DOI:
10.1039/C3DT51689J
(Perspective)
Dalton Trans., 2013,
42, 15826-15834
Fluorescent azobenzenes and aromatic aldimines featuring an N–B interaction
Received 25th June 2013, Accepted 16th August 2013
First published on 6th September 2013
Abstract
Azobenzenes are constituents of the commonly and widely used azo dyes. Many dyes, except for the azo dyes, have been utilized for fluorescent materials. However, there are only a few fluorescent azobenzene derivatives and their fluorescence efficiencies are quite low. The current perspective provides an account of the fluorescent azobenzenes and aromatic aldimines featuring an N–B interaction. Incorporation of the intramolecular N–B interaction by using the bis(pentafluorophenyl)boryl group makes the azobenzenes and aromatic aldimines fluorescent with a range of colours. Some of them fluoresce with extraordinarily high fluorescence quantum yields. Their synthesis, structures, fluorescence properties, and applications are discussed.
1. Introduction
Fluorescent materials have become indispensable tools in the wide field of science and technology. Their applications for various purposes, such as fluorescent probes, fluorescent indicators, fluorescent paint, and light-emitting devices, play important roles in scientific research and our life.1 Fluorescent compounds consisting of organic compounds have the following advantages: fine adjustability of optical properties, ease of synthesis, lightweight, and ease of moulding. Therefore, many types of organic fluorescent compounds have been developed to date.Construction of organic fluorescent molecules requires a π-conjugated structure and a rational method for the processes of both efficient absorption of light and radiative transition. The nitrogen–boron interaction is one of the rational methods to provide fluorescence properties to organic compounds. Boron dipyrromethene (BODIPY) is a well-known example of a fluorescent organic compound featuring the N–B interaction.2 In BODIPY dyes, the N–B interaction fixes the dipyrromethene framework in the coplanar conformation to avoid thermal degradation from the photoexcited state upon irradiation. The N–B interaction also perturbs the orbital energy levels, and the S0 and S1 states correspond to the HOMO (π orbital) and LUMO (π* orbital), respectively. As a result, BODIPY dyes emit intense fluorescence via the S1–S0 relaxation process upon photoirradiation. Thus, this intensely fluorescent fluorophore gains its fluorescent nature by forming N–B dative bonds, while the dipyrromethene free base itself has poor emission characteristics. The N–B dative bond is also a key feature of the boronic acid sugar sensors, and the N–B dative bond changes the fluorescence quenching efficiency of the amine in the aminomethyl fluorophore.3
Azobenzenes are π-conjugated molecules and constituents of commonly and widely used azo dyes and pigments. One of their important features is ease of adjustment of their properties precisely and facilely. They hold the promise of utilization for attractive fluorescent compounds, considering the π-conjugated structures. However, there were only a few examples of fluorescent azobenzenes when we started a project on fluorescent azobenzenes.4 Furthermore, the fluorescence efficiencies of the previous fluorescent azobenzenes were quite low. The above features are also true for diphenylimines. There are several fluorescent compounds including a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double-bond structure utilizing the co-ordination of a nitrogen atom of the heterocyclic ring to the boron atom.5 Fluorescent imines incorporated into a π-conjugated structure are also known to be a part of multi-dentate ligands for metal complexation.6,7 However, there have been only a few fluorescent diphenylimine derivatives with a simple structure.8
N double-bond structure utilizing the co-ordination of a nitrogen atom of the heterocyclic ring to the boron atom.5 Fluorescent imines incorporated into a π-conjugated structure are also known to be a part of multi-dentate ligands for metal complexation.6,7 However, there have been only a few fluorescent diphenylimine derivatives with a simple structure.8
Azobenzenes efficiently undergo photoisomerization in the photoexcited state, and this process is surely a reason for their non-fluorescence.9 Photoisomerization accompanies the change in the direction of the lone pair of one nitrogen atom. Restraining the lone pair from changing direction should prevent the photoisomerization. Therefore, suppression of the photoisomerization movement in the azobenzenes was considered a key to provide them with a fluorescent nature. The authors initially tried to form an N–B dative bond in the azobenzenes during the course of studies on azobenzenes bearing a main-group-element substituent (Fig. 1) and found that 2-[bis(pentafluorophenyl)boryl]azobenzene (1a) fluoresces intensely.10,11 In the 2-borylazobenzenes, the lone pair is used to form an intramolecular N–B interaction and the photoisomerization is inhibited. In addition, the N–B interaction restricts the rotation around the nitrogen–carbon bond and the non-radiative deactivation process is depressed.
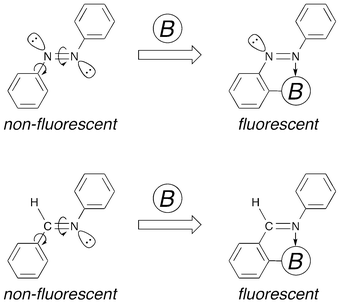 |
| | Fig. 1 Concept of the molecular design to make azobenzenes and aromatic aldimines fluorescent upon photoirradiation by formation of an N–B dative bond to restrict both rotation and isomerization. | |
This perspective aims to provide an overview of the development of fluorescent azobenzenes and aromatic aldimines bearing an N–B interaction as a key interaction and their application to ion sensors.
2. Fluorescent 2-borylazobenzenes and 2,2′-diborylazobenzenes
2.1. Synthesis and structures of fluorescent 2-borylazobenzenes
2-Borylazobenzenes 1a–j were synthesized using the following strategy: first, preparation of 2-iodoazobenzenes bearing functional groups at the 4- and/or 4′-positions, and then introduction of a bis(pentafluorophenyl)boryl group (Fig. 2, Scheme 1, and Table 1).11–14 2,2′-Diborylazobenzenes 2a–d were also synthesized by a similar method from 2,2′-diiodoazobenzenes (Fig. 2, Scheme 1, and Table 2).15 Some of them (1k–o) were obtained by transformation of a functional group on the azobenzene moiety after the introduction of the boryl group.13,14 It is noteworthy that palladium-catalysed cross-coupling reactions, such as the Suzuki–Miyaura cross-coupling reaction, Buchwald–Hartwig amination, and the Sonogashira coupling reaction, are valuable for modifying the brominated 2-borylazobenzenes 1i and 1j, despite the presence of a boryl group in the molecules (Scheme 2).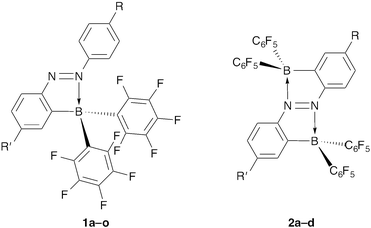 |
| | Fig. 2 2-Borylazobenzenes 1a–o and 2,2′-diborylazobenzenes 2a–d. Substituents R and R′ of 1a–o and 2a–d are shown in Tables 1 and 2, respectively. | |
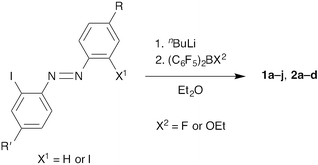 |
| | Scheme 1 Synthesis of 2-borylazobenzenes 1a–j and 2,2′-diborylazobenzenes 2a–d. | |
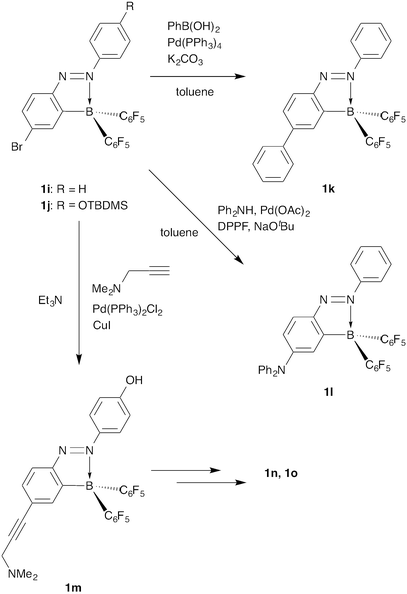 |
| | Scheme 2 Palladium-catalysed cross-coupling reactions of 1i and 1j. | |
Table 1 Substituents at the 4′- and 4-positions (R and R′, respectively), absorption wavelength (λabs), molar absorption coefficient (ε), fluorescence wavelength (λem), fluorescence quantum yield (ΦF), and fluorescence lifetime (τF) of the 2-borylazobenzenes
| | R | R′ | λabs/nm | ε/M−1 cm−1 | λem/nm | ΦF | τF/ns |
|---|
| In hexane. In CH2Cl2. In MeOH. In water. Not available. |
|---|
| 1a | H | H | 386a | 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000a 000a | 503a | 0.20a | 3.82(4)a |
| 1b | OMe | H | 439a | 12000a | 524a | 0.76a | 6.24(5)a |
| 1b | OMe | H | 446b | 14000b | 536b | 0.98b | —e |
| 1c | NMe2 | H | 516a | 55000a | 566a | 0.52a | 6.17(5)a |
| 1d | CF3 | H | 382a | 22000a | 511a | 0.039a | 2.67(3)a |
| 1e | F | H | 390a | 11000a | 504a | 0.29a | —e |
| 1f | OTBDMS | H | 433a | 16000a | 524a | 0.90a | 5.50(2)a |
| 1g | nBu | H | 402a | 22000a | 507a | 0.31a | —e |
| 1h | H | OMe | 443a | 24000a | 538a | 0.048a | <1a |
| 1i | H | Br | 416a | 23000a | 509a | 0.045a | —e |
| 1k | H | Ph | 445a | 22000a | 539a | 0.13a | —e |
| 1l | H | NPh2 | 553a | 28000a | 634a | 0.37a | —e |
| 1n | O(CH2)2N+Me3I− | C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2N+Me3I− CCH2N+Me3I− | 458c | 17000c | 546c | 0.53c | —e |
| 1n | O(CH2)2N+Me3I− | CCCH2N+Me3I− | 456d | 12000d | 552d | 0.10d | —e |
| 1o | O(CH2)2N+Me2(CH2)3SO3− | CCCH2N+Me2(CH2)3SO3− | 468c | 12000c | 555c | 0.14c | —e |
| 1o | O(CH2)2N+Me2(CH2)3SO3− | CCCH2N+Me2(CH2)3SO3− | 460d | 13000d | 545d | 0.092d | —e |
Table 2 Substituents at the 4- and 4′-positions (R and R′, respectively), absorption wavelength (λabs), molar absorption coefficient (ε), fluorescence wavelength (λem), fluorescence quantum yield (ΦF), and fluorescence lifetime (τF) of the 2,2′-diborylazobenzenes in hexane
| | R, R′ | λabs/nm | ε/M−1 cm−1 | λem/nm | ΦF | τF/ns |
|---|
| 2a | H | 521, 552 | 20000, 16000 | 572 | 0.26 | 1.7 |
| 2b | nBu | 547, 582 | 9500, 9900 | 597 | 0.73 | 6.7 |
| 2c | Br | 549, 584 | 52000, 58000 | 602 | 0.39 | 3.7 |
| 2d | OnBu | 603, 646 | 55000, 71000 | 663 | 0.62 | 2.4 |
The molecular structures of all the fluorescent azobenzenes in solution were established by 1H, 11B, 13C, and 19F NMR spectroscopy, and the crystal structures of 1a, 1b, 1e, and 2a were determined by X-ray crystallographic analysis. The 11B NMR spectra show their chemical shifts in the range of −3.2 to −0.1 ppm, indicating the presence of a tetraco-ordinated boron atom in solution because of the N–B dative bond. According to the crystallographic analysis, the NN bond lengths [1.2704(18)–1.292(3) Å] are in the range of a double bond, despite the existence of the dative bond. The N–B dative bonds, whose lengths are in the range of 1.625(2)–1.638(3) Å, make the NN double bond coplanar with one of the two benzene rings of the azobenzene (Fig. 3). In 2,2′-diborylazobenzene 2a, which has two N–B dative bonds, the azobenzene moiety is almost coplanar with the two N–B dative bonds.
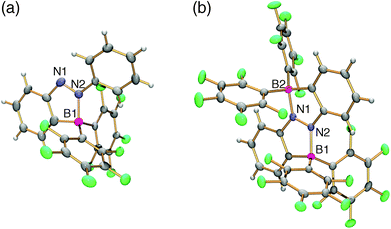 |
| | Fig. 3 ORTEP drawings of azobenzenes (a) 1a and (b) 2a with a thermal ellipsoid plot (50% probability). | |
2.2. Fluorescence properties of 2-borylazobenzenes
2-Bis(pentafluorophenyl)borylazobenzenes 1a–n fluoresce in non-polar organic solvents. The fluorescence colours and intensities depend on the substituents on the benzene rings, R and R′. Fluorescence colours of the 2-borylazobenzenes 1a–o are finely tuned by substitution on the azobenzene moiety and the colour variations are green, yellow, orange, and red (Table 1 and Fig. 4). In azobenzenes 1a–g, the stronger electron-donating group at the 4′-position shows a tendency to give the larger red shift of the emission maximum and the higher fluorescence quantum yield. In particular, azobenzene 1b, which bears an electron-donating methoxy group at the 4′-position, is the most intensely fluorescent azobenzene to date. Its fluorescence quantum yield (ΦF = 0.98 in dichloromethane) is almost 1.00 and extraordinarily high compared with unsubstituted azobenzene (3) (ΦF = 2.53 × 10−5).16 In azobenzenes 1h, 1i, 1k, and 1l, the electron-donating group at the 4-position also gives a red shift of the emission maximum; however, the fluorescence quantum yields are relatively low in comparison with azobenzenes bearing an electron-donating group at the 4′-position. |
| | Fig. 4 Photographs of hexane solutions of azobenzenes 1a–e, 1g, 1h, and 7 (from left to right, top to bottom) under irradiation by UV light. The photographs are reproduced from ref. 13 with permission from John Wiley and Sons. | |
The exceptional fluorescence emission of 2-borylazobenzenes 1a–o was caused by the blocking of photoisomerization of the azobenzene moiety, enhancement of rigidity of the molecular structure, and the change in nature of the transition between the lowest excited state and the ground state from the optically forbidden n–π* transition to the allowed π–π* transition. The change in the transition was confirmed by TD-DFT calculations.11,13 All these factors are provided by the intramolecular N–B interaction, which masks the lone pair of the nitrogen atom of the azo group.
The above-mentioned fluorescent azobenzenes are insoluble in water, and we attempted to improve their solubility in water by incorporation of ionic functional groups. The dimethylaminoethynyl group can be introduced on aromatic rings by the Sonogashira coupling reaction and can easily be quaternized. The quaternized ionic moiety is expected to increase the solubility of the 2-borylazobenzenes in water.17 Azobenzenes 1n and 1o, bearing tetra-alkylammonium and/or sulfonate groups, are slightly dispersible in water (1n, 1.15 g L−1; 1o, 1.19 g L−1), and 1n is even more dispersible in water than in dichloromethane (Fig. 5). They look transparent, like an aqueous solution to the naked eye. They show fluorescence emission in water, although the fluorescence quantum yields in water are lower than those in methanol (Table 1). Their particle sizes were measured using dynamic light scattering, and the results (1n, 212 nm (polydispersity index (PDI) = 1.1) in water, 0.76 nm (PDI = 1.1) in methanol; 1o, 424 nm (PDI = 1.1) in water, 0.73 nm (PDI = 1.1) in methanol) suggest that they aggregate in water and exist as a monomer in methanol. The aggregation caused by the hydrophobic 2-borylazobenzene moiety is considered to provide the differences in emission behaviour between water and organic solvents.
 |
| | Fig. 5 Photographs of 1n in water–dichloromethane under irradiation by (a) ambient light and (b) UV light. The upper and lower layers of the sample are water and dichloromethane, respectively. The photographs are reproduced from ref. 14 with permission from Elsevier. | |
Substituents on the boron atom of the boron-substituted azobenzenes affect their fluorescence behaviours. In the 2-(diarylboryl)azobenzenes, replacement of pentafluorophenyl groups on the boron atom with p-fluorophenyl groups results in loss of the fluorescent property, and the corresponding azobenzene 4 cannot fluoresce (Fig. 6).11 Other 2-borylazobenzenes 5a–c,18 which have a chelate-type diolato ligand on the boron atom, cannot induce fluorescence emission of the azobenzene (Fig. 6). Azobenzenes 6a–c, which bear two hydroxy groups, a pinacolato group, and an o-phenylenediamide group, respectively, no longer hold the N–B dative bond because the Lewis acidity of the boryl groups is weaker than the bis(pentafluorophenyl) group.19 They do not fluoresce naturally. In contrast, the intramolecular N–B dative bond forms if pentafluorophenyl groups bind to the boron atom, even though the boryl group does not bind directly to the benzene ring of the azobenzene. Actually, 2-boryloxyazobenzene 7 fluoresces, but its fluorescence quantum yield (ΦF = 0.033 in hexane) of the red-shifted fluorescence emission (λmax = 576 nm in hexane) is quite low in comparison with 2-borylazobenzene 1a (Fig. 4 and 6).13 The intensely fluorescent, weakly fluorescent, and non-fluorescent azobenzenes so far exemplify the significance of both the existence of strong electron-withdrawing pentafluorophenyl groups on the boron atom and binding of the bis(pentafluorophenyl)boryl group directly to the azobenzene for the intense fluorescence emission from the azobenzene. Using the strong electron-withdrawing bis(pentafluorophenyl)boryl group on the benzene ring in addition to the N–B interaction is important for the intense fluorescence emission from the azobenzene moiety. Replacement of pentafluorophenyl groups with less electron-withdrawing p-fluorophenyl groups changes the HOMO from a π orbital of azobenzene to a π orbital of one of the p-fluorophenyl groups. The S0–S1 transition after the replacement results in a forbidden π–π* transition because of spatial separation, which causes the loss of fluorescence.11,13
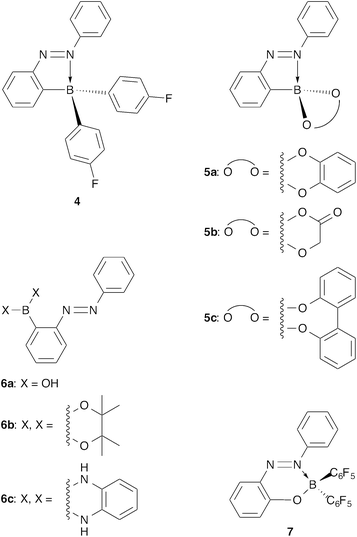 |
| | Fig. 6 Non-fluorescent 2-borylazobenzenes 4–6 and fluorescent 2-boryloxyazobenzene 7. | |
2.3. Fluorescence properties of 2,2′-diborylazobenzenes
2-Borylazobenzenes 1a–o provide only one of two nitrogen atoms of the azo group for the N–B dative bond; however, the other nitrogen atom can be used for an additional dative bond. Furnishing the 2-borylazobenzenes with one more N–B interaction changes their fluorescence properties drastically because of fixation of C–N bond rotation in the azobenzene moiety by two strong N–B interactions. Actually, 2,2′-diborylazobenzenes 2a–d fluoresce in orange or red with their emission maxima at 573–663 nm, showing a large red shift from the emission wavelength of the 2-monoborylazobenzenes (Table 2 and Fig. 7).15 Another notable point of their fluorescence properties is their small Stokes shifts (400 to 630 cm−1). Substituent effects on their fluorescence properties are similar to those of 2-monoborylazobenzenes. |
| | Fig. 7 Photographs of hexane solutions of 2a–d (from left to right) under irradiation by (a) ambient light and (b) UV light. The photographs are reproduced from ref. 15 with permission from John Wiley and Sons. | |
3. Fluorescent boron-substituted aromatic aldimines
Replacement of one nitrogen atom of the azo group (–NN–) in azobenzene (3) with a methine gives another dye molecule, N-benzylideneaniline (8), which contains an imine moiety (–CHN–). The azo and imine compounds differ in thermal stability depending on the substituent on the nitrogen atoms, reactivity towards nucleophiles because of polarization of the CN double bond, and absorption wavelength reflecting the HOMO–LUMO energy gap. However, it resembles azobenzene in several respects—molecular structure, availability through easy organic synthetic reactions, adjustability of its π-conjugated system, and virtually non-fluorescent properties. The similarity of the molecular structures of these compounds motivated the authors to synthesize bis(pentafluorophenyl)boryl-substituted aromatic aldimines, in anticipation of fluorescence characteristics (Fig. 1). This molecular design is based on the structure of fluorescent azobenzene 1a.3.1. Synthesis and structures of fluorescent boron-substituted aromatic aldimines
Aromatic aldimines 9a–h were synthesized by a simple condensation of 2-borylbenzaldehyde 10 and the corresponding amines (Scheme 3 and Table 3).20 All of the aldimines 9a–h have an N–B dative bond in their molecular structures in the solution state and the crystalline state, as evidenced by the chemical shifts in 11B NMR spectra (δB: −3.2 to −0.6) and the N–B bond length (1.605(3)–1.646(3) Å), respectively. These aldimines correspond to Lewis-acid activated aldimines, but they are stable in air. In addition, their CN double bonds (1.285(3)–1.3003(18) Å) are not elongated severely. Similar to the case of 2-borylazobenzenes 1a, the N–B dative bond in their structures works to make their CN double bond coplanar with the benzene ring to which the boron atom binds. In aldimines 9a–d, the aryl group on the nitrogen atom deviates somewhat from coplanarity with the CN double-bond plane in the crystal structures (dihedral angle: 14 to 34°). By contrast, in hydrazone-type compounds 9g and 9h, the dihedral angles (62–73°) are much larger because of the intramolecular π–π stacking between the nitrogen-containing aromatic rings and one of the pentafluorophenyl groups.21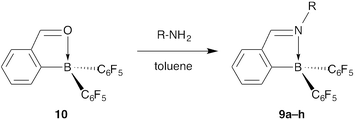 |
| | Scheme 3 Synthesis of the aromatic aldimines 9a–h. Substituent R is shown in Table 3. | |
Table 3 Substituent on the nitrogen atom (R), absorption wavelength (λabs), molar absorption coefficient (ε), fluorescence wavelength (λem), and fluorescence quantum yield (ΦF) of the boron-substituted aromatic aldimines in hexane
| | R | λabs/nm | ε/M−1 cm−1 | λema/nm | ΦF |
|---|
| Excited at λabs unless otherwise noted. Not detectable. Excited at 290 nm. Not available. Excited at 320 nm. |
|---|
| 9a | Ph | 324 | 10000 | 460 | 0.0050 |
| 9b | 4-MeOC6H4 | 366 | 15000 | 466 | 0.074 |
| 9c | 4-Me2NC6H4 | 439 | 25000 | 500, 527 | 0.73 |
| 9d | 2-Anthryl | 442 | 9100 | 497, 531 | 0.39 |
| 9e | nBu | 290 | 13000 | 398 | 0.011 |
| 9f | OMe | 296 | 7600 | —b | —b |
| 9g | 1-Indolyl | 398 | 6800 | 525 (365, 531)c | 0.0092 (—d)c |
| 9h | 9-Carbazolyl | 423 | 2700 | 545 (380, 396, 545)e | 0.0052 (0.020)e |
3.2. Fluorescence properties of boron-substituted aromatic aldimines
Aldimines 9a–d fluoresce blue or green in hexane solution (Table 3 and Fig. 8). The blue emission is a characteristic of the boron-substituted aromatic aldimines. Blue emission has not been achieved in the fluorescent 2-borylazobenzenes so far, because it is difficult to adjust the emission wavelength of the azobenzenes to be short enough, beyond the region of green light. Compared with the 2-borylazobenzenes, aromatic aldimines 9a–d show shorter wavelengths in both absorption and fluorescence spectra because of the larger HOMO–LUMO energy gap in the CN double-bond compounds than in the NN double-bond compounds. The available region of emission wavelengths of the fluorescent azo family has been expanded by adaptation of the azomethine framework. |
| | Fig. 8 Photographs of hexane solution of aromatic aldimines (a) 9a and (b) 9c under irradiation by UV light. The photographs are reproduced from ref. 20 with permission from the American Chemical Society. | |
The fluorescence efficiencies of the boron-substituted aromatic aldimines are much higher than that of non-fluorescent N-benzylideneaniline (8) (ΦF < 0.0001).22 In particular, aldimine 9c, which bears an electron-donating 4-dimethylaminophenyl group on the nitrogen atom of the imine moiety, shows the highest fluorescence quantum yields (0.73) among 9a–d. It is at least 7000 times higher than that of 8. The high fluorescence efficiencies of 9a–d are provided by the intramolecular N–B interaction. The tight N–B interaction gives high rigidity to the molecular structure to suppress the non-radiative relaxation through internal conversion. Furthermore, the non-radiative relaxation path from the lowest singlet excited state, 1(π, π*), via the triplet excited state23 is blocked by the N–B interaction for the following reasons. Masking the lone pair of the nitrogen atom raises the energy levels of all triplet excited states of 3(n, π*), which are responsible for the non-radiative relaxation in N-benzylideneaniline, over the energy level of the 1(π, π*). Therefore, only slow intersystem crossing between 1(π, π*) and the resulting 3(π, π*) is available. As a result, relatively fast radiative relaxation from 1(π, π*) to the ground state occurs in aldimines 9a–d. Thus, the fate of aldimines 9a–d is a radiative relaxation, and they fluoresce. The fluorescence emission mechanism of 9a–d makes a good contrast with that of the 2-borylazobenzenes, in which changing the transition character between the lowest singlet excited state and the ground state is a key for the fluorescent emission.
N-Alkyl aldimine 9e also fluoresces, but its fluorescence is almost invisible because its weak fluorescence emission is mostly in the ultraviolet region. Oxime-type compound 9f does not show fluorescence.
The fluorescence behaviours of hydrazone-type compounds 9g and 9h are interesting. They have two different emission modes: one is based on the transition from the local excited state of the nitrogen-containing aromatic ring moiety to the ground state (HOMO–LUMO + 1 transition), and another is based on the transition from a charge transfer state to the ground state (HOMO–LUMO transition). As a result, they fluoresce with a single emission peak upon excitation at a longer wavelength, while they show dual emission upon excitation at a shorter wavelength, which corresponds to a sufficiently high energy to excite the indole or carbazole moieties (see Table 3).
4. Applications of fluorescent azobenzenes and aromatic aldimines
Fluorescence emission is detectable with high sensitivity, and it is used for various purposes, such as labelling for detection of living organisms and chemo-sensors for toxic materials. The fluorescent boron-substituted azobenzenes and aromatic aldimines are sufficiently stable. The boron-substituted fluorescent azobenzene can be used as a vital fluorescent tracer. Microinjection of azobenzene 1b into Xenopus embryos and observation of the phenotypes revealed its non-toxicity for cells because it did not induce any malformation.13 Actually, we could observe perceptible fluorescence after the development of the cells. The boron-substituted fluorescent azobenzenes and aromatic aldimines show chemical reactivity towards some reagents. Chemical reactions of the boron-substituted azo derivatives are expected to change the fluorescence emission colours or intensity. The fluorescence changes of the fluorescent azobenzenes and aromatic aldimines in response to chemical stimuli are discussed below.4.1. Acid sensing
Some fluorescent azobenzenes show switching of fluorescence behaviours upon receiving chemical stimuli. One example is fluorescence switching of 2-borylazobenzene 1c, which bears a dimethylamino group at the 4′-position, by acid–base reactions. The strong orange fluorescence of 1c in hexane was completely quenched by addition of trifluoroacetic acid (TFA), and the colour change can be clearly observed by the naked eye (Fig. 9).12 The quenched fluorescence was recovered by neutralization of the acidified solution with triethylamine. Therefore, its fluorescence-switching potential by acid–base reactions can be used for a fluorescent pH sensor. The proton-responsive mechanism of 1c is considered to be the same as azo-dye-based pH indicators because the molecular structure of 1c includes that of methyl yellow, 4-(dimethylamino)azobenzene (Scheme 4). Protonation of the azo group of 1c results in a change in the nature of the transition. The allowed π–π* transition on the azobenzene moiety changes to the forbidden transition between the π orbital on a pentafluorophenyl group and the π* orbital on the azobenzene moiety, as confirmed by TD-DFT calculations. This is considered a reason for the fluorescence quenching. |
| | Fig. 9 Photographs of hexane solutions of 1c under irradiation by (a) ambient light and (b) UV light without (left) and with (right) trifluoroacetic acid. The photographs are reproduced from ref. 12 with permission from the Chemical Society of Japan. | |
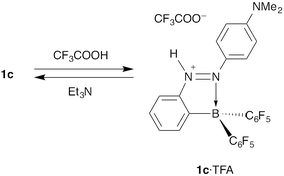 |
| | Scheme 4 Acid–base reactions of azobenzene 1c. | |
4.2. Fluorescence switching invoked by reduction–oxidation reactions
Another example of reaction-based fluorescence switching is the case of 2,2′-diborylazobenzenes 2a–d. Doubling of the N–B interaction provides 2,2′-diborylazobenzenes 2a–d with high electron affinity. As suggested by cyclic voltammetry and DFT calculations, the energy level of the π* orbital (LUMO) in 2b is much lower than those in azobenzenes 3 and 2-borylazobenzene 1a because of two N–B dative bonds. Thus, 2b can easily be reduced to the corresponding radical anion. Single-electron reduction of 2b in THF with decamethylcobaltocene afforded its non-fluorescent azobenzene radical anion 11, accompanying the colour change of the solution from red to blue (Scheme 5).15 Radical anion 11 was readily oxidized by air to give the original fluorescent 2b. Therefore, the fluorescence emission of the 2,2′-diborylazobenzenes is easily quenched and recovered by the redox reactions, and it may be useful for the control of fluorescence emission.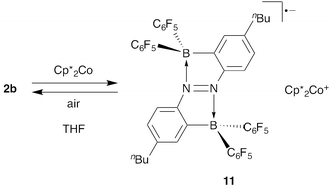 |
| | Scheme 5 Reversible reduction of 2b. | |
4.3. Cyanide ion sensing
The fluorescent boron-substituted aromatic aldimines can be applied to cyanide ion sensing. Although there are several reports on boron-based cyanide ion sensors in which a cyanide ion co-ordinates to the boron atom,24 aldimines 9a and 9d function as a unique cyanide sensor from the viewpoint of a sensing mechanism. Initially, a cyanide ion was anticipated to co-ordinate to the boron atom to kick the N–B interaction out in the reaction of the fluorescent aromatic aldimines with a cyanide ion. However, the reaction of aldimine 9a gave its cyanide adduct 12a, in which a cyanide ion binds to the imine carbon atom instead of by co-ordination to the boron atom (Scheme 6).20,25 Conversion of the CN double bond to a single bond disconnects the π-conjugation and quenches the fluorescence of the aromatic aldimine, performing a turn-off type sensing of cyanide ion.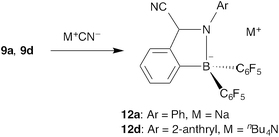 |
| | Scheme 6 Adduct formation of aldimines 9a and 9d with a cyanide ion. | |
N-Anthryl aldimine 9d shows a more drastic change upon addition of a cyanide ion. The fluorescence colour of 9d in benzene changed from yellow to green, with a blue shift of the emission maximum from 547 nm to 500 nm (Fig. 10).26 This fluorescence colour change can be recognized by the naked eye and has potential for application in the ratiometric detection of cyanide ions. In adduct 12d, the π-conjugated system through the imine moiety is disconnected and the π-conjugation of the anthryl group is delocalized only on the anthryl group and the nitrogen atom, causing 2-aminoanthracene-originated fluorescence.
 |
| | Fig. 10 Photographs of a benzene solution of aldimine 9d under irradiation by UV light (a) before and (b) after addition of tetrabutylammonium cyanide. The photographs are reproduced from ref. 26 with permission from the Chemical Society of Japan. | |
5. Conclusions
Incorporation of azobenzene and aromatic aldimines with the intramolecular N–B interaction by using the bis(pentafluorophenyl)boryl group makes these molecules fluorescent. Their fluorescence properties can be easily modified by doubling the N–B interaction or changing the substituents on the benzene rings and the nitrogen atom. Some of them fluoresce with extraordinarily high fluorescence quantum yields. They change their fluorescence properties in response to protons and electrons, and their chemical reactivity shows potential application to fluorescent chemo-sensors. The easily tunable luminescent properties can also be useful for molecular design in applications such as organic light-emitting diodes (OLEDs) and organic light-emitting transistors. This study shows that the π-conjugated molecules, which were considered to be non-fluorescent, can be converted into fluorescent molecules by utilizing the co-ordination of its heteroatom to boron. Actually, some fluorescent aromatic aldimines utilizing an N–B interaction have been developed.27 Many non-fluorescent π-conjugated molecules other than azobenzenes and aromatic aldimines are also anticipated to have fluorescence potential, and they could be applied to some fluorescent materials to provide the fluorescent nature. The method utilizing the intramolecular co-ordination of heteroatom to boron or other elements will be useful for converting non-fluorescent or weakly fluorescent molecules to intensely fluorescent molecules. For example, conversion of aromatic thioketone and simple aromatic ketones such as benzophenone, a phosphorescent molecule, into intensely fluorescent molecules by raising the triplet excited state is an interesting subject from the viewpoint of it being the antithesis of common sense.A popular strategy for molecular design of fluorescent compounds for some applications has been to couple a known fluorescent dye with functional molecules. If we can create new family members of fluorescent dyes by using many π-conjugated structures, we will not need to worry about the difficulty of choosing the correct one among a limited number of fluorescent dyes. We hope that the method of utilizing the intramolecular N–B interaction in this paper is beneficial for people who use fluorescent dyes for practical applications, such as fluorescence chemo-sensors, cell-specific imaging agents, and OLEDs.
Acknowledgements
The authors acknowledge the contributions of their collaborators and co-workers mentioned in the cited references and financial support of the Grants-in-Aid for Scientific Research on Innovative Areas “Coordination Programming” (no. 22108508) and for Young Scientists (A) (no. 22685005) from the Ministry of Education, Culture, Sports, Science and Technology, Japan.Notes and references
- B. Valeur, Molecular Fluorescence, Wiley-VCH, Weinheim, 2002 Search PubMed; J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- A. Treibs and F.-H. Kreuzer, Liebigs Ann. Chem., 1968, 718, 208 CrossRef CAS; A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef PubMed; R. Ziessel, G. Ulrich and A. Harriman, New J. Chem., 2007, 31, 496 RSC; G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184 CrossRef PubMed.
- T. D. James, K. Sandanayake and S. Shinkai, J. Chem. Soc., Chem. Commun., 1994, 477 RSC.
- Y. Wakatsuki, H. Yamazaki, P. A. Grutsch, M. Santhanam and C. Kutal, J. Am. Chem. Soc., 1985, 107, 8153 CrossRef CAS; M. Shimomura and T. Kunitake, J. Am. Chem. Soc., 1987, 109, 5175 CrossRef; M. Ghedini, D. Pucci, G. Calogero and F. Barigelletti, Chem. Phys. Lett., 1997, 267, 341 CrossRef; M. Ghedini, D. Pucci, A. Crispini, I. Aiello, F. Barigelletti, A. Gessi and O. Francescangeli, Appl. Organomet. Chem., 1999, 13, 565 CrossRef; I. Aiello, M. Ghedini and M. La Deda, J. Lumin., 2002, 96, 249 CrossRef; M. Han and M. Hara, J. Am. Chem. Soc., 2005, 127, 10951 CrossRef PubMed; M. R. Han and M. Hara, New J. Chem., 2006, 30, 223 RSC.
- S.-F. Liu, Q. Wu, H. L. Schmider, H. Aziz, N.-X. Hu, Z. Popovic and S. Wang, J. Am. Chem. Soc., 2000, 122, 3671 CrossRef CAS; C. A. Jask, D. J. H. Emslie, M. J. D. Bosdet, W. Piers, T. S. Sorensen and M. Parvez, J. Am. Chem. Soc., 2006, 128, 10885 CrossRef PubMed; A. Wakamiya, T. Taniguchi and S. Yamaguchi, Angew. Chem., Int. Ed., 2006, 45, 3170 CrossRef PubMed; A. Job, A. Wakamiya, G. Kehr, G. Erker and S. Yamaguchi, Org. Lett., 2010, 12, 5470 CrossRef PubMed; Y. Kubota, H. Hara, S. Tanaka, K. Funabiki and M. Matsui, Org. Lett., 2011, 13, 6544 CrossRef PubMed; D. Li, H. Zhang, C. Wang, S. Huang, J. Guo and Y. Wang, J. Mater. Chem., 2012, 22, 4319 RSC; Y. Kubota, S. Tanaka, K. Funabiki and M. Matsui, Org. Lett., 2012, 14, 4682 CrossRef PubMed; S. Shimizu, T. Iino, Y. Araki and N. Kobayashi, Chem. Commun., 2013, 49, 1621 RSC; J.-S. Lu, S.-B. Ko, N. R. Walters, Y. Kang, F. Sauriol and S. Wang, Angew. Chem., Int. Ed., 2013, 52, 4544 CrossRef PubMed.
- P. Wang, Z. Hong, Z. Xie, S. Tong, O. Wong, C.-S. Lee, N. Wong, L. Hung and S. Lee, Chem. Commun., 2003, 1664 RSC.
- K. Morishige, J. Inorg. Nucl. Chem., 1978, 40, 843 CrossRef.
- J. Xie, J. Qiao, L. Wang, J. Xie and Y. Qiu, Inorg. Chim. Acta, 2005, 358, 4451 CrossRef CAS PubMed; L. Chen, J. Qiao, J. Xie, L. Duan, D. Zhang, L. Wang and Y. Qiu, Inorg. Chim. Acta, 2009, 362, 2327 CrossRef PubMed.
- H. Rau, Angew. Chem., Int. Ed. Engl., 1973, 12, 224 CrossRef CAS; A. Cembran, F. Bernardi, M. Garavelli, L. Gagliardi and G. Orlandi, J. Am. Chem. Soc., 2004, 126, 3234 CrossRef PubMed.
- N. Kano and T. Kawashima, TCI Mail, 2007, 136, 2 CAS.
- J. Yoshino, N. Kano and T. Kawashima, Chem. Commun., 2007, 559 RSC.
- J. Yoshino, N. Kano and T. Kawashima, Chem. Lett., 2008, 37, 960 CrossRef CAS.
- J. Yoshino, A. Furuta, T. Kambe, H. Itoi, N. Kano, T. Kawashima, Y. Ito and M. Asashima, Chem.–Eur. J., 2010, 16, 5026 CrossRef CAS PubMed.
- H. Itoi, T. Kambe, N. Kano and T. Kawashima, Inorg. Chim. Acta, 2012, 381, 117 CrossRef CAS PubMed.
- N. Kano, A. Furuta, T. Kambe, J. Yoshino, Y. Shibata, T. Kawashima, N. Mizorogi and S. Nagase, Eur. J. Inorg. Chem., 2012, 1584 CrossRef CAS.
- T. Fujino, S. Yu. Arzhantsev and T. Tahara, J. Phys. Chem. A, 2001, 105, 8123 CrossRef CAS.
- S.-L. Niu, G. Ulrich, P. Retailleau, J. Harrowfield and R. Ziessel, Tetrahedron Lett., 2009, 50, 3840 CrossRef CAS PubMed; S.-L. Niu, G. Ulrich, R. Ziessel, A. Kiss, P.-Y. Renard and A. Romieu, Org. Lett., 2009, 11, 2049 CrossRef PubMed.
- N. Kano, J. Yoshino and T. Kawashima, Org. Lett., 2005, 7, 3909 CrossRef CAS PubMed; J. Yoshino, N. Kano and T. Kawashima, Tetrahedron, 2008, 64, 7774 CrossRef PubMed.
- K. Ishihara, in Lewis Acid Reagents: A Practical Approach, ed. H. Yamamoto, Oxford University Press, New York, 1999, pp. 31–64 CrossRef CAS PubMed; J. A. Plumley and J. D. Evanseck, J. Phys. Chem. A, 2009, 113, 5985 CrossRef CAS PubMed; B. L. Durfey and T. M. Gilbert, Inorg. Chem., 2011, 50, 7871 CrossRef PubMed.
- J. Yoshino, N. Kano and T. Kawashima, J. Org. Chem., 2009, 74, 7496 CrossRef CAS PubMed.
- G. W. Coates, A. R. Dunn, L. M. Henling, J. W. Ziller, E. B. Lobkovsky and R. H. Grubbs, J. Am. Chem. Soc., 1998, 120, 3641 CrossRef CAS; C. Dai, P. Nguyen, T. B. Marder, A. J. Scott, W. Clegg and C. Viney, Chem. Commun., 1999, 2493 RSC; V. R. Vangala, A. Nangia and V. M. Lynch, Chem. Commun., 2002, 1304 RSC.
- M. Belletete and G. Durocher, Can. J. Chem., 1982, 60, 2332 CrossRef CAS.
- M. I. Knyazhanskii, M. B. Stryukov and V. I. Minkin, Opt. Spectrosc., 1972, 33, 484 Search PubMed.
- R. Badugu, J. R. Lakowicz and C. D. Geddes, J. Am. Chem. Soc., 2005, 127, 3635 CrossRef CAS PubMed; C.-W. Chiu, Y. Kim and F. P. Gabbaï, J. Am. Chem. Soc., 2009, 131, 60 CrossRef PubMed.
- S. Madhu, S. K. Basu, S. Jadhav and M. Ravikanth, Analyst, 2013, 138, 299 RSC.
- J. Yoshino, N. Kano and T. Kawashima, Bull. Chem. Soc. Jpn., 2010, 83, 1185 CrossRef CAS.
- Z. García-Hernández and F. P. Gabbaï, Z. Naturforsch., B: Chem. Sci., 2009, 64b, 1381 Search PubMed; B. Neue, R. Frohlich, B. Wibbeling, A. Fukazawa, A. Wakamiya, S. Yamaguchi and E.-U. Wurthwein, J. Org. Chem., 2012, 77, 2176 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2013 |
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence