 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hemicellulose hydrolysis catalysed by solid acids†
Piera Demma
Carà
abc,
Mario
Pagliaro
b,
Ahmed
Elmekawy
d,
David. R.
Brown
d,
Peter
Verschuren
a,
N. Raveendran
Shiju
*a and
Gadi
Rothenberg
a
aVan't Hoff Institute for Molecular Sciences, University of Amsterdam, P.O. Box 94157, 1090GD Amsterdam, The Netherlands. E-mail: n.r.shiju@uva.nl; Web: http://hims.uva.nl/hcsc
bIstituto per lo Studio dei Materiali Nanostrutturati, CNR, via U. La Malfa 153, 90146 Palermo, Italy
cDipartimento Sistemi Agro-Ambientali, Universitèa degli Studi di Palermo, viale delle Science, 90128 Palermo, Italy
dMaterials and Catalysis Research Centre, Dept. of Chemical and Biological Sciences, University of Huddersfield, Queensgate HD1 3DH, UK
First published on 3rd April 2013
Abstract
Depolymerising hemicellulose into platform sugar molecules is a key step in developing the concept of an integrated biorefinery. This reaction is traditionally catalysed by either enzymes or homogeneous mineral acids. We compared various solid catalysts for hemicellulose hydrolysis, running reactions in water, under neutral pH and relatively mild temperature and pressure (120 °C and 10 bar) conditions. Sulphonated resins are highly active, but they leach out sulphonic groups. Sulphonated silicas are less active, but more stable. They have weakly and strongly bound sites and the strongly bound ones do not leach. Zeolites are moderately active and stable. Among them, H-ferrierite especially, despite its small pores, exhibited high activity as well as good recyclability.
1. Introduction
Switching from crude oil to biomass as the source for chemicals is the big challenge we chemists face in the 21st century.1 In that respect, the main research thrust must be directed at lignocellulose, the most abundant fraction of biomass. Lignocellulose is made up of ca. 50% cellulose, 30% hemicellulose and 20% lignin. Although cellulose and lignin are the subject of many studies,2–23 there are very few reports on hemicellulose conversions.24–33 Here we are particularly interested in the hemicellulose xylan, which is composed of different C5 and C6 sugar units (xylose, arabinose, mannose, glucose, and galactose) linked by β(1–4) glycosidic bonds.25 Xylan is non-edible and abundant, so its hydrolysis into sugars has important implications for both the food and the chemical sectors. Selective hydrolysis of xylan would produce monosugars, which are both a primary food source as well as renewable raw materials for making valuable chemicals.25,27Traditionally, hemicellulose hydrolysis is catalyzed by enzymes and mineral acids. The problem is that these routes incur separation/recycling difficulties, expensive catalyst recovery and corrosion problems. Efficient catalysis over solid acids in water can avoid all these problems, as it is carried out at neutral pH and catalyst/product separation is simple.34 Moreover, a stable solid catalyst can be reused, giving an economic advantage, especially in large-scale processing.35 Recently, Dhepe and Sahu reported a one-pot process for the conversion of solid hemicellulose into monomers using solid acid catalysts in aqueous media.27 They showed that solid acid catalysts could hydrolyze hemicellulose in a one-pot method to produce xylose and arabinose with 41% yield. Also, the conversion of arabinogalactans into sugars over ion-exchange resins has been reported.24 Arabinose was the primary product, followed by the release of galactose without further degradation of the monomers. Here we report the efficient hydrolysis of hemicellulose xylan using a number of solid acid catalysts. By testing various materials (sulfonated resins, sulfonated silicas and zeolites), we found that the zeolite H-ferrierite, despite its small pores, is well suited for catalyzing xylan hydrolysis under mild conditions.
2. Experimental section
2.1 Materials and instrumentation
Unless stated otherwise, chemicals were purchased from commercial sources and used as received. Beechwood xylan (Sigma Aldrich) was used as the hemicellulose substrate. Amberlyst 70 and Amberlyst 35 were purchased from Rohm and Haas; H-ZSM5 zeolites in protonic form with a nominal Si/Al ratio of 50, 80 and 100 and zeolite Y with a nominal Si/Al ratio of 5.1 were purchased from Zeolyst International; Faujasite in sodium form and ferrierite in ammonium form (a nominal Si/Al ratio of 55) were purchased from Shell. D5082 and D5081 are sulphonated hypercrosslinked polystyrene resins and were supplied in bead form by Purolite Ltd.X-ray diffraction (XRD) patterns were measured on a Rigaku miniflex diffractometer. Data were recorded in the 2θ range of 3–90° with an angular step size of 0.05° and a counting time of 5° min−1. The N2 adsorption–desorption isotherms were measured at 77 K on a Micromeritics ASAP-2020 after evacuation at 200 °C for 5 h. Surface areas were calculated by the BET method. The ammonia adsorption flow calorimetry system is based on a Setaram 111 DSC with an automated gas flow and switching system and a mass spectrometer (Hiden HPR20) for sampling the downstream gas flow.36,37 The sample (10–25 mg) was held on a glass frit in a vertical silica sample tube and activated at 150 °C under a dried nitrogen flow (5 ml min−1) for 5 h. After activation, the sample temperature was maintained at 150 °C and 1 ml pulses of the probe gas (1% ammonia in nitrogen) at atmospheric pressure were injected at regular intervals into the carrier gas stream. The ammonia concentration downstream of the sample was monitored continuously by mass spectroscopy. Pulse intervals were chosen so as to ensure that the ammonia concentration in the carrier gas returned to zero, allowing the DSC baseline to stabilise. The net amount of ammonia irreversibly adsorbed from each pulse was determined by comparing the MS signal with that recorded through a control experiment using a blank sample tube. The net heat released by each pulse was calculated from the thermal DSC curve.
All catalytic experiments were carried out in a Parr 5000 multi-autoclave system. Samples were analysed by size-exclusion chromatography (SEC), using PL aquagel-OH 30 8 μm and PL aquagel-OH MIXED 8 μm columns in series, a refractive index detector (RID-10A, Shimadzu) and ultrapure water (0.5 ml min−1) as a mobile phase.
2.2 Procedure for catalyst synthesis
Sulfonic acid functionalized silica gel was synthesized via a modification of a published procedure.38 Silica gel (2 g; Biosolve 60 Å, 0.063–0.200 mm) was added to toluene (20 ml). The mixture was heated to 115 °C and stirred for 1 h. Then, 3-mercaptopropyltrimethoxysilane (10.0 mmol, 2.0 g) was added, under stirring at the same temperature. After 24 h the solid was filtered, washed with water (5 × 20 ml) and then dried in air. The dry cake was then re-suspended in 35 ml of 33% H2O2(aq) in a closed vessel and stirred for 1 h at 60 °C (elevated temperature is advantageous39). The solid was then filtered, washed with water, and suspended in 35 ml of 10% H2SO4(aq). After stirring for 1 h at room temperature, the sulfonic acid catalyst was filtered, washed with water (4 × 20 ml) and dried at 110 °C for 16 h.The Faujasite (zeolite Y) in sodium form was exchanged into its ammonium form by dispersing it in 1 M NH4NO3(aq) (20 ml g−1 zeolite). The suspension was heated at 80 °C for 3 h, after which the zeolite was filtered, washed with water (4 × 20 ml) and dried for 6 h at 120 °C. Finally, the zeolite was converted from the ammonium form to the protonic form by heating to 500 °C at 5 °C min−1 and calcining at this temperature for 4 h. The NH4-ferrierite was similarly converted into its protonic form.
2.3 General procedure for catalytic hydrolysis of xylan
Beechwood xylan (0.1 g) and a solid acid catalyst (0.1 g, 0.33 equiv. H+ sites per sugar–sugar bond) were added to 10 ml of deionized water and placed in a stainless steel autoclave reactor at 120 °C under a pressure of 10 bar argon. Substrate conversion and product yields were quantified by SEC-HPLC analysis. Catalyst reusability was tested by first isolating the solid catalyst by filtration or decantation, then washing it with 20 ml of water and 20 ml of acetone. The zeolites were calcined at 500 °C for 4 h while the resins, because of their instability at high temperature, and the sulfonated silica gel catalysts were dried overnight at 120 °C. After the workup each catalyst was re-tested with a fresh reactant mixture. Conversions were higher than the total amount of sugar products. However, we could not detect the additional products using our HPLC system; so here we report only the amounts of xylose and arabinose.3. Results and discussion
One notable difference between crude oil and biomass as a starting material is the much greater variation in biomass feedstock quality. Because of this, projecting the results from one study to another should be done cautiously, keeping in mind the characteristics of the respective feed batches.The nature of hemicelluloses also depends on the type of wood.25 The most important hemicelluloses in softwoods (coniferous trees) are galactoglucomannans and arabinoglucuronoxylans. Furthermore, softwoods contain arabinogalactan, xyloglucans, and other glucans. The most important hardwood hemicellulose is xylan, especially O-acetyl-4-O-methylglucuronoxylan, amounting to about 80–90% of the hardwood hemicelluloses. Fig. 1 shows the X-ray diffraction pattern of “our” beechwood xylan. The broad peak at 20° indicates some short-range order in the amorphous polymeric structure of the hemicellulose. The magnitude of this short-range order or semi-crystallinity can change from batch to batch and thus can affect the reactivity of the sample.
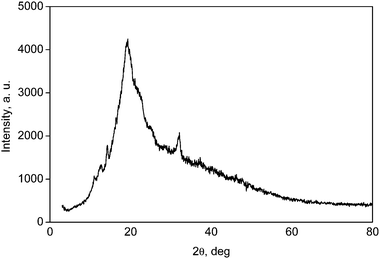 | ||
| Fig. 1 X-ray diffraction pattern of xylan from beechwood. | ||
In a typical reaction, beechwood xylan and solid acid catalyst were mixed in deionized water at 120 °C under 10 bar argon. Though the weight ratio between xylan and catalyst is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the ratio between the H+ sites of the catalyst and the sugar–sugar bond of the xylan is much lower (e.g. 0.255 mmol of H+ in the case of Amberlyst 70 for 0.75 mmol of sugar bonds). Reaction progress was monitored by SEC-HPLC (see the Experimental section for detailed procedures), and the yield was calculated considering the MW of the xylan as 132 g mol−1.
:1, the ratio between the H+ sites of the catalyst and the sugar–sugar bond of the xylan is much lower (e.g. 0.255 mmol of H+ in the case of Amberlyst 70 for 0.75 mmol of sugar bonds). Reaction progress was monitored by SEC-HPLC (see the Experimental section for detailed procedures), and the yield was calculated considering the MW of the xylan as 132 g mol−1.
3.1 Sulfonic acid functionalized polystyrene resins
Fig. 2 shows the kinetics of xylose and arabinose formation by xylan hydrolysis using Amberlyst 70. The resin has a concentration of 2.55 mmol H+ g−1. Under our conditions it releases 0.255 mmol of H+ per 0.75 mmol sugar–sugar bonds indicating a catalytic reaction. The hydrolysis was fast, yielding more than 60% of xylose and arabinose in 2 h. After 4 h the yield of sugar monomers stabilised at 76%.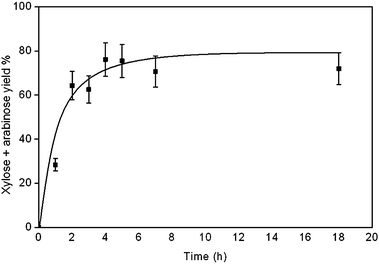 | ||
| Fig. 2 Time-resolved yield of xylose and arabinose using Amberlyst 70. Reaction conditions: xylan (0.1 g); Amberlyst 70 (0.1 g); water (10 ml); 120 °C, 10 bar (Ar). | ||
Control experiments confirmed that only trace amounts of sugar monomers were obtained in the absence of a catalyst (Fig. 3). The xylan breaks into high molecular weight fragments and oligomers under these conditions. Using homogeneous H2SO4 (0.036 M) yielded 74% of sugar monomers. The sulphonated resins Amberlyst 70 and Amberlyst 35 both gave similar catalytic performance, with ca. 80% yield of xylose and arabinose. D5081 and D5082 gave slightly lower yields of sugar monomers (typically 55–70%, see Fig. 3), probably due to the weaker acid sites than the Amberlyst resins.40 This is consistent with the relatively large separation between acid groups and lower interactions between neighbouring sulphonic acid groups. Earlier studies showed that D5082 has stronger acid sites than D5081, which explains the observed activity trend.41
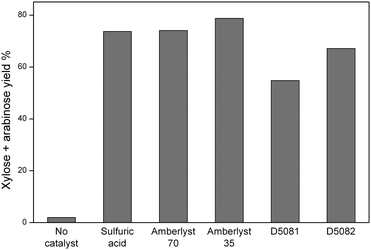 | ||
| Fig. 3 Yield of xylose and arabinose using different resins, compared with H2SO4 and blank reaction. Reaction conditions: xylan (0.1 g); catalyst (0.1 g); water (10 ml); 120 °C, 10 bar Ar, 4 h. | ||
We then carried out catalyst recycling studies using Amberlyst 70 and D5082 resins (Fig. 4). A significant decrease in the yield of xylose and arabinose was observed after the first recycling, indicating rapid catalyst deactivation. This deactivation continued with further recycling tests.
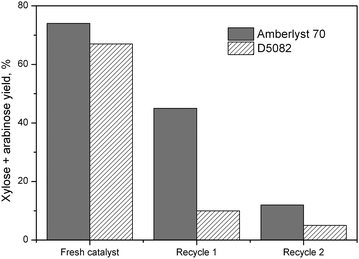 | ||
| Fig. 4 Yield of xylose and arabinose after the recycling test using Amberlyst 70 and purolite D5082. Reaction conditions: xylan (0.1 g); catalyst (0.1 g); water (10 ml); 120 °C, 10 bar Ar, 4 h. | ||
We envisage two pathways for the deactivation of sulfonic acid resins. One is organic fouling. By-products such as humin can be trapped in the polymeric matrix, masking the active sites. In such cases, calcining the catalyst can reverse the deactivation. Another option is via leaching of the sulfonic acid groups, especially above 150 °C. Such leaching results in a permanent loss of catalytic activity.
To confirm or exclude the leaching of the sulfonic acid groups, we carried out hot filtration tests, comparing the reaction progress before and after filtering the solid catalyst (Fig. S1, ESI†). These tests confirmed that both Amberlyst 70 and purolite D5082 undergo leaching of sulfonic groups. We therefore moved to two other types of solid acid catalysts: sulphonated silica gel and proton-exchanged zeolites.
3.2 Sulfonic acid functionalized silica gel
Fig. 5 shows the kinetics of xylan hydrolysis over silica gel functionalized with sulfonic acid groups. | ||
| Fig. 5 Combined yield of xylose and arabinose using sulfonated silica gel. Reaction conditions: xylan (0.1 g); Amberlyst 70 (0.1 g); water (10 ml); 140 °C, 10 bar (Ar). | ||
After 3 h, complete conversion of xylan was observed with more than 50% xylose and arabinose yield. No improvement in sugar yields was observed thereafter. Also in this case, we tested the recyclability of the solid catalyst.
The combined yield of xylose and arabinose decreased after the first recycling, but then remained stable at around 25% in subsequent recycling experiments (Fig. 6). This shows that sulfonated silica gel has two types of sulfonic groups: loosely bound ones that leach into solution, and strongly bound ones (most probably inside the pores) that remain available for catalysis also after recycling.41
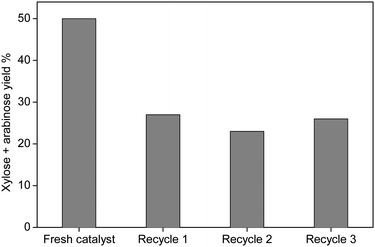 | ||
| Fig. 6 Yield of xylose and arabinose after the recycling test using sulfonated silica gel. Reaction conditions: xylan (0.1 g); catalyst (0.1 g); water (10 ml); 140 °C, 10 bar (Ar), 3 h. | ||
3.3 Zeolites
Fig. 7 shows the hydrolysis of xylan catalyzed by three different proton-exchanged zeolites: H-Faujasite (HY), HZSM-5 and H-ferrierite. HZSM-5 with Si/Al ratios 50 and 80 gave 33% and 29% sugar yields respectively. Despite its larger pores, HY gave a lower yield (20%). Surprisingly, H-ferrierite gave the highest yield (43%) of xylose and arabinose. Hemicellulose is too large to enter the ferrierite pores. Hence, the initial depolymerisation must take place on the external surface of the catalyst. Possibly, H-ferrierite has stronger acid sites on its external surface compared to HY and H-ZSM-5, resulting in higher activity.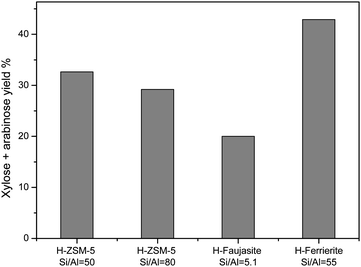 | ||
| Fig. 7 Yield of xylose and arabinose using different zeolites as catalysts. Reaction conditions: xylan (0.1 g); catalyst (0.1 g); water (10 ml); 120 °C, 20 h, 10 bar (Ar). | ||
We then carried out further experiments using ferrierite. Increasing the temperature to 140 °C gave faster reactions with yields comparable to those obtained at 120 °C (Fig. S2, ESI†). Optimal yields were obtained after 4 h, and longer reaction times led to coke formation. We then ran tests at 160 °C, but this caused xylan degradation (Fig. S3, ESI†). Further experiments were therefore conducted at 140 °C for 4 h.
We also ran reactions with lower ferrierite/xylan ratios (Fig. S4, ESI†). The yield of xylose and arabinose was 31%, considerably higher than the blank reaction, when the catalyst amount was decreased from 0.1 g to 0.025 g. This shows that ferrierite efficiently catalyses the conversion of xylan. Triple recycling tests of the ferrierite showed only a marginal decrease in activity when the spent catalyst was washed with water and calcined in static air (Fig. S5, ESI†).
We evaluated the total acidity of the samples by ammonia adsorption microcalorimetry (Fig. 8). The initial differential heat of NH3 adsorption was the highest for H-ferrierite, indicating stronger acid sites than HY and HZSM-5. Though an arbitrary method, averaging the values above a reasonable heat of adsorption provides an indication of acid strength. The average enthalpies above 85 kJ mol−1 are 122.4 kJ mol−1, 107.6 kJ mol−1 and 99.3 kJ mol−1 for H-ferrierite, HZSM-5 and HY respectively. This indicates again that our H-ferrierite sample has strong acid sites. The BET surface areas of H-ferrierite (305 m2 g−1) and HZSM-5 (300 m2 g−1) were similar, so the higher initial activity of ferrierite could be attributed to its higher acidic strength. Though microcalorimetry cannot distinguish between external and internal acid sites, the results give an indication of overall acidic strength. Interestingly, ferrierite showed exceptional performance in butene skeletal isomerization. It is a very selective and stable catalyst, resulting in high yields of isobutene at relatively low temperatures without the need for a diluted feed.42 The SEM images of zeolites are given in ESI† (Fig. S6). The samples are aggregates of particles. HY consists of aggregates of 200–500 nm size particles. H-ferrierite is composed of flakes of sizes of a few hundred nm. HZSM-5 has a particle size of around 100–300 nm.
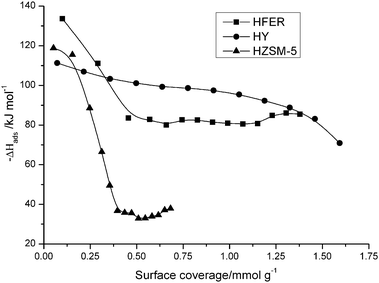 | ||
| Fig. 8 NH3 adsorption microcalorimetry data H-ferrierite, HY, and HZSM-5 zeolites, showing the heat of NH3 adsorption as a function of the surface coverage. | ||
4. Conclusions
Beechwood xylan is readily hydrolysed to a mixture of monomeric sugars and dimer/oligomer byproducts in the presence of a solid acid catalyst. The catalytic activity varies considerably depending on the solid acid type. Acidic ion exchange resins rapidly lose their activity due to leaching of sulfonic acid groups. Sulfonated silica gels are better, despite showing a considerable loss of activity after the first reaction run. The proton-exchanged zeolite ferrierite affords the best results in terms of both activity and catalyst stability. We attribute this to its relatively high acid strength, as evaluated by NH3 adsorption microcalorimetry.Notes and references
-
B. Kamm, P. R. Gruber and M. Kamm, Biorefineries – Industrial Processes and Products, Wiley-VCH, Weinheim, 2006 Search PubMed
.
- Z. Strassberger, S. Tanase and G. Rothenberg, Eur. J. Org. Chem., 2011, 5246–5249 CrossRef CAS
- S. Van de Vyver, L. Peng, J. Geboers, H. Schepers, F. de Clippel, C. J. Gommes, B. Goderis, P. A. Jacobs and B. F. Sels, Green Chem., 2010, 12, 1560–1563 RSC
- A. Takagaki, C. Tagusagawa and K. Domen, Chem. Commun., 2008, 5363–5365 RSC
- P. L. Dhepe and A. Fukuoka, ChemSusChem, 2008, 1, 969–975 CrossRef CAS
- A. Fukuoka and P. L. Dhepe, Angew. Chem., Int. Ed., 2006, 45, 5161–5163 CrossRef CAS
- A. Fukuoka and P. L. Dhepe, Chem. Rec., 2009, 9, 224–235 CrossRef CAS
- T. Minowa, F. Zhen and T. Ogi, J. Supercrit. Fluids, 1998, 13, 253–259 CrossRef CAS
- A. Onda, T. Ochi and K. Yanagisawa, Green Chem., 2008, 10, 1033–1037 RSC
- R. Palkovits, K. Tajvidi, J. Procelewska, R. Rinaldi and A. Ruppert, Green Chem., 2010, 12, 972–978 RSC
- J. Pang, A. Wang, M. Zheng and T. Zhang, Chem. Commun., 2010, 46, 6935–6937 RSC
- R. Rinaldi, R. Palkovits and F. Schueth, Angew. Chem., Int. Ed., 2008, 47, 8047–8050 CrossRef CAS
- R. Rinaldi and F. Schueth, ChemSusChem, 2009, 2, 1096–1107 CrossRef CAS
- S. Suganuma, K. Nakajima, M. Kitano, D. Yamaguchi, H. Kato, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2008, 130, 12787–12793 CrossRef CAS
- N. Villandier and A. Corma, Chem. Commun., 2010, 46, 4408–4410 RSC
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS
- A. Corma, O. de la Torre and M. Renz, Energy Environ. Sci., 2012, 5, 6328–6344 CAS
- P. Gallezot, Catal. Today, 2011, 167, 31–36 CrossRef CAS
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS
- J. C. Serrano-Ruiz, D. J. Braden, R. M. West and J. A. Dumesic, Appl. Catal., B, 2010, 100, 184–189 CrossRef CAS
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC
- M. Rose and R. Palkovits, Macromol. Rapid Commun., 2011, 32, 1299–1311 CrossRef CAS
- M. Hara, Energy Environ. Sci., 2010, 3, 601–607 CAS
- B. T. Kusema, G. Hilmann, P. Maki-Arvela, S. Willfor, B. Holmbom, T. Salmi and D. Y. Murzin, Catal. Lett., 2011, 141, 408–412 CrossRef CAS
- P. Mäki-Arvela, T. Salmi, B. Holmbom, S. Willför and D. Y. Murzin, Chem. Rev., 2011, 111, 5638–5666 CrossRef
- R. Sahu and P. L. Dhepe, ChemSusChem, 2012, 5, 751–761 CrossRef CAS
- P. L. Dhepe and R. Sahu, Green Chem., 2010, 12, 2153–2156 RSC
- E. I. Guerbuez, S. G. Wettstein and J. A. Dumesic, ChemSusChem, 2012, 5, 383–387 CrossRef CAS
- C. Li, M. Zheng, A. Wang and T. Zhang, Energy Environ. Sci., 2012, 5, 6383–6390 CAS
- E. S. Kim, S. Liu, M. M. Abu-Omar and N. S. Mosier, Energy Fuels, 2012, 26, 1298–1304 CrossRef CAS
- R. Xing, W. Qi and G. W. Huber, Energy Environ. Sci., 2011, 4, 2193–2205 CAS
- Y. Ogaki, Y. Shinozuka, T. Hara, N. Ichikuni and S. Shimazu, Catal. Today, 2011, 164, 415–418 CrossRef CAS
- R. Xing, A. V. Subrahmanyam, H. Olcay, W. Qi, G. P. van Walsum, H. Pendse and G. W. Huber, Green Chem., 2010, 12, 1933–1946 RSC
- K. Wilson, D. J. Adams, G. Rothenberg and J. H. Clark, J. Mol. Catal., 2000, 159, 309–314 CrossRef CAS
- N. R. Shiju, G. Rothenberg and D. R. Brown, Top. Catal., 2010, 53, 1217–1223 CrossRef CAS
- M. Bandyopadhyay, N. R. Shiju and D. R. Brown, Catal. Commun., 2010, 11, 660–664 CrossRef CAS
- N. R. Shiju, M. AnilKumar, W. F. Hoelderich and D. R. Brown, J. Phys. Chem. C, 2009, 113, 7735–7742 CAS
- P. F. Siril, N. R. Shiju, D. R. Brown and K. Wilson, Appl. Catal., A, 2009, 364, 95–100 CrossRef CAS
- E. Cano-Serrano, G. Blanco-Brieva, J. M. Campos-Martin and J. L. Fierro, Langmuir, 2003, 19, 7621–7627 CrossRef CAS
- E. Andrijanto, E. A. Dawson and D. R. Brown, Appl. Catal., B, 2012, 115–116, 261–268 CrossRef CAS
- R. Ciriminna, L. M. Ilharco, A. Fidalgo, S. Campestrini and M. Pagliaro, Soft Matter, 2005, 1, 231–237 RSC
- A. C. Butler and C. P. Nicolaides, Catal. Today, 1993, 18, 443–471 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cy20838a |
| This journal is © The Royal Society of Chemistry 2013 |