 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photo-isomerisation of alkenyl complexes of platinum(II): structural, spectroscopic, kinetic and computational investigations†
Carmen R.
Barone
a,
Cecilia
Coletti
b,
Ruth J.
McQuitty
c,
Nicola J.
Farrer
c,
Giuseppe
Lorusso
a,
Luciana
Maresca
*a,
Alessandro
Marrone
b,
Giovanni
Natile
a,
Concetta
Pacifico
a,
Simon
Parsons
d,
Nazzareno
Re
b,
Peter J.
Sadler
c and
Fraser J.
White
d
aDipartimento di Chimica, Università degli Studi “Aldo Moro” di Bari, Via E. Orabona 4, I-70125 Bari, Italy. E-mail: luciana.maresca@uniba.it
bDipartimento di Farmacia, Università degli Studi “Gabriele d'Annunzio”, Via dei Vestini 31, I-06100 Chieti, Italy
cDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK
dSchool of Chemistry, The University of Edinburgh, King's Building, West Mains Road, Scotland, EH9 3JJ, UK
First published on 20th February 2013
Abstract
In this work UVA and blue light have been used to study photo-isomerisation about the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond in complexes of the type [PtCl(–CHCHAr)(tmeda)] [Ar = C6H5, (E)-2a; 4-CH3O–C6H4, (E)-2b; 3-NO2–C6H4, (E)-2c; and 3-CH3O–C6H4, (E)-2d]. The progress of the reaction has been monitored by NMR spectroscopy following irradiation of the NMR sample. The NMR data have been complemented with X-ray diffractometric analysis of compounds (E)-2a–c and (Z)-2a. The kinetic data clearly indicate that a monomolecular mechanism is operating with the energy of the irradiating light influencing the rate of isomerisation but not the equilibrium composition, which is only slightly in favour of the Z isomer. DFT and TD-DFT theoretical investigations have been carried out to elucidate the nature of the main electronic transitions in the UV-Vis region and the mechanism of the photo-isomerisation reaction appears to proceed through a CC bond twist process similar to that involved in purely organic molecules such as stilbene. In the Z isomer, one ortho proton of the phenyl group can come close to platinum (Pt⋯Hortho distance of 2.632 Å in (Z)-2a). In the case of 2c, the difference in chemical shift between the two ortho protons varies from 3.30 ppm in the Z isomer, where interaction with Pt is possible, to 0.60 ppm in the E isomer, where such interaction cannot take place. The analysis of the DFT orbitals indicates that the most shifted Hortho is that with a greater positive charge, pointing to an H-bond type of interaction.
C double bond in complexes of the type [PtCl(–CHCHAr)(tmeda)] [Ar = C6H5, (E)-2a; 4-CH3O–C6H4, (E)-2b; 3-NO2–C6H4, (E)-2c; and 3-CH3O–C6H4, (E)-2d]. The progress of the reaction has been monitored by NMR spectroscopy following irradiation of the NMR sample. The NMR data have been complemented with X-ray diffractometric analysis of compounds (E)-2a–c and (Z)-2a. The kinetic data clearly indicate that a monomolecular mechanism is operating with the energy of the irradiating light influencing the rate of isomerisation but not the equilibrium composition, which is only slightly in favour of the Z isomer. DFT and TD-DFT theoretical investigations have been carried out to elucidate the nature of the main electronic transitions in the UV-Vis region and the mechanism of the photo-isomerisation reaction appears to proceed through a CC bond twist process similar to that involved in purely organic molecules such as stilbene. In the Z isomer, one ortho proton of the phenyl group can come close to platinum (Pt⋯Hortho distance of 2.632 Å in (Z)-2a). In the case of 2c, the difference in chemical shift between the two ortho protons varies from 3.30 ppm in the Z isomer, where interaction with Pt is possible, to 0.60 ppm in the E isomer, where such interaction cannot take place. The analysis of the DFT orbitals indicates that the most shifted Hortho is that with a greater positive charge, pointing to an H-bond type of interaction.
Introduction
Metal alkenyl complexes of late transition elements have been known for several decades and their importance is associated with metal-mediated reactions involving unsaturated hydrocarbons.1 With reference to species having the metal directly bound to a vinyl carbon (also called metal–vinyl compounds), they are generally obtained as a result of: (i) nucleophilic addition to a coordinated alkyne or a metal vinylidene complex,2,3 (ii) insertion of an alkyne, or sometimes an allene, into a metal–hydrogen,4–10 metal–carbon,11,12 or even metal–sulphur bond,13 (iii) oxidative addition to low valent metal species.14–17 In the formation of metal alkenyl complexes, the presence of substituents on the unsaturated carbons can lead to different isomers whose yield depends upon their kinetic preference and subsequent possible isomerisation process.2,6,8,11 An interesting example of how the regio-selectivity can be governed by steric effects has been reported.18Some years ago we found that alkenyl platinum(II) species could rapidly be formed by deprotonation of a coordinated styrene in cationic complexes of the type [PtCl(η2-CH2CHAr)(tmeda)]+, 1, (tmeda = N,N,N′,N′-tetramethyl-1,2-diaminoethane). The reaction of proton abstraction, promoted by poorly-coordinating bases (such as tertiary aliphatic amines or inorganic carbonate), was completely regio-selective and led to the unique formation of the E isomer [PtCl(E-CHCHAr)(tmeda)] [Ar = C6H5, (E)-2a; 4-CH3O–C6H4, (E)-2b; and 3-NO2–C6H4, (E)-2c]. The observed selectivity appears to have a kinetic origin since in the cationic precursor the abstracted proton (the one trans to the aromatic ring) is the most exposed to the solvent and, hence, the most easily accessed by the attacking base.19
In chlorinated solvents, the E isomer partially isomerises into the corresponding Z form. Thus we decided to investigate the mechanism of the isomerisation process to see if the presence of a metal atom (platinum in the present case) directly bound to an olefinic carbon would influence a transformation very much alike to the classic cis–trans isomerisation of a stilbene molecule. In our system the isomerisation is extremely slow in the dark but is catalyzed by visible light. In the case of (E)-2a and (E)-2b, some unidentified decomposition products were formed in solution over prolonged period of time, the formation of which was less evident in the photo-activated process. We, therefore, decided to study the E–Z isomerisation process using a broad band light source.
UV and visible radiation has been widely used in chemistry to initiate chemical reactions or to carry out photochemical processes. Particularly challenging is the possibility of inducing specific reactions by using excitation radiation of a given wavelength and, indeed, lasers and other light sources have been used not only in conventional photochemistry, but also as a mean to induce specific effects and produce selective photochemical reactions.20
In this work UVA and blue light have been used to study a conventional process: the photo-isomerisation about a CC double bond. Thus, the progress of the reaction has been monitored by NMR spectroscopy following irradiation of the NMR sample.21 This type of isomerisation generally causes only minor changes in the UV-Vis spectrum; in contrast, large differences can be observed in the chemical shifts of some NMR signals belonging to the two isomers, allowing the ratio between isomers to be precisely evaluated by recording the NMR spectrum (1D 1H NMR in the present case).
A second focus of the present work was a better understanding of the type of interaction occurring between the platinum centre and the phenyl ortho protons in the Z isomer which, as in the case of compound (Z)-2c, can cause a downfield shift greater than 3 ppm and ten times greater for one ortho proton over the other. The NMR and kinetic data have been complemented with the X-ray diffractometric analysis of compounds (E)-2a–c and (Z)-2a and with DFT and TD-DFT (time-dependent density functional theory) theoretical investigations which allowed us to shed light on the mechanism of the photo-isomerisation process as well as on the nature of the interaction between the platinum centre and the phenyl ortho protons in the Z isomer.
Results
The complexes
The complexes [PtCl(E-CHCHAr)(tmeda)] (Ar = C6H5, (E)-2a; 4-CH3O–C6H4, (E)-2b; 3-NO2–C6H4, (E)-2c; and 3-CH3O–C6H4, (E)-2d, see Chart 1) were prepared by the reaction of triethylamine with the corresponding parent cationic complex as previously described for compounds (E)-2a–c.19 Complex (E)-2d is new and we decided to also include in our series the complex containing 3-CH3O-styryl since a meta-methoxy substituent on the phenyl ring would behave as a pure, though weak, electron-withdrawing group since its +M effect does not affect the ipso carbon atom.22 Thus 2d can be placed somewhat in between 2a and 2c, as far as the electronic characteristics are concerned.
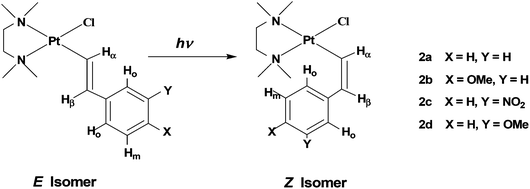 | ||
| Chart 1 Sketches of isomers E and Z with identification of alkenyl protons and numbering of the complexes. | ||
Complexes (E)-2c and (E)-2d (both containing an electron-withdrawing group on the phenyl ring) appear to be more stable in chloroform solution than (E)-2a and (E)-2b and, after exposure to sunlight for several hours, a E/Z ratio close to 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 was reached without any detectable formation of decomposition products.
:50 was reached without any detectable formation of decomposition products.
Photo-isomerisation of (E)-2a and (E)-2b
Compounds 2 were obtained as unique reaction products (1H NMR and elemental analyses); however, ESI-MS spectra, recorded under low cone voltage, of samples of (E)-2a and (E)-2b, prepared according to the reported procedures (see ref. 19), did show the presence of trace amounts (undetectable in 1H NMR and eluded by elemental analysis) of [PtCl{η1-CH(Ar)CH2NEt3}(tmeda)]+ and/or isomeric [PtCl{η1-CH2CH(Ar)NEt3}(tmeda)]+, which had been formed by amine (NEt3) addition to the η2 olefin in the parent cationic complex [PtCl{η2-CH2CH(Ar)}(tmeda)]+. To avoid the possibility that these impurities, during the irradiation process, could affect product formation, (E)-2a and (E)-2b were purified by conventional column chromatography which eliminated the cationic species as confirmed by ESI-MS runs (always under low cone voltage).
Absorption (UV-Vis) spectra in CDCl3 (Fig. S1†) reveal significant absorption bands for (E)-2a and (E)-2b at wavelengths <380 nm, with an absorption maximum close to 280 nm which has been assigned by the TD-DFT calculations to an essentially π–π* transition (see below). Moreover, the absorption spectra in CDCl3 do not show significant changes upon E–Z isomerisation. However, the E and Z isomers of a given alkenyl have well separated 1H NMR signals which allow an accurate evaluation of their relative concentrations, in particular the signal of the high-field vinylic proton in the E isomers (HβE) and that of the low field ortho-phenyl proton in the Z form (HoZ) (see Chart 1 and Fig. 1 for alkenyl proton identification scheme). Therefore we decided to follow the isomerisation process by placing the samples in NMR tubes and irradiating with continuous wave (CW) light of wave length 351 or 420 nm. These wavelengths fall in the tail of the UV absorption band, ensuring a low absorption and a sufficient penetration of the radiation into the NMR sample. For the same reason (penetration of the radiation into the sample), the complex concentration was also kept rather low (8–13 mM).
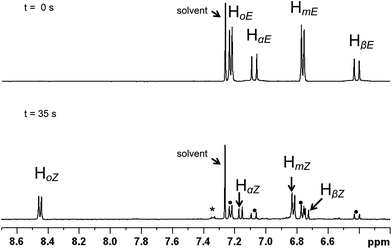 | ||
| Fig. 1 Aromatic region of the 1H NMR spectrum of 2b in CDCl3 (13 mM). Top: prior to irradiation (pure E isomer). Bottom: after irradiation to the PSS (λ = 351 nm, 35 min); apart from resonances of the Z isomer, residual signals of the E form are also indicated (•). The final spectrum suggests that there may be a small amount of photo-decomposition (*). | ||
For both compounds, (E)-2a and (E)-2b, an E/Z ratio of ca. 38/62 was reached at equilibrium. The energy of the irradiating light did not affect the equilibrium composition, but influenced the rate of isomerisation. The photostationary state (PSS), defined as the state for which the forward and the back reactions have equal rates, was reached after ca. 30 and 120 min of irradiation at 351 and 420 nm, respectively. On the basis of a first order transformation of one isomer into the other, the integrated rate law for the PSS model is given by eqn (1) which represents the rate law for the reaction:
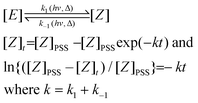 | (1) |
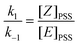 | (2) |
![Photo-isomerisation of (E)-2b (λ = 351 nm). Top: plot of percentage of Z isomer (Z%) versus time of irradiation (s). Bottom: plot of −ln{([Z]PSS − [Z]t)/[Z]PSS} against time (s).](/image/article/2013/DT/c3dt32354d/c3dt32354d-f2.gif) | ||
| Fig. 2 Photo-isomerisation of (E)-2b (λ = 351 nm). Top: plot of percentage of Z isomer (Z%) versus time of irradiation (s). Bottom: plot of −ln{([Z]PSS − [Z]t)/[Z]PSS} against time (s). | ||
X-ray structure
Crystals suitable for X-ray analysis were obtained for (E)-2a–c (Fig. S3–S5 and Table S1†) and (Z)-2a (Fig. 3). Relevant bond distances and angles are reported in Table 1, where calculated values for E and Z isomers of 2a–d have been included (see Quantum chemical calculations, later on in this section). In all molecules, platinum(II) has the usual square-planar coordination geometry, when the alkenyl carbon, the chloride, and the tmeda nitrogen atoms are taken into account. As expected, the chelated diamine penta-atomic ring is puckered to accommodate the tetrahedral geometry of the nitrogen and carbon atoms. The distances between the metal centre and the two tmeda nitrogen atoms are significantly different, owing to the stronger trans-influence of the σ carbon with respect to the chlorido ligand (the Pt–N(2) bond trans to the alkenyl carbon being, on average, 0.1 Å longer than that trans to the chlorido ligand). The Cα–Cβ and Cβ–Cipso bond lengths are typical of a double and a single bond, respectively; indicating, though, that there is little conjugation between the olefin double bond and the π system of the phenyl ring.![Drawing of complex [PtCl{Z-CHCHC6H5}(tmeda)], (Z)-2a, showing the position of the ortho hydrogen apical on the Pt coordination plane (Pt⋯H2ortho distance = 2.632 Å and Pt⋯Hortho–Cortho (ψ) angle = 138°). The ellipsoids enclose 30% probability.](/image/article/2013/DT/c3dt32354d/c3dt32354d-f3.gif) | ||
| Fig. 3 Drawing of complex [PtCl{Z-CHCHC6H5}(tmeda)], (Z)-2a, showing the position of the ortho hydrogen apical on the Pt coordination plane (Pt⋯H2ortho distance = 2.632 Å and Pt⋯Hortho–Cortho (ψ) angle = 138°). The ellipsoids enclose 30% probability. | ||
| (E)-2a | (Z)-2a | (E)-2b | (Z)-2b | (E)-2c | (Z)-2c | (E)-2d | (Z)-2d | |
|---|---|---|---|---|---|---|---|---|
| Pt1–N1 | 2.068(4) | 2.074(7) | 2.069(3) | 2.071(7) | ||||
| 2.074(4) | ||||||||
| 2.072(4) | ||||||||
| [2.107] | [2.106] | [2.107] | [2.108] | [2.107] | [2.109] | [2.107] | [2.106] | |
| Pt1–N2 | 2.164(4) | 2.161(7) | 2.179(3) | 2.160(7) | ||||
| 2.185(4) | ||||||||
| 2.180(4) | ||||||||
| [2.222] | [2.223] | [2.224] | [2.223] | [2.282] | [2.285] | [2.222] | [2.221] | |
| Pt1–Cl1 | 2.298(1) | 2.305(2) | 2.3021(8) | 2.304(2) | ||||
| 2.305(1) | ||||||||
| 2.302(1) | ||||||||
| [2.284] | [2.288] | [2.283] | [2.284] | [2.282] | [2.281] | [2.283] | [2.287] | |
| Pt1–C8 | 1.984(4) | 1.977(9) | 1.992(3) | 1.990(9) | ||||
| 1.983(5) | ||||||||
| 1.977(4) | ||||||||
| [1.974] | [1.975] | [1.975] | [1.975] | [1.971] | [1.971] | [1.973] | [1.974] | |
| C7–C8 | 1.315(7) | 1.34(1) | 1.312(5) | 1.33(1) | ||||
| 1.322(7) | ||||||||
| 1.330(6) | ||||||||
| [1.339] | [1.341] | [1.339] | [1.339] | [1.339] | [1.340] | [1.339] | [1.341] | |
| C1–C7 | 1.479(6) | 1.48(1) | 1.479(4) | 1.46(1) | ||||
| 1.481(7) | ||||||||
| 1.475(6) | ||||||||
| [1.467] | [1.467] | [1.467] | [1.467] | [1.464] | [1.464] | [1.467] | [1.467] | |
| N1–Pt1–N2 | 84.8(2) | 84.5(3) | 84.05(1) | 83.8(3) | ||||
| 84.4(2) | ||||||||
| 84.5(2) | ||||||||
| [84.03] | [83.98] | [84.04] | [83.95] | [84.04] | [84.00] | [84.04] | [84.00] | |
| Cl1–Pt1–C8 | 90.4(1) | 89.8(3) | 89.9(1) | 89.3(3) | ||||
| 92.2(1) | ||||||||
| 91.7(1) | ||||||||
| [90.17] | [90.45] | [90.29] | [89.89] | [90.71] | [90.91] | [90.44] | [90.52] | |
| N2–Pt1–Cl1 | 92.5(2) | 91.9(2) | 91.8(1) | 92.5(2) | ||||
| 91.8(1) | ||||||||
| 93.0(1) | ||||||||
| [91.85] | [91.76] | [91.85] | [91.87] | [91.80] | [91.80] | [91.85] | [91.66] | |
| C8–Pt1–N1 | 92.4(2) | 93.9(3) | 93.8(1) | 94.3(3) | ||||
| 91.9(2) | ||||||||
| 90.8(2) | ||||||||
| [93.96] | [93.78] | [93.83] | [94.26] | [93.43] | [93.24] | [93.67] | [93.77] | |
| C7–C8–Pt1 | 127.4(4) | 132.9(7) | 132.7(3) | 129.6(7) | ||||
| 131.7(4) | ||||||||
| 128.1(4) | ||||||||
| [127.13] | [131.5] | [127.03] | [127.72] | [126.98] | [129.78] | [127.20] | [131.90] | |
| C8–C7–C1 | 127.5(5) | 129.5(8) | 125.9(3) | 127.0(9) | ||||
| 124.4(5) | ||||||||
| 127.1(4) | ||||||||
| [127.06] | [129.0] | [127.43] | [126.73] | [126.66] | [128.11] | [127.06] | [129.85] |
There are three independent molecules in the asymmetric unit of (E)-2a (Fig. S3†); the only differences among them are in the values of the dihedral angle between the coordination and the olefin plane (φ1) and between the olefin plane and the phenyl ring (φ2) (the coordination plane is the mean plane defined by the four donor atoms and the olefin plane is that containing the Pt–Cα–Cβ–Cipso alkenyl back-bone). The values of φ1 and φ2, for all structurally characterized molecules, are reported in Table 2, where also the calculated values of the two angles for E and Z isomers of 2a–d have been included.
| Complex | φ 1 | φ 2 |
|---|---|---|
| (E)-2a | 49.7–86.1–89.8 | 25.8–5.8–23.2 |
| [78.2] | [10.2] | |
| (E)-2b | 83.9 | 19.9 |
| [78.2] | [2.8] | |
| (E)-2c | 72.3 | 15.1 |
| [88.7] | [7.3] | |
| (E)-2d | [79.6] | [1.9] |
| (Z)-2a | 87.3 | 5.1 |
| [83.7] | [26.9] | |
| (Z)-2b | [81.7] | [25.4] |
| (Z)-2c | [83.8] | [18.1] |
| (Z)-2d | [80.2] | [8.1] |
The angle φ1 in most cases lies above 80° and therefore close to the value of 90° which should minimize the steric interactions between the alkenyl and the cis ligands. In two cases, presumably depending upon crystal packing effects, it exhibits a somewhat lower value, being ∼70° in (E)-2c and ∼50° in one of the molecules of (E)-2a.
The angle φ2, which gives a measure of the co-planarity of the alkenyl moiety and the phenyl ring is usually small (<26°) and in two cases is not higher than 6°. However, there is no significant evidence of conjugation between the olefin double bond and the aromatic π system judging from the values of the Cα–Cβ and Cβ–Cipso bond lengths.
In (E)-2b, however, there is conjugation between the oxygen lone pair of the methoxy substituent on C4 and the phenyl π system, which is illustrated by the short value of the Me–O and O–Ph bonds (1.437(3) and 1.385(4) Å, respectively) and the large value of the Me–O–Ph angle (116.6(3)°). Seemingly the oxygen atom has sp2 hybridization and the O–Ph bond some double-bond character. A similar situation was found in the parent cationic complex [PtCl{η2-CH2CH(C6H4-4-OMe)}(tmeda)]+,22 and it was also previously described for some iminoether complexes of platinum.23 An oxygen atom adjacent to a double bond often contributes to the π system.
In this context it is worth noting the case of compound (Z)-2a (Fig. 3) for which the dihedral angle of the phenyl ring with respect to the olefin plane is very small (φ2 = 5°) while the olefin and the coordination planes are almost orthogonal (φ1 = 87°). In such a situation the phenyl ring is also practically perpendicular to the metal coordination plane (making an angle of 89°). This arrangement brings one ortho-proton of the aromatic ring in close contact with the platinum atom: the Pt⋯Hortho distance being 2.632 Å (Pt⋯Cortho = 3.4012(4) Å and Pt⋯Hortho–Cortho angle = 138°). Moreover, in (Z)-2a the platinum atom has a pyramidal distortion and is displaced by 0.0343(1) Å from the coordination plane towards the interacting hydrogen. In all other complexes investigated here the metal atom either exhibits a tetrahedral distortion [(E)-2a,b], or it lies approximately on the coordination plane [displaced by only 0.0034(4) Å in (E)-2c].
NMR spectroscopy
Significant NMR parameters for both E and Z isomers of 2a–d are summarized in Table 3. In the 1H NMR spectra of all compounds, the methyl groups on the tmeda nitrogen atoms give rise to two sharp singlets with significant differences in their chemical shifts and 3JPt,H coupling constants. These differences are caused by the different trans influences of the two ligands opposite to the two nitrogens. Thus, the two methyls on each nitrogen are magnetically equivalent implying fast rotation around the Pt–Cα bond. This feature is fully supported by the variability of the φ1 angles found in the solid state structure of (E)-2a, described in the preceding section.| Complex | Hortho | Hmeta | Hpara |
|---|---|---|---|
| (E)-2a | 7.27 | 7.18 | 7.01 |
| (Z)-2a | 8.50 | 7.24 | 7.10 |
| (E)-2b | 7.18 | 6.74 | — |
| (Z)-2b | 8.43 | 6.80 | — |
| (E)-2c | 8.10, 7.50 | 7.30 | 7.85 |
| (Z)-2c | 11.10, 7.80 | 7.35 | 7.95 |
| (E)-2d | 6.85, 6.88 | 7.10 | 6.59 |
| (Z)-2d | 8.46, 7.90 | 7.13 | 6.70 |
On going from the E to the Z isomers, the most significant differences observed in the 1H NMR spectra are: (i) the smaller 3JH,H between the two vinyl protons which in the Z isomer are cis disposed with respect to the olefinic double bond, and (ii) the chemical shifts of the two ortho protons of the phenyl ring which undergo a considerable downfield shift. When the two ortho protons are equivalent (this is the case of compounds 2a and 2b where the substituent on the phenyl ring is in the para position), the shift, in the Z isomer, is ∼1.3 ppm downfield with respect to the corresponding resonance in the E form. When the two ortho protons are not equivalent (this is the case of compounds 2c and 2d where the substituent on the phenyl ring is in the meta position), the downfield shifts are 1.61 and 1.02 ppm for compound (Z)-2d and 3.0 and 0.30 ppm for compound (Z)-2c (in both cases the more shifted proton is that close to the substituent). Therefore, the difference in chemical shift between the two ortho protons is much more marked in (Z)-2c (nitro substituent) than in (Z)-2d (methoxy substituent) with a downfield shift which is 10 times greater for one ortho proton than for the other. It is possible to conclude that in the case of (Z)-2c the residence time in the proximity of the metal atom (which causes the downfield shift) is much greater for one proton than for the other. This is most likely a consequence of the electron-withdrawing properties of the nitro group which increases the partial positive charge on the adjacent ortho proton. As a consequence, this proton can form a hydrogen bond type interaction with the metal centre much greater than that of the other ortho proton which is much further from the nitro group. It is also to be noted that the mean downfield shift of the ortho protons in the Z isomer, with respect to the E isomer, is in the range 1.23–1.31 ppm in three out of the four complexes examined and becomes 1.65 ppm in the nitro-derivative, further evidence of a strong Pt⋯H interaction in the last case.
Quantum chemical calculations
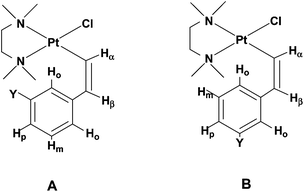 | ||
| Chart 2 Sketches of the two possible conformers of (Z)-2c and (Z)-2d. | ||
A marked increase of the charge on the Hortho is observed on going from the E to the Z isomer. This charge increase in the Z isomer is particularly high for the H atom next to the electron-withdrawing –NO2 substituent in (Z)-2c. Parallel to this high charge increase, we observe a shorter Pt⋯Hortho distance, 2.461 Å in (Z)-2c, to be compared with 2.738 Å in (Z)-2a; both effects being diagnostic of a strong Pt⋯Hortho hydrogen bond interaction. The presence of such an interaction is further supported by the comparison of the relative energies of the two possible conformers of the Z isomers (with either of the two non-equivalent ortho hydrogens interacting with the platinum centre). In (Z)-2c the conformer A, where the metal atom interacts with the Hortho closer to the electron-withdrawing –NO2 substituent, is significantly lower in energy (by 2.9 kJ mol−1; 7.0 kJ mol−1 in gas phase) than conformer B; while in (Z)-2d the two conformers are almost isoenergetic (they differ by only 0.2 kJ mol−1; 0.7 kJ mol−1 in the gas phase). The larger stability of conformer A in (Z)-2c also explains why the difference in chemical shift between the two ortho protons is so marked (Δδ of 3.30 in (Z)-2c to be compared with a Δδ of 0.56 ppm in (Z)-2d, Table 3) with the proton closer to the substituent shifting at lower field. Indeed, in the case of (Z)-2c conformer A, where the platinum atom interacts with the most positively-charged ortho hydrogen, is much more populated than conformer B; in contrast, in the case of (Z)-2d the two conformers are almost equally populated.
The nature of the Pt⋯Hortho interaction in the Z isomers was also investigated by performing a natural bond orbital (NBO) analysis which, through a perturbation theory approach in terms of bond orbitals, allows unravelling of specific donor–acceptor interactions involving metal centred orbitals (empty or filled) and two-centred C–H orbitals (bonding or antibonding) (Table S4†). The analysis shows that the most energetically important contribution to the Pt⋯Hortho interaction is a donation from the filled dz2 orbital of the metal to the empty σ* orbital of C–Hortho. Such an interaction has already been proposed as the main theoretical feature of M⋯H–C hydrogen bond-like interactions.24 An additional but minor donation can come from a filled π(Pt–Cl) orbital (comprising an important metal dxz component) and a chlorine lone pair to the empty σ*(C–Hortho) antibonding orbital. Moreover, the metal dz2/dxz to σ*(C–Hortho) donation increases on going from (Z)-2a, (Z)-2b or (Z)-2d to (Z)-2c and is significantly higher for conformer A, where the most positively-charged Hortho is closer to the Pt atom, further confirming the hydrogen-bond nature of the Pt⋯Hortho interaction.
C) → π*(phenyl) contribution. Three much weaker transitions of lower energy, falling at 286 (S3), 290 (S2) and 309 (S1) nm, with mainly π(CC) → d or d → d character, contribute to the absorption shoulder around 300 nm. Note that the two most intense transitions of (E)-2a, S4 and S6, correspond to the two lowest excited states of stilbene and have mainly the same character, respectively π(CC) → π*(CC) and π(CC) → π*(phenyl), and similar excitation energies.
To shed some light on the first steps along the E/Z photo-isomerisation pathway, a TD-DFT relaxed scan of the potential energy surface (PES) for the excited S4 state of (E)-2a was performed as a function of the Pt–CαCβ–Hβ torsion angle around the alkenyl CC double bond (ϕ varied from 180 to 105°). The results are reported in Fig. S8† and show how, on decreasing ϕ from 180° to 105°, there is a slow decrease of S4 which intersects the excited states S1–S3 and becomes the lowest excited state for ϕ below ca. 150°. Angles below 105° were not considered due to the well-known instability of the TD-DFT approach near the conical intersection with the ground state. This picture was confirmed by a full geometrical optimization of the lowest triplet state, not showing any stability problem, which leads to the expected structure with ϕ = 90° and a broken alkenyl CC double bond.
Thus the obtained results suggest, for the E/Z photo-isomerisation process, the traditional CC bond twist mechanism characterized for the analogous purely organic stilbene molecule.25 Upon photo-excitation, the excited state relaxes out of the FC region toward partially twisted structures of the E-isomer on the S4 surface and reaches the S4/S0 conical intersection, thus isomerizing to the ground state Z-isomer. The presence of three curve crossings with S1–S3 could slow down the relaxation to the conical intersection with S0, thus allowing part of the excited state population to quickly decay back to the ground state via fluorescence. However the latter possibility was not investigated since it cannot be adequately approached with the TD-DFT method.
Discussion
Deprotonation versus nucleophilic addition
The formation of alkenyl complexes by deprotonation of a η2-bonded olefin represents an interesting synthetic procedure, which, however, depends very much upon steric effects. The η2-bonded olefin in compounds of type 1 is highly electrophilic and usually reacts with a base to give nucleophilic addition.22,26,27 The steric bulk of triethylamine, used as a base, produces severe retardation of the nucleophilic addition (only a tiny amount of addition product is formed) allowing the alternative process of olefin deprotonation to become dominant. The deprotonation involves the hydrogen most exposed to the solvent (that lying trans to the phenyl ring) and not the most acidic one which, in free styrene, is that on the carbon bearing the phenyl group.28 The pivotal role of the steric bulk in this type of reaction is also supported by the observation that, in cationic complexes of type 1, the addition of a tertiary aminic function to the olefin has been observed exclusively in the case of un-substituted ethene29 and that, in the case of NHEt2, the product of addition to the cationic complexes 1a and 1b sees, at equilibrium, a large preference for the anti-Markovnikov isomer.27Solid-state and solution structure of the alkenyl complexes
X-ray parameters of (E)-2a–c and (Z)-2a are in agreement with literature data for platinum alkenyl complexes.30,31 In our series of complexes the metal-to-carbon distances are just below 2 Å and are in line with those reported for other platinum-to-carbon σ bonds opposite a donor of similar trans influence (an aminic nitrogen in our case).30 Similarly, the trans influence of an alkenyl ligand is found to be practically identical to that of an alkyl group (Pt–N distances trans to Pt–Cα (Table 1) in the range 2.16–2.18 Å as found for the bond between platinum and the tmeda nitrogen trans to a sp3-hybridized carbon atom).27 The lack of conjugation between the vinyl group and the π system of an aromatic substituent appears to be quite general31 notwithstanding the quasi co-planarity of the alkenyl and phenyl moieties observed in several cases in the solid state. In contrast, there is conjugation between the oxygen lone-pair of a methoxy substituent and the phenyl π-system fully confirming previous observations.22,23Of particular interest is the very short Pt⋯Hortho distance observed in compound (Z)-2a (2.632 Å), accompanied by a pyramidal distortion of the square-planar coordination and a significant displacement of the platinum atom from the coordination plane towards the phenyl ortho proton, while the Pt⋯H–C angle (ψ) is ca. 140°. The values of M⋯H distance and ψ angle have been assumed as useful criteria to distinguish between agostic and non-agostic M⋯H–X interactions.24,32–35 In particular, agostic interactions (which involve two electrons and three centres)32 are characterized by a relatively short M⋯H distance (1.8–2.3 Å) and a ψ angle value in the range 90–140°. Unlike agostic interactions, hydrogen bond interactions (which involve four electrons and three centres)34,35 exhibit a relatively longer M⋯H distance (2.3–2.9 Å) and a larger ψ angle which can range from 160–180° for stronger bonds to 140–160° for weaker bonds, thus approaching the upper value of the range reported for agostic interactions. However, the values of crystallographic parameters alone might not be sufficient to evaluate the type of interaction between a metal and a hydrogen atom lying in its proximity, whereas solution NMR data are much more informative in this respect.
A most relevant feature resulting from the NMR investigation is the downfield shift experienced by the ortho phenyl protons upon E to Z isomerisation. In principle this downfield shift could be ascribed to the magnetic anisotropy of the metal centre. Large downfield shifts are typical of protons in a pseudo-axial position above the platinum atom in a square-planar complex.36 However, the difference in downfield shift between the two ortho phenyl protons in 2d and in 2c cannot be due to a generic effect of the magnetic anisotropy of the metal core, nor to an agostic interaction (which typically produce an upfield shift), but points to a type of hydrogen bond interaction. The possibility for the platinum atom to act as H-bond acceptor, also in neutral complexes, is now well documented.35 In the alkenyl complexes here described, we are in the fortunate case of a phenyl group with the two ortho protons having different partial positive charges depending upon the electron-withdrawing strength of the adjacent substituent. The greater the positive charge accumulated on the hydrogen atom, the greater is its downfield shift. In the case of (Z)-2c (nitro substituent) the downfield shift for the more positive ortho proton is ten times greater than for the other ortho proton, indicating that the greater the positive charge of the hydrogen, the more exclusive is its interaction with the metal centre; this is typical of the H-bond type interaction. There is no detectable JPt,H coupling resulting from such a Pt⋯H interaction, which could be essentially electrostatic. It must be pointed out that, even in compound (Z)-2c, where the Pt⋯Hortho interaction is very strong, in solution the alkenyl moiety can freely rotate about the Pt–Cα bond as indicated by the equivalence of the methyls on each nitrogen of the tmeda ligand.
The nature of the Pt⋯Hortho interaction is fully supported by the results obtained with the performed calculations. In particular the good agreement between experimental and calculated value of the Pt⋯Hortho distance in (Z)-2a gives complete significance to the calculated values of all other Z isomers setting an important H-bond interaction between the metal and the Hortho protons of the phenyl group. However, even in the case of (Z)-2c; where there is a very strong H-bond interaction with one Hortho, the free rotation around the Cβ–Cipso bond is not prevented in solution (as already pointed out in the previous paragraph). Moreover, this picture is also supported by the calculated charges on the Hortho, which are consistent with the experimental 1H NMR shifts. Indeed, the analysis of the DFT orbitals indicates that the most shifted Hortho is that with greater positive charge, pointing to an H-bond type of interaction.
Isomerisation around the double bond
The E/Z isomerisation around the CC double bond, which was another focus of this work, is a process which is seldom observed in alkenyl metal complexes, where the presence of different forms generally stems from different reaction mechanisms rather than from a post-formation isomerisation process.6
There are cases, however, in which, starting from a kinetically-determined isomeric composition of the alkenyl, a new composition, under thermodynamic control, has been obtained by administering some energy as heat2,10 or light (as in our case), or by electrochemical oxidative catalysis (the E+ → Z+ transformation appears to be faster than that occurring between neutral molecules).37
In our case the equilibrium composition was only slightly in favour of the Z isomer and the kinetic data clearly indicate that a monomolecular mechanism is operating. As expected, the energy of the irradiating light influences the rate of isomerisation but not the equilibrium composition.
As far as the mechanism of the isomerisation process is concerned, TD-DFT calculations indicate that it proceeds through the traditional CC bond twist process operating in purely organic molecules, like stilbene.
Experimental
Reagents and methods
Reagents and solvents were commercially available and used as received without further purification. Elemental analyses were performed with a CHN Eurovector EA 3011.1H and 13C NMR spectra were recorded on a 300 MHz Mercury Varian, DPX-WB 300 and 500 Avance Bruker instruments equipped with probes for inverse detection and with z gradient for gradient-accelerated spectroscopy. 1H and 13C NMR spectra were referenced to TMS; the residual proton signal of the solvent was used as internal standard. 1H/13C inversely detected gradient-sensitivity enhanced hetero-correlated 2D NMR spectra for normal coupling (INVIEAGSSI) was acquired using standard Bruker automation programs and pulse sequences. Each block of data was preceded by eight dummy scans. The data were processed in the phase-sensitive mode.
ESI-MS spectra were recorded on a Platform II Mass spectrometer (Micromass, Manchester, U.K.).
UV-Vis electronic absorption spectra were obtained on a Varian Cary 300 UV-Vis spectrophotometer equipped with a temperature control block using 1 cm path-length cuvettes. All spectra were baseline-corrected using pure solvent as a background. Data were processed using Microsoft Office Excel.
The complexes
The alkenyl complexes [PtCl(η1-CαH–CβHAr)(tmeda)], 2, (Ar = C6H5, (E)-2a; 4-CH3O–C6H4, (E)-2b; 3-NO2–C6H4, (E)-2c; and 3-CH3O–C6H4, (E)-2d) were prepared according to the procedure described in ref. 19. Analytical data for the yet un-reported 2d, isolated as pure E isomer, are hereafter listed. Elemental analysis calcd (%) for C15H25ClN2OPt (479.9): C 37.54, H 5.25, N 5.84; found: C 37.25, H 5.31, N 5.70. NMR (CDCl3, 294 K): δ(1H) = 2.76 and 2.92 (s, 12 H), CH3N (tmeda); 2.60 and 2.80 (m, 4 H), CH2N (tmeda); 3.78 (s, 3 H), –C6H4OCH3; 6.42 and 7.32 (d, 2H, 3JH–Hcis = 15 Hz) –CαH–CβH–; aromatic ring: 6.59 (dd, 1H) Hp; 6.85 (s, 1 H) and 6.88 (dd, 1 H) Ho; 7.10 (m, 1H) Hm.Prior to light irradiation experiments (E)-2a and (E)-2b samples were chromatographed on silica gel, using dichloromethane–ethyl acetate, 9:1 v/v, as eluent. All manipulations were carried out by shielding the complexes from light.
Combined irradiation-NMR experiments
Solutions of compound (E)-2a and (E)-2b (600 μL, 13 mM in CDCl3) in a 5 mm NMR tube were irradiated in a Luzchem 1CH2 photo-reactor (Hitachi FL8BL), fitted with B UVA (λmax = 351 nm) or Luzchem LZC-420 (λmax = 420 nm) bulbs at 310 K. The power of the light source was measured with an A.W. Sperry SLM-110 power meter and an International Light Technologies ILT 1400-A photometer using a UVA#28880 or a W#12664 filter in the case of lower and higher wavelength, respectively. At regular intervals the samples were removed and analysed by 1H NMR spectroscopy.In both sets of experiments, the spectroscopic analyses took approx. 15 min to be completed. When not being irradiated the solutions were handled under low lighting conditions and no change in speciation was observed on this timescale in the absence of the light source. All 1H NMR spectra were acquired with 32 scans on a Bruker DRX-500 NMR spectrometer (frequency width was 20000 Hz (20 ppm) pulse angle was 30°, the relaxation delay 1 s). The data were analysed using Topspin (version 2.1). Integration of proton signals HoZ, HoE, HαZ and HαE was used to determine the ratios of the different isomers.
X-ray investigations
Diffraction data were collected: for (E)-2a, (Z)-2a, and (E)-2b on a Bruker Smart Apex-II diffractometer equipped with an Oxford Cryosystems low-temperature device operating at 150 K; and for (E)-2d on an Bruker AXS X8 APEX CCD system equipped with a four-circle Kappa goniometer and a 4K CCD detector (in this case data collection was performed at room temperature). MoKα radiation was used in all cases. Corrections for systematic errors were performed with SADABS program.38 The structures were solved using DIRDIF,39 SHELXS,40 or SIR2004;41 and they were refined by full matrix least squares against F2 and all data (SHELXL).40 All calculations and molecular graphics were carried out using PARST97,42 WinGX,43 and Mercury CSD 2.0.44 All non-H atoms were refined with anisotropic displacement parameters, and H-atoms were included in ideal positions. Symmetry checks on structure (E)-2a, which contains three molecules in the asymmetric unit, do not indicate any missed translational operation.Computational methods
The molecular structures of the ground states of the (E) and (Z)-2a complexes were initially optimized at the DFT level using the B3LYP45 BH-HLYP,46 PBE,47 and PBE048 functionals. The energies of the 20 lowest lying singlet-singlet energy transitions for (E)-2a were calculated at the optimized geometry with the time dependent density functional theory (TD-DFT)49 employing the same exchange-correlation functionals. The comparison of the optimized geometries with the experimental X-ray geometry, see Table S2,† shows a better agreement for the PBE0 functional. The comparison of the calculated UV-Vis spectra (only that for the PBE0 is reported in Fig. S7†) with the experimental spectrum in Fig. S1,† also shows that the PBE0 functional can better reproduce the experimental features (e.g. 277 vs. 280 nm for the maximum with a shift of only 3 nm, instead of about 10 nm for all other functionals). We therefore employed the PBE0 functional for further calculations on the ground state of all considered alkenyl Pt(II) complexes (E) and (Z)-2a–d, and on the excited states of (E)-2a.Reaction enthalpies and free energies for the E/Z isomerisation were estimated at this level of theory for all Pt alkenyl complexes. For 2c and 2d species two conformations per metal complex, differing by a 180° rotation of the phenyl ring around the Cβ–Cipso bond (C7–C1 in Fig. 3), were considered. The rotation is expected to be almost free at room temperature. The reported enthalpies and free energies were computed by using the lowest energy conformation for each isomer. The free energy differences between these two conformations in complexes 2-c and 2-d were used to perform a Boltzmann-weighted average of chemical shifts (vide infra).
The structures obtained at the DFT and TD-DFT levels were confirmed to be energy minima by calculating the harmonic vibrational frequencies using numerical differences. The Karlsruhe triple-ζ basis sets augmented with one set of polarization functions, designed for small core ECPs (def2-TZVP) were used.50 All calculations have been done with TURBOMOLE.51 The effect of chloroform solvent was taken into account by COSMO polarisable continuum model (ε = 4.9), as implemented in TURBOMOLE.51
Further calculations on the PBE0 optimized geometries of alkenyl complexes were performed by using Gaussian 03.52 Natural bond orbitals (NBO) analyses, Mulliken atomic charges and isotropic shielding constants were calculated by using PBE0 (PBE1PBE in the Gaussian 03 implementation) and double-ζ basis sets: the all electron 6-31G(d,p) basis set was used for C, N, O, Cl and H atoms,53 whereas the LANL2DZ basis set, comprising the LANL2 effective core potential for the inclusion of relativistic effects, was used for the Pt atom.54
1H chemical shifts of (E) and (Z)-2a–d complexes were calculated by using the gauge-independent atomic orbital (GIAO) method.55 The chemical shifts were obtained by subtracting the reference isotropic shielding of TMS protons calculated at the same PBE0 level of theory. Both GIAO and NBO calculations were carried out in the presence of a polarizable continuum simulating the chloroform solution (ε = 4.9) by means of the CPCM method56 implemented in Gaussian 03.
Conclusions
The alkenyl complexes of platinum(II), examined in the present investigation, can have E or Z configuration. The ground state energy of the two isomers is quite similar and, in chlorinated solvents, the [Z]/[E] ratio at equilibrium is only slightly above 1. The E forms are kinetically favoured and rather stable at room temperature; however, by heating or by irradiation with UVA and blue light, the thermodynamic equilibrium between E and Z isomers can be reached.The isomerisation process most probably occurs through the classical CC bond twist mechanism operating in organic molecules such as stilbenes.
In the Z isomers an H-bond can be established between an ortho hydrogen of the phenyl ring and the platinum centre whose strength parallels the Mulliken charge of the hydrogen atom.
Acknowledgements
We are grateful to Prof. Ferdinando Scordari and Dr Ernesto Mesto (University of Bari, Italy) for the X-ray diffraction data collection (compound 2c). We thank the EPSRC (grant no EP/G006792/1 and studentship for RJM); ERC (grant no 247450); AWM/ERDF/Science City; the University Consortium for Research in Chemistry of Metal Ions in Biological Systems (C.I.R.C.M.S.B.) for their support for this work.Notes and references
- T. L. Gianetti, N. C. Tomson, J. Arnold and R. G. Bergman, J. Am. Chem. Soc., 2011, 133, 14904–14907 CrossRef CAS; D. Vasu, A. Das and R.-S. Liu, Chem. Commun., 2010, 46, 4115–4117 RSC; L.-B. Han, Y. Ono, Q. Xu and S. Shimada, Bull. Chem. Soc. Jpn., 2010, 83, 1086–1099 CrossRef.
- D. L. Reger and P. J. McElligot, J. Am. Chem. Soc., 1980, 102, 5293–5294 CrossRef.
- M. J. Cowley, J. M. Lynam, R. S. Moneypenny, A. C. Whitwood and A. J. Wilson, Dalton Trans., 2009, 9529–9542 RSC.
- H. Werner, M. A. Esteruelas and H. Otto, Organometallics, 1986, 5, 2295–2299 CrossRef CAS.
- M. R. Torres, A. Vegas, A. Santos and J. Ross, J. Organomet. Chem., 1986, 309, 169–177 CrossRef CAS.
- H. C. Clark, G. Ferguson, A. B. Goel, E. G. Janzen, H. Ruegger, P. Y. Siew and C. S. Wong, J. Am. Chem. Soc., 1986, 108, 6961–6972 CrossRef CAS.
- C. Allevi, L. Garlaschelli and M. C. Malatesta, J. Chem. Soc., Dalton Trans., 1988, 1949–1955 RSC.
- J. A. Cabeza, I. del Rio, S. Garcia-Granda, L. Martinez-Mendez and E. Pérez-Carreno, Chem.–Eur. J., 2005, 29, 6040–6052 CrossRef.
- J. A. Cabeza, I. del Rio, J. M. Fernandez-Colina, E. Pérez-Carreno and D. Vazquez-Garcìa, Organometallics, 2010, 29, 4818–4828 CrossRef CAS.
- S. L. Chatwin, M. F. Mahon, T. J. Prior and M. K. Whittlesey, Inorg. Chim. Acta, 2010, 363, 625–632 CrossRef CAS.
- J. M. Huggins and R. G. Bergman, J. Am. Chem. Soc., 1979, 101, 4410–4411 CrossRef CAS.
- F. Balegroune, P. Braunstein, T. M. Gomes Carneiro, D. Grandjean and D. Matt, J. Chem. Soc., Chem. Commun., 1989, 582–584 RSC.
- H. Kuniyasu, K. Takekawa, F. Yamashita, K. Miyafuji, S. Asano, Y. Takai, A. Ohtaka, A. Tanaka, K. Sugoh, H. Kurosawa and N. Kambe, Organometallics, 2008, 27, 4788–4802 CrossRef CAS.
- B. E. Mann, B. L. Shaw and N. I. Tucker, J. Chem. Soc. A, 1971, 2667–2673 RSC.
- T. W. Bell, D. M. Haddleton, A. McCamley, M. G. Partridge, R. N. Perutz and H. Willner, J. Am. Chem. Soc., 1990, 112, 9212–9226 CrossRef CAS.
- K. J. Cavell and H. Jin, J. Chem. Soc., Dalton Trans., 1995, 4081–4089 RSC.
- M. A. Bennett, L. Kwan, A. D. Rae, E. Wenger and A. C. Willis, J. Chem. Soc., Dalton Trans., 2002, 226–233 RSC.
- A. Hamze, O. Provot, J.-D. Brion and M. Alami, J. Organomet. Chem., 2008, 693, 2789–2797 CrossRef CAS.
- G. Lorusso, G. Boccaletti, N. G. Di Masi, F. P. Fanizzi, L. Maresca and G. Natile, Eur. J. Inorg. Chem., 2004, 4751–4754 CrossRef CAS.
- S. Kimel and S. Speiser, Chem. Rev., 1977, 77, 437–472 CrossRef CAS; J. Svoboda and B. König, Chem. Rev., 2006, 106, 5413–5430 CrossRef; M. Fagnoni, D. Dondi, D. Ravelli and A. Albini, Chem. Rev., 2007, 107, 2725–2756 CrossRef; N. Hoffmann, Chem. Rev., 2008, 108, 1052–1103 CrossRef.
- H. Kornweitz, M. Brith and H. Friedmann, J. Photochem., 1980, 102, 5293–5294 Search PubMed; K. M. Tait, J. A. Parkinson, S. P. Bates, W. J. Ebenezer and A. C. Jones, J. Photochem. Photobiol., A, 2003, 154, 179–188 CrossRef CAS; S. W. Magennis, F. S. Mckay, A. C. Jones, K. M. Tait and P. J. Sadler, Chem. Mater., 2005, 17, 2059–2062 CrossRef.
- C. R. Barone, R. Cini, E. Clot, O. Eisenstein, L. Maresca, G. Natile and G. Tamasi, J. Organomet. Chem., 2008, 693, 2819–2827 CrossRef CAS.
- R. Cini, P. A. Caputo, F. P. Intini and G. Natile, Inorg. Chem., 1995, 34, 1130–1137 CrossRef CAS.
- D. Braga, F. Grepioni and E. Tedesco, Organometallics, 1997, 16, 1846–1856 CrossRef CAS; Y. Zhang, J. C. Lewis, R. G. Bergman, J. A. Ellman and E. Oldfield, Organometallics, 2006, 25, 3515–3519 CrossRef.
- V. Molina, M. Merchàn and B. O. Roos, J. Phys. Chem. A, 1997, 101, 3478–3487 CrossRef CAS; M. J. Bearpark, F. Bernardi, S. Clifford, M. Olivucci, M. A. Robb and T. Vreven, J. Phys. Chem. A, 1997, 101, 3841–3847 CrossRef; R. Improta and F. Santoro, J. Phys. Chem. A, 2005, 109, 10058–10067 CrossRef.
- F. P. Fanizzi, L. Maresca, G. Natile and C. Pacifico, Gazz. Chim. Ital., 1994, 124, 137–142 Search PubMed; L. Maresca and G. Natile, Inorg. Chim. Acta, 1999, 285, 301–304 CrossRef CAS.
- G. Lorusso, N. G. Di Masi, L. Maresca, C. Pacifico and G. Natile, Inorg. Chem. Commun., 2006, 9, 500–503 CrossRef CAS; G. Lorusso, C. R. Barone, N. G. Di Masi, L. Maresca, C. Pacifico and G. Natile, Eur. J. Inorg. Chem., 2007, 2144–2150 CrossRef.
- M. Meot-Ner and S. A. Kafafi, J. Am. Chem. Soc., 1988, 110, 6297–6303 CrossRef CAS.
- A. Tiripicchio, M. Tiripicchio-Camellini, L. Maresca, G. Natile and G. Rizzardi, Cryst. Struct. Commun., 1979, 8, 689–693 Search PubMed; L. Maresca and G. Natile, J. Chem. Soc., Dalton Trans., 1982, 1903–1906 RSC; G. Annibale, L. Maresca, G. Natile, A. Tiripicchio and M. Tiripicchio-Camellini, J. Chem. Soc., Dalton Trans., 1982, 1587–1591 RSC.
- M. K. Cooper, P. J. Guerny, H. J. Goodwin and M. McPartlin, J. Chem. Soc., Dalton Trans., 1982, 757–764 RSC; H. C. Clark, G. Ferguson, A. B. Goel and B. L. Rhul, Organometallics, 1984, 3, 15–17 CrossRef CAS.
- C. J. Cardin, D. J. Cardin, H. E. Parge and A. C. Sullivan, J. Chem. Soc., Dalton Trans., 1986, 2315–2320 RSC; A. R. Siedle, W. B. Gleason and R. A. Newmark, Organometallics, 1986, 5, 1969–1975 CrossRef CAS.
- M. Brookhart and M. L. H. Green, J. Organomet. Chem., 1983, 250, 395–408 CrossRef CAS; M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef.
- D. M. Roe, P. M. Bailey, K. Moseley and P. M. Maitlis, J. Chem. Soc., Chem. Commun., 1972, 1273–1274 RSC; A. Albinati, C. Arz and P. S. Pregosin, Inorg. Chem., 1987, 26, 508–513 CrossRef CAS; A. Albinati, P. S. Pregosin and F. Wombacher, Inorg. Chem., 1990, 29, 1812–1817 CrossRef; W. Yao, O. Eisenstein and R. H. Crabtree, Inorg. Chim. Acta, 1997, 254, 105–111 CrossRef; R. H. Crabtree, O. Eisenstein, G. Sini and E. Peris, J. Organomet. Chem., 1998, 567, 7–11 CrossRef.
- L. Brammer, J. M. Charnock, P. L. Goggin, R. J. Goodfellow, A. Guy Orpen and T. Koetzle, J. Chem. Soc., Dalton Trans., 1991, 1789–1798 RSC; S. G. Kazarian, P. A. Hamley and M. Poliakoff, J. Am. Chem. Soc., 1993, 115, 9069–9079 CrossRef CAS; J. M. Casas, L. R. Falvello, J. Forniés and A. Martìn, Inorg. Chem., 1996, 35, 6009–6014 CrossRef; L. Brammer, Dalton Trans., 2003, 3145–3157 RSC.
- A. Albinati, F. Lianza, P. S. Pregosin and B. Müller, Inorg. Chem., 1994, 33, 2522–2526 CrossRef CAS; R. Cini, F. P. Fanizzi, F. P. Intini and G. Natile, Inorg. Chim. Acta, 1996, 251, 111–118 CrossRef; A. Martìn, J. Chem. Educ., 1999, 76, 578–583 CrossRef; S. Rizzato, J. Bergès, S. A. Mason, A. Albinati and J. Kozelka, Angew. Chem., Int. Ed., 2010, 49, 7440–7443 CrossRef.
- R. G. Miller, R. D. Stauffer, D. R. Fahey and D. R. Parnell, J. Am. Chem. Soc., 1974, 92, 1511–1521 CrossRef CAS; W. I. Sundquist, D. P. Bancroft and S. J. Lippard, J. Am. Chem. Soc., 1990, 112, 1590–1596 CrossRef; L. Maresca, C. Pacifico, M. C. Pappadopoli and G. Natile, Inorg. Chim. Acta, 2000, 304, 274–282 CrossRef; Y. Liu, C. Pacifico, G. Natile and E. Sletten, Angew. Chem., Int. Ed., 2001, 40, 1226–1228 CrossRef.
- R. H. Philp Jr. and D. L. Reger, Organometallics, 1989, 8, 1714–1718 CrossRef.
- G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Germany, 1996 Search PubMed.
- P. T. Beurskens, G. Beurskens, W. P. Bosman, R. de Gelder, S. Garcia-Granda, R. O. Gould, R. Israel and J. M. M. Smits, The DIRDIF96 Program System, University of Nijmegen, Nijmegen, The Netherlands, 1996 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef.
- M. C. Burla, M. Camalli, B. Carrozzini, G. L. Cascarano, G. Giacovazzo, G. Polidori and R. Spagna, SIR2002: the program, J. Appl. Crystallogr., 2003, 36, 1103 CrossRef CAS.
- M. Nardelli, PARST: a system of FORTRAN routines for calculating molecular structure parameters from results of crystal structure analyses, Comput. Chem., 1983, 7, 95–97 CrossRef CAS; M. Nardelli, PARST: a system of FORTRAN routines for calculating molecular structure parameters from results of crystal structure analyses, J. Appl. Crystallogr., 1995, 28, 659 CrossRef.
- L. J. Farrugia, WinGX suite for small-molecule single-crystal crystallography, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Enzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS; M. Enzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS; F. Furche and R. Ahlrichs, J. Chem. Phys., 2002, 117, 7433–7447 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef CAS; F. Weigend, M. Häser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143–152 CrossRef.
- TURBOMOLE V6.0 2009, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007, available from http://www.turbomole.com.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman Jr., J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision C.02), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- P. Hariharan and J. A. Pople, Chem. Phys. Lett., 1972, 16, 217–219 CrossRef CAS; V. A. Rassolov, M. A. Ratner, J. A. Pople, P. C. Redfern and L. A. Curtiss, J. Comput. Chem., 2001, 22, 976–984 CrossRef.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS; W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef; P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef.
- F. London, J. Phys. Radium, 1937, 8, 397–409 CrossRef CAS; R. McWeeny, Phys. Rev., 1962, 126, 1028–1034 CrossRef; R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef; J. L. Dodds, R. McWeeny and A. J. Sadlej, Mol. Phys., 1980, 41, 1419–1430 CrossRef; K. Wolinski, J. F. Hilton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Pdf file: Fig. S1–S8 and Tables S1–S5. CCDC 904374 [(E)-2a], 904375 [(Z)-2a], 904377 [(E)-2b] and 904378 [(E)-2c]. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3dt32354d |
| This journal is © The Royal Society of Chemistry 2013 |