 679 |
| cap.g | 111 | 120 | 97 | |
| Net power output (MWe) | ref.g | 549 | 550 | 758 |
| cap.g | 445 | 559 | 614 | |
| Net plant efficiency (LHV, %) | ref.g | 41.8 | 41.2 | 45.2 |
| cap.g | 30.0 | 29.4 | 36.6 | |
| Efficiency penalty (%-points)h | 11.8 | 11.8 | 8.6 | |
| Capital costs (USD per kWe) | ref.g | 1412 | 1900 | 1922i |
| cap.g | 2407 | 3534 | 2739 | |
| LCOE (USD per MW h) | ref.g | 56 | 61 | 75 |
| cap.g | 89 | 104 | 103 | |
| Cost of CO2 avoided (USD per tCO2) | 41 | 69 | 49 | |
A spray tower absorber is normally used for CO2 scrubbing, as it can accommodate both aqueous (NH4)2CO3/NH4HCO3 and slurries containing solid NH4HCO3. A second absorber–stripper system is introduced atop the main absorber to remove NH3 carried by vent gas because, even though the absorber is kept at 1 °C, the amount of ammonia carried in the vent gas may still be several times the acceptable environmental standard.37
The rich solvent or slurry is pressurised from 1–2 bar to ∼30 bar and then heated from 10 to ∼130 °C. The heating favours the reverse of reaction (6) and the increased pressure reduces the evaporation of ammonia and water. The stripper exhausts CO2 at 130 °C and 30 bar. The heat recycled from the CO2 stream can be used to heat the vent gas from the absorber from 15 to 55 °C, which reduces visible plume formation and prevents ammonia odours from reaching the ground. As CO2 is already pressurised to 30 bar when released, less energy is needed for final compression to 120 bar.
| M2CO3(s) + CO2(g) + H2O(g) ⇌ 2MHCO3(s) | (7) |
The stoichiometric CO2 carrying capacities of Na2CO3 and K2CO3 reach 9.43 and 7.23 mol CO2 per kg sorbent, respectively, making this technology promising for PCC processes. However, they have to be loaded on an inert substrate in order to enhance their attrition resistance before they can be used in large-scale adsorbers, and this substantially decreases the CO2 capture capacity (by ∼70 to 90%).47 A wide range of support materials has been reported; these include alumina, activated carbon, zeolite, magnesium oxide and silica.48–50
Na2CO3 supported on alumina is the alkali-metal carbonate most commonly applied in PCC because of its proven stability in multiple applications. When this sorbent is used, the regenerator temperature must be limited to 150 °C to avoid permanent deactivation (via the formation of NaAl(CO3)(OH)2), despite that higher temperatures are more favourable for the endothermic decarbonation (eqn (7)).36 Research Triangle Institute (RTI, in NC, USA) developed a Na2CO3/Al2O3 sorbent with an active carbonate fraction of 10–40 wt%, a BET surface area of 100–120 m2 g−1 and a DI (Davison Attrition Index) of 12, and applied it in a demonstration PCC process in 2007.26 This technology captured >90% CO2 in the pilot project. Although no detailed schematic was given in the techno-economic study published by RTI,26Fig. 4 shows the process and highlights the major energy- and chemical-consuming parts.
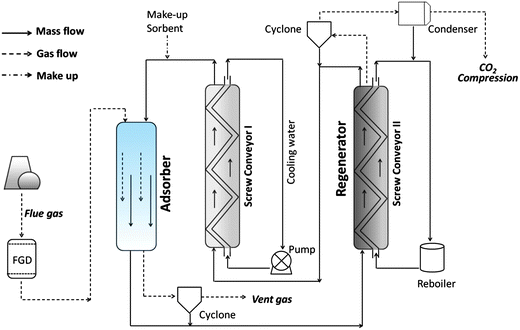 | ||
| Fig. 4 Schematic flow of dry-carbonate PCC process. FGD = flue gas desulfurisation. | ||
Neither fixed-bed nor dense-phase fluidised-bed systems are suitable for the dry carbonate process because they produce excess heat during the exothermic adsorption (eqn (7)); this significantly increases the bed temperature and thus mitigates the CO2 capture capacity.51 A concurrent entrained-bed adsorber is usually employed and the “gravity-based” design causes no apparent decrease in the pressure drop of gas flow, thus reducing the energy required by blowers.
The desulphurised gas (comprised mainly of CO2, H2O, and N2) and cooled sorbent flow concurrently through the adsorber and their contact removes most of the CO2. The gas flow passes through a cyclone and is vented to atmosphere. The CO2-loaded sorbents from the bottom of the entrained bed are transported by an up-flow screw conveyor and, simultaneously, steam generated from a reboiler or separated from the power plant is introduced into the hollow-shaft screws. The sorbents are heated to 120 °C, releasing CO2 that passes through another cyclone before final compression. The condensed water is reused in the reboiler.
Another screw conveyor with the same design is used to cool and transport the refreshed sorbents before they are returned to the top of the adsorber. The cooling medium is domestic water at ambient temperature. Due to degradation, the dry carbonate sorbent is continuously compensated, with the water make-up, to the adsorber (Fig. 4).
| Literature | 25,26a | Key measures | ref.b | cap.b |
|---|---|---|---|---|
| a The original cost data was in USD2005.b ‘ref.’ refers to the measures for the reference plant without CO2 capture, and ‘cap.’ refers to those of the target plant with PCC.c Not applicable.d Cost of CO2 avoided was obtained from eqn (2). | ||||
| Organisation | EPRI, RTI | CO2 emissions (kg per MW h) | 774 | 84 |
| Location | USA | Net power output (MWe) | 462 | 381 |
| Fuel type | Illinois # 6 | Net plant efficiency (LHV) (%) | 42.6 | 32.2 |
| Plant type | SCPC | Efficiency penalty (%-points) | —c | 10.4 |
| Plant size, gross | 500 MWe | Capital costs (USD per kWe) | 1418 | 2150 |
| Capacity factor | 65% | LCOE (USD per MW h) | 65 | 88 |
| CO2 capture rate | 90% | Cost of CO2 avoided (USD per tCO2) | —c | 34d |
A gas-separation membrane is a thin layer, normally less than half a micrometer thick, through which selective transports occur via solution-diffusion. The driving force for gas separation is usually a pressure gradient. The key parameters56 describing a membrane material are defined in eqn (8) and (9).
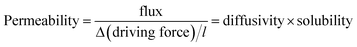 | (8) |
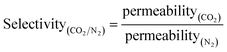 | (9) |
The Barrer, equal to 10−9 mm3 (STP) mm−2 s−1 mm Hg−1, is a common unit of permeability. In this study, for specific membranes, permeance (obtained by dividing permeability by the thickness of membrane (l)) is used, and is expressed in GPU, i.e. gas permeance unit (10−10 mm3 (STP) mm−2 s−1 mm Hg−1).
Generally, a more selective membrane enables a more efficient process that needs less driving force and thus decreases energy costs; also, if flux is higher, a smaller membrane area is required, and the capital requirements are lower.64 Therefore, membrane selectivity and permeance are key to the cost of a membrane-separation process, although they also need to be balanced against factors such as the durability and mechanical integrity of the membrane.54
Current commercial membranes, mostly based on polymeric materials, are applied in the natural gas and chemical industries to separate H2–CH4, CO2–CH4, O2–N2 and N2–CH4 mixtures, but are generally less effective in separating CO2 and N2, which have similar diffusivity values (the kinetic molecular diameters of CO2 and N2 are 0.33 and 0.36 nm, respectively56). The commercial glassy polymer used for gas separation is slightly more selective than the commercial rubbery polymer, but suffers from insufficient permeance; whereas the converse is true for the rubbery polymer (Fig. 5). This produces a lower bound in the Robeson-type plot, below which insufficient trade-offs between permeance and selectivity occur.
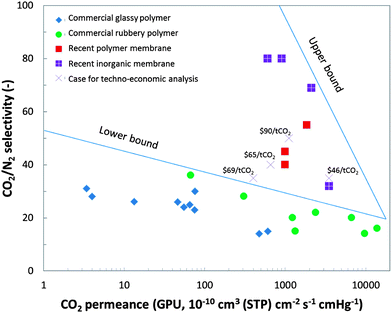 | ||
| Fig. 5 Robeson-type plot of CO2/N2 selectivity versus CO2 permeance for promising PCC membranes and commercial polymer membranes. The data for commercial gas separation membranes is taken from Tables 4 (glassy) and 5 (rubbery) of article,56 and the GPU data is calculated from the original Barrer data for a 0.1 μm thick membrane. The recent polymer membranes for PCC are from articles,57–59 the recent inorganic membranes for PCC are from articles,60–63 the data sources for the techno-economic cases are those summarised in Table 4 (vide infra), and the numbers above the ‘×’ labels give the cost of CO2 avoided. | ||
The polar bonds in CO2 are more attracted to the polar groups in membranes than N2 is, providing an approach to improving the solubility selectivity of CO2 over N2. Recent polymer membranes with modified structures have shown much better permeance–selectivity trade-offs beyond the lower bound, but are still restrained within an empirical upper bound supposed by Robeson52 (Fig. 5). Three representative polymers are marked in Fig. 5, including the cardo polyimide hollow-fibre membrane produced by the RITE Institute (Japan),57 the Polaris™ membrane of MTR Inc. (USA)58 and the ultra-thin membrane based on the Polyactive® PBT–PEO (PBT = poly(butylene terephthalate), PEO = poly(ethylene oxide)) block copolymer manufactured by GKSS Research Centre Geesthacht GmbH (Germany).59
Fig. 5 also shows permeance–selectivity relationships for recently developed microporous inorganic membranes, which are significantly more selective than solution-diffusion polymers. However, the sol–gel-based synthesis of silica and zeolite membranes normally means a high capital cost that will be exacerbated by the difficulties in reproducibly fabricating defect-free selective layers (e.g. SAPO-34 zeolite, with a regular pore size of 0.38 nm (ref. 63)). Additional limitations may be attributed to the poor hydrothermal stability of both pure and functionalised silicas and silicates.61 Comparatively, polymeric membranes are more promising for cheap, mass manufacture, easy quality control and hydrothermal stability,53 and their poorer selectivity might be counteracted using a multi-stage design.
Considering the low CO2 concentration (∼14 mol%) in flue gas at ambient pressure, a superb CO2/N2 selectivity (>200),55 an extra-large membrane area (∼150 × 106 m2)65 and a high-power compressor that maintained the flue gas pressure at >10 bar (ref. 58) would be required to achieve 90% CO2 removal and 95% pure captured CO2 stream from a 1000 MWe power plant. The resulting high unit price and total capital investments for membrane modules, as well as the high energy consumption, are far from practical.
Both parametric studies65–67 and completed projects68,69 have addressed this challenge, and Fig. 6 displays an economically feasible design that is abstracted from these studies. In it, the treated wet gas that emerges from the desulfurisation and particle-removal units must be compressed to create a pressure gradient between the feed and permeate sides of the first membrane module (M1, typically a cross-flow membrane). Generally, the lower a single module's stage-cut (defined as the molar ratio of permeate to feed flow), the less membrane area and compression energy are required. In practice, if the overall CO2 stage-cut for a multi-stage process is 90%, that of M1 varies between 50 and 70%.68,69
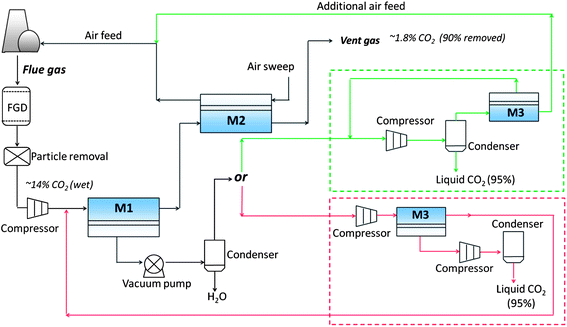 | ||
| Fig. 6 Economically feasible process design schematic for membrane post-combustion CO2 capture. M = membrane module, FGD = flue gas desulfurisation. | ||
M2 is a counter-flow module that uses combustion air as a sweep gas, generating a “free” driving force to extract CO2 from its feed flow, which in this case is the retentate flow from M1. The stage-cut of M2 depends upon the design of M1. In any case, its retentate will satisfy the requirement for emission to atmosphere (∼1.8 mol% CO2, ∼90% of original feed CO2 removed). The small fraction of CO2 carried through the membrane by the sweep air will enter the coal combustor, making a loop.
The design of module M3 also depends on the stage-cut of M1. A lower CO2 stage-cut (50%) at M1 gives a higher-concentration CO2 permeate (85 mol% after H2O condensation) that can be delivered directly to a compressor that liquefies CO2 directly. The overhead unwanted species (CO2, N2, O2, etc.) pass a small membrane module (M3, cross-flow). The retentate of this module makes up the combustion feed air; whereas the permeate enters a loop and is returned for liquefaction. Comparatively, a higher CO2 stage-cut (70%) at M1 gives only 65% CO2 in the permeate stream; this cannot be liquefied without further purification. So, after compression to ∼5 bar, the flow will be further separated with the ∼95% pure CO2 permeate that is ready for liquefaction. The retentate will rejoin the CO2 capture process as feed to M1.
With the multistage design, the membrane area required for 90% CO2 removal and a 95% pure CO2 stream can generally be decreased to a few million m2 or less.58,65 However, the need to drive compressors, vacuum pumps, blowers, pretreatment devices and other auxiliary equipment contributes significant cost due to energy consumption, which reaches ∼20% of the power generated.58,69,70
Several field-development membrane-separation projects have been completed, and well-studied techno-economic analyses exist (Table 4), although no pilot-scale integration into an operating power plant has been described. Cases 10, 12 and 13 consider three completed PCC membrane projects from DOE/NETL (USA), who have developed hollow-fibre Ni–silica membranes (NSF,71), spiral-wound Polaris™ membranes (MTR,68) and hollow-fibre polycarbonate membranes (RTI,69); Case 11 is a techno-economic study by IEF-3 Germany,67,70 who applied a membrane developed by GKSS.59
| Literature | Case 10 | Case 11 | Case 12 | Case 13 | |
|---|---|---|---|---|---|
| 71a | 67,70b | 68c | 69d | ||
| a This is Case 3 in Table 6.1 of ref. 71; the original cost data was in USD2000.b The two-stage membrane configuration from ref. 70 (1st: 6.37 × 106 m2, 2nd: 0.08 × 106 m2) is adopted in this study to achieve 90% CO2 capture rate; data for the reference plant is from ref. 67 and the cost data are calculated according to the method used by the author; the original cost data was in Euro and the year is assumed the same as submission time, 2009.c This is Case 5A (MTI membrane) of ref. 68; the original cost data was in USD2007.d The original cost data was in USD2007.e The suffix ‘ref.’ refers to the measures of the reference plants without CO2 capture, and ‘cap.’ refers to those of the target plants with PCC.f Data is not found in the original publication.g With respect to the reference case.h Calculated using eqn (2). | |||||
| Organisation | NSF | IEF-3 | MTR | RTI | |
| Location | USA | EU | USA | USA | |
| Coal type | Illinois#6 | Kleinkopje | Illinois#6 | Illinois#6 | |
| Steam turbine type | SCPC | SCPC | SCPC | Sub-PC | |
| Plant size (gross, MWe) | 491 | 600 | 580 | 583 | |
| Capacity factor (%) | 65 | 70 | 85 | 85 | |
| Designed CO2 capture rate (%) | 90 | 90 | 90 | 90 | |
| CO2 emissions (kg per MW h) | ref.e | 776 | —f | 760 | 807 |
| cap.e | 108 | —f | 87 | 80 | |
| Net power output (MWe) | ref.e | 462 | 550 | 550 | 550 |
| cap.e | —f | —f | 461 | 398 | |
| Net plant efficiency (LHV, %) | ref.e | 42.6 | 45.9 | 41.4 | 38.7 |
| cap.e | 30.6 | 34.5 | 34.4 | 26.5 | |
| Efficiency penalty (%-points)g | 12.0 | 11.4 | 7.0 | 12.2 | |
| Capital costs (USD per kWe) | ref.e | 1804 | 1407 | 1727 | 1624 |
| cap.e | 2551 | 2770 | 2627 | 2093 | |
| LCOE (USD per MW h) | ref.e | 72 | 51 | 62 | 67 |
| cap.e | 115 | 98 | 93 | 117 | |
| Cost of CO2 avoided (USD per tCO2) | 65 | 90 | 46 | 69h | |
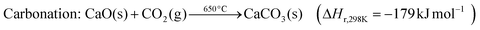 | (10) |
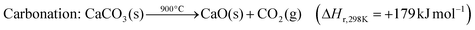 | (11) |
The exothermic carbonation reaction, which takes place around 650 °C, produces excess heat that could be integrated into the power plant or used in a separate steam cycle.74 The fluidised bed reactor (FBR) is a suitable technology for the carbonation–calcination cycle because of its mature design and because it favours gas–solid contact at a uniform bed temperature; in addition, FSB has been well-established and widely deployed on a large scale.74–76
CaO has a stoichiometric CO2 carrying capacity of 17.8 mol CO2 per kg sorbent, and it has further advantages such as its availability in Nature (as limestone) and low cost. However, the performance of CaO derived from calcined lime is intrinsically restrained by a substantial decay of CO2 carrying capacity over multiple cycles; the lowest residual activity has been reported ∼10%.12 The major reasons for this include the sintering of active CaO, its poisoning by sulphation/sulphidation reactions with SOx, and sorbent loss due to attritions.77–79 Even so, the CO2 carrying ability of a degraded CaO sorbent is competitive with amine-based solvents or sorbents. Furthermore, continuous make-up of CaO from cheap, naturally available sources increases the stable residual reactivity to 23%.80
Further adjustments can also be made to address the problems of CaO capacity and material loss. Firstly, the low residual reactivity of CaO is less of a problem under realistic conditions when steam is present in the flue gases81,82 because of the demonstrated significant increases in sorbent activity measured under realistic conditions of CO2 adsorption/desorption.83 Secondly, a more stable CaO sorbent could be achieved by tailoring the structure84–86 or doping with inert materials;87–91 the latter also enhances resistance to attrition. More notably, a powdered-coal power plant retrofitted with calcium looping PCC could take advantage of the fine CaO particles lost to attrition and purging from FBRs by integrating them into cement production. This can substantially decrease the operating costs from sorbent loss,92 as has been demonstrated experimentally.93 This is why most of the relevant techno-economic models have assumed the cost of sorbents to be zero (vide infra).16,80,94
Shimizu's PCC process for applying calcium looping has been widely adopted in subsequent investigations.80 A CFB (circulating fluidised bed) carbonator and an oxy-fuel CFB calciner are typically considered. The flue gas fully contacts the refreshed sorbents and loses CO2 in the carbonator at ∼650 °C before it is vented to the atmosphere; the calciner runs as a fluidised-bed combustor directly burning powdered coal in pure oxygen that is injected from an air separation unit (ASU). CO2 is released for compression treatment and the sorbents are regenerated. Promising results have been achieved in an EU-based industrial pilot-scale project, “CaOling”, which has demonstrated long-life-time (>1500 h) capture capacities of >90% and >95% for CO2 and SO2, respectively, using two interconnected CFBs at operating continuously at the MWe scale.95
The primary concerns surrounding high-temperature CO2 capture involve heat demands and integration. Fortunately, the current configuration has been designed to optimally integrate the excess heat into new steam cycles, which can potentially control the energy penalty at an economically acceptable level. This conclusion is mainly based on studies from four active research groups: those of Romeo,13,16,80,98 Grasa,99,100 Epple,97,101,102 and Zhai.94 A state-of-the-art process design, abstracted from these works, is shown in Fig. 7, and the technical and economical performance of this process is summarised in Table 5.
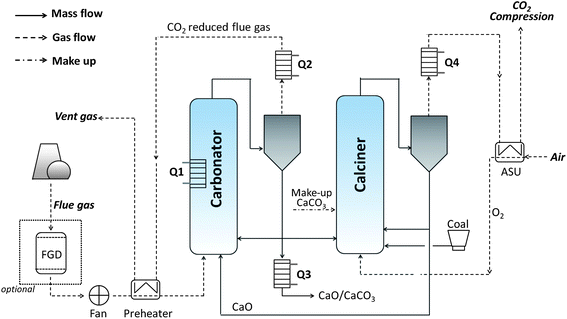 | ||
| Fig. 7 Schematic of calcium looping PCC process. FGD = Flue Gas Desulphurisation unit, ASU = Air Separation Unit. | ||
| Literature | Case 14 | Case 15 | Case 16 | Case 17 | Case 18 | |
|---|---|---|---|---|---|---|
| 16a | 80b | 94c | 96d | 97d | ||
| a The best estimate of the original report is adopted rather than the optimistic or pessimistic sets; the original cost data was in Euro2005.b The original cost data was in Euro2007.c This is Case 4 of the original publication; the original cost data was in Euro2009.d Neither Case 17 nor 18 gave economic estimates, but they are included in this review to show the efficiency performance of a larger power plant.e The suffix ‘ref.’ refers to the measures of the reference plants without CO2 capture, and ‘cap.’ refers to those of the target plants with PCC.f With respect to the reference case. | ||||||
| Organisation | CSIC | CIRCE | NCEPU/ETC | Stuttgart | TUD | |
| Location | EU | EU | China/EU | EU | EU | |
| Coal type | Lignite | Lignite | Lignite | Bitcoal | Bitcoal | |
| Steam turbine type | SCPC | SCPC | SCPC | SCPC | SCPC | |
| Plant size (gross, MWe) | 500 | 450 | 600 | 1100 | 1100 | |
| Capacity factor (%) | 80 | 80 | 80 | — | — | |
| Designed CO2 capture rate (%) | 86 | 85 | 85 | 88 | 87 | |
| CO2 emissions (kg per MW h) | ref.e | 795 | 784 | 784 | — | — |
| cap.e | 135 | 123 | 116 | — | — | |
| Net power output (MWe) | ref.e | 500 | 428 | 600 | 1052 | 1052 |
| cap.e | 748 | 621 | 846 | 1533 | 1583 | |
| Net plant efficiency (LHV, %) | ref.e | 43.0 | 45.0 | 40.6 | 45.6 | 45.6 |
| cap.e | 35.6 | 37.0 | 36.8 | 39.2 | 42.8 | |
| Efficiency penalty (%-points)f | 7.4 | 8.0 | 3.8 | 6.4 | 2.8 | |
| Capital costs (USD per kWe) | ref.e | 1530 | 1819 | 1816 | — | — |
| cap.e | 2136 | 2634 | 3110 | — | — | |
| LCOE (USD per MW h) | ref.e | 46 | 57 | 53 | — | — |
| cap.e | 58 | 73 | 82 | — | — | |
| Cost of CO2 avoided (USD per tCO2) | 18 | 24 | 44 | — | — | |
As noted in Fig. 7, no separate FGD unit is necessary for PCC based on calcium looping, as CaO is the desulphurisation agent applied in most pulverised-coal power plants. The CaO saved by omitting FGD can therefore be counted as part of the make-up sorbent without increasing the overall sorbent consumption; moreover, the absence of FGD decreases the capital costs and energy input. Also importantly, the cost of sorbents is usually assumed to be zero because the sorbents and sorption products (CaO, CaCO3 and CaSO4) can all feed cement-producing equipment, generating value.16,98
Nevertheless, to drive the endothermic calcination reaction of CaCO3 (eqn (11)), the calciner is fed with coal and pure oxygen and thus consumes a large quantity of fuel and electricity; this is the major source of energy penalty and incremental cost. For example, the energy penalty from ASU (∼6% decrease in net efficiency) is even larger than that from another major energy consumer—the CO2 compressor (∼5%).16 Thus, the ASU and CO2 conditioning alone cause efficiency to drop by >10%. However, heat recovery from the oxy-fuel FDB combustor and carbonator running at high temperature is practically simple, and this is likely to offset the large energy input.100
The basic heat-integration technology that has been designed for this process can be described in four parts:99,101 firstly, the exothermic CFB carbonator gives much more heat than is required to maintain the carbonation temperature (Q1, Fig. 7); secondly, the CO2-poor flue gas emitted from the cyclone at ∼600 °C generates steam cycles (Q2) for extra electricity output when it is cooled prior to being vented; thirdly, the hot CaO/CaCO3 flow leaving the carbonator cyclone carries recyclable heat (Q3); and finally, the CO2-rich gas cleaned by the cyclone of the calciner exits as a very hot gaseous flow (>800 °C) that provides additional heat (Q4) for steam turbines. Yang et al.94 have quantified the advantage of integrating heat recovery into a powdered-coal power plant with calcium looping PCC at an 82% decrease in energy penalty and 50% drop in the electricity costs.
The CaO/CO2 ratio and make-up flow rate are also important to the efficiency and economic performance of this system. Increasing these quantities can improve the binding of CO2via carbonation, but can also raise the solid circulation and heat demand during calcination. Calculations have shown that a high CaO/CO2 molar ratio (∼5) should be adopted, but that purging percentages should be strictly restrained (<5%) to minimise the cost of CO2 avoided.98 The exemption is the case that integrates the purged materials into a cement works, which likely demands a higher purge rate.74
4 Trend estimates
We used the techno-economic parameters from Cases 1–18 (Tables 1–5) to compare key measures across the five technologies (Table 6) reviewed in this paper. In particular, we have calculated the relative changes between the reference and retrofitted plants, as these are most useful for cross-technique comparisons. These contrasts, in terms of relative changes in net power out, net plant efficiency, capital investment and levelised cost of electricity, are highlighted in Fig. 8, and 9 shows the costs of CO2 avoided through each of the pathways discussed here.| MEA | CAP | AMC | Mem.a | CaOb | CaOc | ||
|---|---|---|---|---|---|---|---|
| a Mem. = membrane.b Based on Cases 14–16.c Based on Cases 17 and 18.d Calculated as the average of values from individual cases.e The suffix ‘ref.’ refers to the measures of the reference plants without CO2 capture, and ‘cap.’ refers to those of the target plants with PCC.f The relative changes were calculated for each individual case, then averaged.g Not applicable. | |||||||
| Plant size (gross, MWe)d | 595 | 619 | 500 | 564 | 517 | 1100 | |
| Net power output (MWe)d | ref.e | 569 | 619 | 462 | 528 | 509 | 1052 |
| cap.e | 455 | 539 | 381 | 430 | 738 | 1558 | |
| Relative size change (%)f | −20.0 | −12.9 | −17.5 | −18.6 | +44.5 | +48.1 | |
| Designed CO2 capture rate (%)d | 89.0 | 86.7 | 90 | 90 | 85.3 | 87.5 | |
| CO2 emissions (kg per MW h)d | ref.e | 807 | 799 | 774 | 781 | 788 | —g |
| cap.e | 112 | 109 | 84 | 92 | 125 | —g | |
| Net CO2 capture rate (%)f | 86.1 | 86.2 | 89.1 | 88.2 | 84.2 | —g | |
| Net plant efficiency (LHV, %)d | ref.e | 42.1 | 42.7 | 42.6 | 42.2 | 42.9 | 45.6 |
| cap.e | 30.6 | 32.0 | 32.2 | 31.5 | 36.5 | 41.0 | |
| Net efficiency penalty (%-points)d | 11.5 | 10.7 | 10.4 | 10.7 | 6.4 | 4.6 | |
| Relative efficiency change (%)f | −27.4 | −25.3 | −24.4 | −25.4 | −14.8 | −10.1 | |
| Capital costs (USD per kWe)d | ref.e | 1688 | 1745 | 1418 | 1640 | 1722 | —g |
| cap.e | 3054 | 2893 | 2150 | 2510 | 2627 | —g | |
| Relative capital costs change (%)f | +80.9 | +66.4 | +51.6 | +54.8 | +51.9 | —g | |
| LCOE (USD per MW h)d | ref.e | 57 | 64 | 65 | 63 | 52 | —g |
| cap.e | 100 | 99 | 88 | 106 | 71 | —g | |
| Relative LCOE change (%)f | +75.1 | +55.5 | +36.2 | +69.6 | +36.3 | —g | |
| Cost of CO2 avoided (USD per tCO2)e | 61 | 53 | 34 | 68 | 29 | —g | |
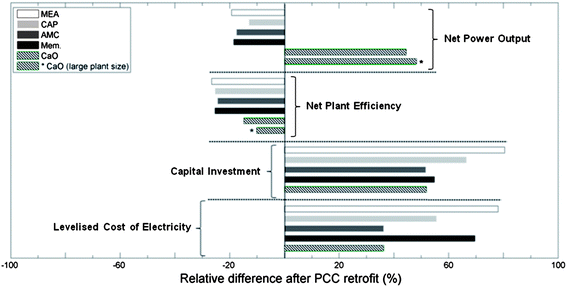 | ||
| Fig. 8 Relative changes in the key techno-economic indices of a pulverised-coal power plant after PCC retrofit. Average for each technology derived from the relevant techno-economic cases presented in Tables 1–6. Mem. = membrane. | ||
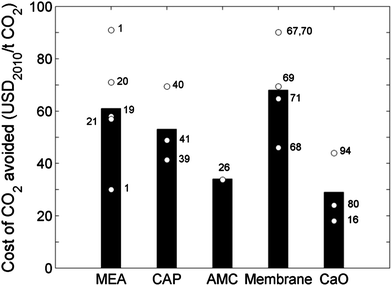 | ||
| Fig. 9 Cost of CO2 avoided for the five PCC technologies considered here. Bars represent average values derived from the relevant techno-economic cases presented in Tables 1–5; circles represent individual cases, which are labelled with reference number. | ||
For Cases 1–16, for which comprehensive techno-economic analyses were available, only marginal variations were observed in the average plant size (gross), net CO2 capture rate and plant efficiency across the five technologies; these fell in the ranges of 500–600 MWe, 84–89%, and 41–43%LHV, respectively. This increases the reliability of the technology comparisons.
All five PCC processes consume energy in order to regenerate the solvents, adsorbents or driving forces for permeation, to facilitate other auxiliary equipment to maintain the mass or gaseous streams and to compress the captured CO2 into a form suitable for sequestration. Specifically, the stripping of CO2 to regenerate MEA, CAP, AMC, and the compression (or evacuation) of membrane modules, all depend on steam extraction from the boilers of the main power plant, which causes a significant (up to 20%) decrease in the net power output; whereas in calcium looping, which uses a high-temperature adsorbent, the decarbonation of CO2-loaded sorbents is integrated to an additional coal combustor, enabling a big rise in the net power output (∼45% increase). Despite that this increase in net power output is partially attributable to the boosting of the actual plant size, calcium-looping-based PCC is also superior to the other processes in terms of net efficiency. MEA-, CAP-, AMC- and membrane-based PCCs all result in net efficiency penalties of more than 10 percentage points in lower heat value, whereas calcium looping can cost as little as 6.4 percentage points. The relative decrease in net efficiency show the same trend, with the CaO process resulting in a decrease of 14.8% (cf. >24% for any other process).
Cases 17 and 18 only give energy balances, without economic data. They are included here to demonstrate that doubling the designed size of a calcium-looping PCC-retrofitted power plant is likely to improve its efficiency performance. These plants gave a 48.1% increase in net power output and an efficiency penalty of 4.6%-points, a relative decrease of just 10.1% compared to the reference plant without PCC.
There is rough agreement among the estimates of initial capital investment required for reference plants without PCC (Table 5), but the capital increase due to retrofitting PCC varies greatly depending on the technology. The MEA- and CAP-based processes need significantly higher capital investments than the other three (Fig. 8); increasing the capital cost of a pulverised-coal power plant by 85 or 66.4%, respectively. This is mainly because of the high cost of designing and building scrubbers and strippers to withstand these corrosive sorbents. Membrane modules are comparatively easy to set up. The total capital costs of these systems depend heavily on the area of the applied membrane, which can be effectively reduced using a multistage design (vide supra). Therefore, membrane PCC needs a low capital supplement, 54.8%. Alkali-metal-carbonate adsorption takes advantage of screw conveyors to motivate mass flows and complete the CO2 capture and release. This design keeps the extra capital cost down to 51.6%, though such screw conveyors are unlikely to be successful on a large scale. Calcium looping PCC shows a capital requirement (51.9%) similar to those of membrane- and alkali-metal-carbonate-based PCC processes. In the calcium-looping process, the CFB calciner and carbonator also contribute to the total equipment costs; however, unlike the scrubber-stripper design, the calciner is also an oxyfuel combustor and the carbonator accommodates highly exothermic reactions, so the heat from both apparati can facilitate supercritical steam turbines and generate more electricity. Thus, the calciner and carbonator contribute to both the capital increment and net power output, which markedly decreases the extra capital requirement compared with MEA and CAP.
The cost of electricity exhibits a trend similar to that of the capital costs, except that the LCOE for membrane-based PCC is much higher than those of AMC- and CaO-based processes. This is due to the large amount of energy needed to drive the multiple stages of membrane separations, and results in the second-highest LCOE increase, ∼70%. MEA gives the most expensive electricity as well, that is, 78.1% higher than no-capture products. Chilled ammonia offers some improvement, increasing the electricity price by only 55.5% due to its relatively low capital investments and energy demands. CaO and AMC are again the winners when LCOE is considered, as both give a low increment of ∼36%. However, CaO is likely better than AMC in this respect because of its conservative estimate and scalable potential.
As shown in Fig. 9, calcium looping exhibits the lowest cost of CO2 avoided—29 USD2010 per tCO2. This is because the oxyfuel calciner centralises its own CO2 and that of the main power plant, which results in more CO2 avoided and thus a lower cost per unit of CO2 avoided (refer to eqn (2)). Membrane-based PCC has the highest cost of CO2 avoided (68 USD2010 per tCO2) because it consumes a lot of extra electricity, decreasing the amount of CO2 avoided. The other three technologies show intermediate performances.
5 Conclusions
Calcium looping provides an economic pathway for post-combustion CO2 capture from conventional pulverised-coal-fired power plants. It shows a competitive ability to decrease the efficiency penalty and lower the costs of PCC. It does not require steam from the main plant turbines and can be retrofitted to existing plants, which also takes the advantages of mature CFB combustor designs. The availability of cheap CaO from abundant natural sources, the avoidance of FGD and the integration of wastes into the cement industry also contribute to its promise as a near-term alternative to the current MEA-based PCC processes. However, it should be noted that the goal set by DOE for less than 20% increase in LCOE with >90% CO2 capture has yet to be met. Furthermore, calcium-looping techniques are still challenged by the rapid decay in CO2 carrying capacity over long cycles. Future work should focus on the development of more stable CaO-based sorbents and on engineering a more efficient process design with optimal heat integration.Glossary
| AMC | Alkali-metal carbonate |
| ASU | Air separation unit |
| CAP | Chilled ammonia process |
| CCS | Carbon capture and sequestration |
| CEPCI | Chemical engineering plant cost index |
| CFB | Circulating fluidised bed |
| CIRCE | Centro de Investigación de Recursos, Spain |
| CMU | Carnegie Mellon University |
| CSIC | Instituto Nacional del Carbón, Spain |
| CSIRO | Commonwealth Scientific and Industrial Research Organisation, Australia |
| DCC | Direct contact cooler |
| DOE | Department of Energy, USA |
| EPRI | Electric Power Research Institute |
| ETC | Energy Technology Centre, Cranfield University, UK |
| GPU | Gas permeance unit |
| FBR | Fluidised bed reactor |
| FGD | Flue gas desulphurisation |
| IEA | International Energy Agency |
| IEF-3 | Institute of Energy Research – Fuel Cells, Germany |
| IPCC | Intergovernmental panel on climate change |
| LCOE | Levelised cost of electricity |
| LHV | Lower heating value |
| MEA | Monoethanolamine |
| MTR | Membrane Technology and Research, Inc. |
| MW h | Megawatt hour |
| MWe | Megawatt electrical |
| NCEPU | North China Electric Power University |
| NETL | National Energy Technology Laboratory, DOE |
| NSF | NSF Center for Micro-engineering Materials, The University of Mexico |
| OECD | Organisation of Economic Co-operation and Development |
| PC | Pulverised coal |
| PCC | Post-combustion capture |
| R&D | Research & Development |
| ref. | Reference case |
| RTI | Research Triangle Institute |
| SCPC | Super critical pulverised coal (plant) |
| STP | Standard temperature and pressure |
| Sub-PC | Subcritical pulverised coal (plant) |
| TNO | TNO Science and Industry, The Netherlands |
| TPRI | Thermal Power Research Institute, China |
| TUP | Technische Universität Darmstadt, Germany |
| USCPC | Ultra super critical pulverised coal (plant) |
| USD | U.S. dollar |
Acknowledgements
The authors are grateful for the financial support of the Australian Research Council, Australian National Low Emissions Research And Development Fund and Xstrata Coal. We also thank Drs Tamara L. Church and Xia Zhong for their assistance.Notes and references
- N. Dave, T. Do, D. Palfreyman, P. H. M. Feron, S. Xu, S. Gao and L. Liu, Energy Procedia, 2011, 4, 1869–1877 CrossRef.
- D. Singh, E. Croiset, P. L. Douglas and M. A. Douglas, Energy Convers. Manage., 2003, 44, 3073–3091 CrossRef CAS.
- B. Metz, O. Davidson, H. C. de Coninck, M. Loos and L. Meyer, IPCC Special Report on Carbon Dioxide Capture and Storage, Cambridge University Press, Cambridge, UK and New York, NY, USA, 2005, p. 442 Search PubMed.
- A. S. Bhown and B. C. Freeman, Environ. Sci. Technol., 2011, 45, 8624–8632 CrossRef CAS.
- IEA, Energy Technology Perspectives 2010, Scenarios & Strategies to 2050, OECD/IEA, Paris, France, 2010 Search PubMed.
- Z. H. Lee, K. T. Lee, S. Bhatia and A. R. Mohamed, Renewable Sustainable Energy Rev., 2012, 16, 2599–2609 CrossRef CAS.
- J. D. Figueroa, T. Fout, S. Plasynski, H. McIlvried and R. D. Srivastava, Int. J. Greenhouse Gas Control, 2008, 2, 9–20 CrossRef CAS.
- P. Markewitz, W. Kuckshinrichs, W. Leitner, J. Linssen, P. Zapp, R. Bongartz, A. Schreiber and T. E. Muller, Energy Environ. Sci., 2012, 5, 7281–7305 CAS.
- M. Wang, A. Lawal, P. Stephenson, J. Sidders and C. Ramshaw, Chem. Eng. Res. Des., 2011, 89, 1609–1624 CrossRef CAS.
- DOE/NETL, Cost and Performance Baseline for Fossil Energy Plants, Volume 1: Bituminous Coal and Natural Gas to Electricity, 2010 Search PubMed.
- DOE, Carbon Capture and Sequestration Systems Analysis Guidelines, U.S. Department of Energy, 2005 Search PubMed.
- N. MacDowell, N. Florin, A. Buchard, J. Hallett, A. Galindo, G. Jackson, C. S. Adjiman, C. K. Williams, N. Shah and P. Fennell, Energy Environ. Sci., 2010, 3, 1645–1669 CAS.
- P. Lisbona, A. Martínez, Y. Lara and L. M. Romeo, Energy Fuels, 2009, 24, 728–736 CrossRef.
- C. M. Quintella, S. A. Hatimondi, A. P. S. Musse, S. F. Miyazaki, G. S. Cerqueira and A. de Araujo Moreira, Energy Procedia, 2011, 4, 2050–2057 CrossRef.
- M. J. Tuinier, H. P. Hamers and M. van Sint Annaland, Int. J. Greenhouse Gas Control, 2011, 5, 1559–1565 CrossRef CAS.
- J. C. Abanades, G. Grasa, M. Alonso, N. Rodriguez, E. J. Anthony and L. M. Romeo, Environ. Sci. Technol., 2007, 41, 5523–5527 CrossRef CAS.
- A. Bandyopadhyay, Clean Technologies and Environmental Policy, 2011, vol. 13, pp. 269–294 Search PubMed.
- M. Finkenrath, Cost and Performance of Carbon Dioxide Capture from Power Generation, IEA Energy Papers, no. 2011/05, OECD Publishing, 2011 Search PubMed.
- E. S. Rubin, C. Chen and A. B. Rao, Energy Policy, 2007, 35, 4444–4454 CrossRef.
- EPRI, Updated Cost and Performance Estimates for Clean Coal Technologies Including CO2 Capture – 2009, Electric Power Research Institute, Palo Alto, California, 2009 Search PubMed.
- M. R. M. Abu-Zahra, J. P. M. Niederer, P. H. M. Feron and G. F. Versteeg, Int. J. Greenhouse Gas Control, 2007, 1, 135–142 CrossRef CAS.
- X. Gang, D. Liqiang, Z. Mingde, Y. Yongping, L. Ji, L. Le and C. Haizhan, Proceedings of 2010 International Conference on Electrical and Control Engineering (ICECE), Wuhan, 2010 Search PubMed.
- OANDA Historical Currency Rate, http://www.oanda.com/currency/historical-rates/.
- CEPCI, in Chemical Engineering, 2012, vol. 119, ch. 72, p. 72 Search PubMed.
- EPRI, Evaluation of Innovative Fossile Fuel Power Plants with CO2 Removal – Technical Report, Electric Power Research Institute, 2000 Search PubMed.
- T. O. Nelson, D. A. Green, P. Box, R. P. Gupta, G. Henningsen and B. S. Turk, Carbon Dioxide Capture from Flue Gas Using Dry Regenerable Sorbents – Final Report, Rsearch Triangle Institute International, 2009 Search PubMed.
- M.-O. Schach, R. Schneider, H. Schramm and J.-U. Repke, Ind. Eng. Chem. Res., 2010, 49, 2363–2370 CrossRef CAS.
- S. Chi and G. T. Rochelle, Ind. Eng. Chem. Res., 2002, 41, 4178–4186 CrossRef CAS.
- N. Dave, T. Do, D. Palfreyman and P. H. M. Feron, Chem. Eng. Res. Des., 2011, 89, 1625–1638 CrossRef CAS.
- IEA, CO2 Capture Ready Plants, 2007/4, IEA Greenhouse Gas R&D Programme (IEA GHG), 2007 Search PubMed.
- A. N. M. Peeters, A. P. C. Faaij and W. C. Turkenburg, Int. J. Greenhouse Gas Control, 2007, 1, 396–417 CrossRef CAS.
- R. Hoefnagels, Master thesis, Utrecht University, 2008.
- D. Zhang, M. Xu and A. Li, Asia-Pac. J. Chem. Eng., 2012, 7, S201–S208 CrossRef CAS.
- X. Gang, L. Shoucheng, Y. Yongping, L. Le and C. Haizhan, Proceedings of 2011 Asia-Pacific Power and Energy Engineering Conference (APPEEC), Wuhan, 2011 Search PubMed.
- M. Simmonds, P. Hurst, M. B. Wilkinson, S. Reddy and S. Khambaty, The Second Annual Conference on Carbon Sequestration Alexandria, VA, USA, 2003 Search PubMed.
- S. C. Lee, H. J. Chae, S. J. Lee, Y. H. Park, C. K. Ryu, C. K. Yi and J. C. Kim, J. Mol. Catal. B: Enzym., 2009, 56, 179–184 CrossRef CAS.
- A. Kothandaraman, Ph.D. thesis, Massachusetts Institute of Technology, 2010.
- E. Gal, US Pat., 2008/0072762, 2006.
- NETL, Chilled Ammonia-Based Wet Scrubbing for Post-Combustion CO2 Capture, The National Energy Technology Laboratory, DOE, USA, 2007 Search PubMed.
- P. Versteeg and E. S. Rubin, Int. J. Greenhouse Gas Control, 2011, 5, 1596–1605 CrossRef CAS.
- G. Valenti, D. Bonalumi and E. Macchi, Fuel, 2012, 101, 74–83 CrossRef CAS.
- V. Darde, K. Thomsen, W. J. M. van Well and E. H. Stenby, Int. J. Greenhouse Gas Control, 2010, 4, 131–136 CrossRef CAS.
- G. Valenti, D. Bonalumi and E. Macchi, Energy Procedia, 2009, 1, 1059–1066 CrossRef CAS.
- G. Ordorica-Garcia, S. Wong and J. Faltinson, Final Report: CO2 Supply from the Fort McMurray Area, Alberta Research Council Inc., Edmonton, Alberta, Canada, 2009 Search PubMed.
- N. K. Solie, Chilled Ammonia Process for Post-Combustion CO2 Capture, http://www.alstom.com Search PubMed.
- Y. Liang, D. P. Harrison, R. P. Gupta, D. A. Green and W. J. McMichael, Energy Fuels, 2004, 18, 569–575 CrossRef CAS.
- A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2011, 51, 1438–1463 CrossRef.
- V. Sharonov, A. Okunev and Y. Aristov, React. Kinet. Catal. Lett., 2004, 82, 363–369 CrossRef CAS.
- S. C. Lee, B. Y. Choi, T. J. Lee, C. K. Ryu, Y. S. Ahn and J. C. Kim, Catal. Today, 2006, 111, 385–390 CrossRef CAS.
- S. Lee and J. Kim, Catal. Surv. Asia, 2007, 11, 171–185 CrossRef CAS.
- T. O. Nelson, L. J. I. Coleman, D. A. Green and R. P. Gupta, Energy Procedia, 2009, 1, 1305–1311 CrossRef CAS.
- L. M. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- K. Ramasubramanian and W. S. W. Ho, Curr. Opin. Chem. Eng., 2011, 1, 47–54 CrossRef CAS.
- W. J. Koros and R. Mahajan, J. Membr. Sci., 2000, 175, 181–196 CrossRef CAS.
- J. P. Van Der Sluijs, C. A. Hendriks and K. Blok, Energy Convers. Manage., 1992, 33, 429–436 CrossRef CAS.
- W. J. Koros, G. K. Fleming, S. M. Jordan, T. H. Kim and H. H. Hoehn, Prog. Polym. Sci., 1988, 13, 339–401 CrossRef CAS.
- S. Kazama, S. Morimoto, S. Tanaka, H. Mano, T. Yashima, K. Yamada and K. Haraya, in Greenhouse Gas Control Technologies 7, Elsevier Science Ltd, Oxford, 2005, pp. 75–82 Search PubMed.
- T. C. Merkel, M. Zhou and R. W. Baker, J. Membr. Sci., 2012, 389, 441–450 CrossRef CAS.
- Y. Wilfredo, C. Anja, W. Jan and P. Klaus-Viktor, Nanotechnology, 2010, 21, 395301 CrossRef.
- G. Xomeritakis, N. G. Liu, Z. Chen, Y. B. Jiang, R. Köhn, P. E. Johnson, C. Y. Tsai, P. B. Shah, S. Khalil, S. Singh and C. J. Brinker, J. Membr. Sci., 2007, 287, 157–161 CrossRef CAS.
- G. Xomeritakis, C. Y. Tsai, Y. B. Jiang and C. J. Brinker, J. Membr. Sci., 2009, 341, 30–36 CrossRef CAS.
- H. Guo, G. Zhu, H. Li, X. Zou, X. Yin, W. Yang, S. Qiu and R. Xu, Angew. Chem., Int. Ed., 2006, 118, 7211–7214 CrossRef.
- S. Li and C. Q. Fan, Ind. Eng. Chem. Res., 2010, 49, 4399–4404 CrossRef CAS.
- G. Q. Lu, J. C. Diniz da Costa, M. Duke, S. Giessler, R. Socolow, R. H. Williams and T. Kreutz, J. Colloid Interface Sci., 2007, 314, 589–603 CrossRef CAS.
- L. Zhao, E. Riensche, R. Menzer, L. Blum and D. Stolten, J. Membr. Sci., 2008, 325, 284–294 CrossRef CAS.
- A. Hussain and M.-B. Hägg, J. Membr. Sci., 2010, 359, 140–148 CrossRef CAS.
- L. Zhao, E. Riensche, L. Blum and D. Stolten, J. Membr. Sci., 2010, 359, 160–172 CrossRef CAS.
- NETL, DOE/NETL-2012/1557 Final Report: Current and Future Technologies for Power Generation with Post-Combustion Carbon Capture, 2012 Search PubMed.
- L. Toy, A. Kataria and R. P. Gupta, Final Technical Report to DOE: CO2 Capture Membrane Process for Power Plant Flue Gas, RTI International, 2012 Search PubMed.
- L. Zhao, E. Riensche, L. Blum and D. Stolten, Energy Procedia, 2011, 4, 629–636 CrossRef CAS.
- C. J. Brinker, G. K. Xomeritakis, C.-Y. A. Tsai and Y. B. Jiang, Final Report to DOE: Novel Dual-Functional Membrane for Controlling Carbon Dioxide Emissions from Fossil Fuel Power Plants, NSF Center for Micro-engineering Materials, The University of New Mexico, Albuquerque, NM 87106, 2009 Search PubMed.
- E. J. Anthony, E. M. Bulewicz and L. Jia, Prog. Energy Combust. Sci., 2007, 33, 171–210 CrossRef CAS.
- T. Shimizu, T. Hirama, H. Hosoda, K. Kitano, M. Inagaki and K. Tejima, Chem. Eng. Res. Des., 1999, 77, 62–68 CrossRef CAS.
- C. C. Dean, J. Blamey, N. H. Florin, M. J. Al-Jeboori and P. S. Fennell, Chem. Eng. Res. Des., 2011, 89, 836–855 CrossRef CAS.
- J. C. Abanades, Chem. Eng. J., 2002, 90, 303–306 CrossRef CAS.
- J. C. Abanades, E. J. Anthony, J. Wang and J. E. Oakey, Environ. Sci. Technol., 2005, 39, 2861–2866 CrossRef CAS.
- V. Manovic and E. J. Anthony, Ind. Eng. Chem. Res., 2009, 48, 8906–8912 CrossRef CAS.
- J. Wang, V. Manovic, Y. Wu and E. J. Anthony, Appl. Energy, 2010, 87, 1453–1458 CrossRef CAS.
- P. Sun, J. R. Grace, C. J. Lim and E. J. Anthony, AIChE J., 2007, 53, 2432–2442 CrossRef CAS.
- L. M. Romeo, J. C. Abanades, J. M. Escosa, J. Paño, A. Giménez, A. Sánchez-Biezma and J. C. Ballesteros, Energy Convers. Manage., 2008, 49, 2809–2814 CrossRef CAS.
- F. Donat, N. H. Florin, E. J. Anthony and P. S. Fennell, Environ. Sci. Technol., 2011, 46, 1262–1269 CrossRef.
- V. Manovic and E. J. Anthony, Ind. Eng. Chem. Res., 2010, 49, 9105–9110 CrossRef CAS.
- B. Gonzalez, M. J. Al-Jeboori and P. S. Fennell, 4th IEAGHG Network Meeting – High Temperature Solid Looping Cycles 21–22nd, August 2012, Tsinghua University, Beijing, 2012 Search PubMed.
- Z. Yang, M. Zhao, N. H. Florin and A. T. Harris, Ind. Eng. Chem. Res., 2009, 48, 10765–10770 CrossRef CAS.
- N. H. Florin and A. T. Harris, Energy Fuels, 2008, 22, 2734–2742 CrossRef CAS.
- N. H. Florin and A. T. Harris, Chem. Eng. Sci., 2009, 64, 187–191 CrossRef CAS.
- P. S. Fennell, J. F. Davidson, J. S. Dennis and A. N. Hayhurst, J. Energy Inst., 2007, 80, 116–119 CrossRef CAS.
- M. Zhao, X. Yang, T. L. Church and A. T. Harris, Environ. Sci. Technol., 2012, 46, 2976–2983 CrossRef CAS.
- Z.-s. Li, N.-s. Cai, Y.-y. Huang and H.-j. Han, Energy Fuels, 2005, 19, 1447–1452 CrossRef CAS.
- V. Manovic and E. J. Anthony, Ind. Eng. Chem. Res., 2009, 48, 8906–8912 CrossRef CAS.
- M. Zhao and A. T. Harris, 241st ACS National Meeting and Exposition, Anaheim, CA, USA, 2011 Search PubMed.
- J. Blamey, E. J. Anthony, J. Wang and P. S. Fennell, Prog. Energy Combust. Sci., 2010, 36, 260–279 CrossRef CAS.
- C. C. Dean, D. Dugwell and P. S. Fennell, Energy Environ. Sci., 2011, 4, 2050–2053 CAS.
- Y. Yang, R. Zhai, L. Duan, M. Kavosh, K. Patchigolla and J. Oakey, Int. J. Greenhouse Gas Control, 2010, 4, 603–612 CrossRef CAS.
- Spanish National Research Council/CSIC-INCAR, CaOling Workshop, http://www.caoling.eu/documentos/AbanadesCSICPresentationWorkshop19042012.pdf, Oviedo, 19th of April 2012 Search PubMed.
- C. Hawthorne, M. Trossmann, P. Galindo Cifre, A. Schuster and G. Scheffknecht, Energy Procedia, 2009, 1, 1387–1394 CrossRef CAS.
- J. Ströhle, A. Lasheras, A. Galloy and B. Epple, Chem. Eng. Technol., 2009, 32, 435–442 CrossRef.
- L. M. Romeo, Y. Lara, P. Lisbona and J. M. Escosa, Chem. Eng. J., 2009, 147, 252–258 CrossRef CAS.
- I. Martínez, R. Murillo, G. Grasa and J. C. Abanades, AIChE J., 2011, 57, 2599–2607 CrossRef.
- I. Martínez, R. Murillo, G. Grasa and J. C. Abanades, Energy Procedia, 2011, 4, 1699–1706 CrossRef.
- J. Ströhle, A. Galloy and B. Epple, Energy Procedia, 2009, 1, 1313–1320 CrossRef.
- A. Lasheras, J. Ströhle, A. Galloy and B. Epple, Int. J. Greenhouse Gas Control, 2011, 5, 686–693 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |