 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selectivity of the Fischer–Tropsch process: deviations from single alpha product distribution explained by gradients in process conditions
Katrina D.
Kruit
a,
David
Vervloet
a,
Freek
Kapteijn
b and
J. Ruud
van Ommen
*a
aProduct & Process Engineering, Delft University of Technology, Faculty of Applied Sciences, Julianalaan 136, 2628 BL Delft, The Netherlands. E-mail: j.r.vanommen@tudelft.nl
bCatalysis Engineering, Delft University of Technology, Faculty of Applied Sciences, Julianalaan 136, 2628 BL Delft, The Netherlands
First published on 10th May 2013
Abstract
Non-ASF product distributions in Fischer–Tropsch synthesis can occur with a gradient in process conditions at the particle or reactor scale, leading to a gradient in chain growth probability α. Weighted summation of local product distributions gives the proper, non-ASF product distribution.
The Fischer–Tropsch (FT) process, in which syngas is converted to hydrocarbons, is a key conversion step in one of the most important routes to alternative fuels and chemicals. Various models exist for the selectivity of this reaction. The simplest and most widely used is the Anderson–Schulz–Flory (ASF) model,1 where the FT reaction is modeled as an addition polymerization reaction with chain growth probability α. The resulting product distribution, depicted as a plot of the logarithmic molar fraction versus the carbon number (‘ASF-plot’), is a straight line.
However, deviations from this ASF product distribution are regularly observed. Commonly reported deviations from the ASF model are the C1 (methane) and C2 (ethane/ethene) values. Typically, C1 is formed in a greater quantity than that predicted by the ASF distribution, while a much lower amount of C2 is usually measured.2 Another deviation from the ASF product distribution shape that is encountered in practice is a chain-length-dependent chain growth probability. Instead of a linear trend in an ‘ASF-plot’ (cf.Fig. 1c), the product distribution shows a double chain growth probability or a curved distribution.3,4 The fractions of heavier hydrocarbons are in some cases lower than expected based on the ASF model5 but in most cases they are higher.2,4,6–13
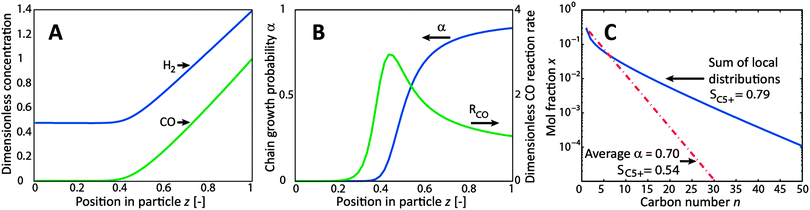 | ||
| Fig. 1 Results of the reaction-diffusion model for the spherical catalyst particle at T = 488 K, the gas phase molar ratio H2/COin = 1.8, dcat = 0.5 mm, FYS = 2, P = 30 bar. z = 0 at the centre and z = 1 at the surface of the particle. (A) Liquid H2 and CO concentration profile in the catalyst particle normalized to CO concentration at the external particle surface, (B) profile of chain growth probability and CO conversion rate, (C) resulting product distributions calculated from local product distributions and from average α. | ||
The cause of these positive deviations from ASF product distributions is a disputed topic. It has been attributed to flaws in experimentation,8–10 the existence of multiple chain growth mechanisms11 or multiple different active sites on the catalyst.4,12 However, more recent work assumes that olefin readsorption and incorporation is responsible.2,6,7,13
In this communication we propose an alternative explanation for non-ASF product distributions which is nevertheless based on a single-chain-growth-probability model. Our explanation is based on the process condition dependence of α, where we assume a variable-α model that takes into account the local reactor temperature and the syngas ratio.14 It has previously been demonstrated that gradients in the process conditions may lead to selectivity gradients.3,8,14 Temperature and concentration gradients are the most likely variables that cause variations in selectivity. These gradients can take place on the particle scale due to diffusion limitations inside the catalyst particle, as well as on the reactor scale, e.g. due to heat transport limitations in a packed bed reactor.8,15 We will show that these gradients in process variables can result in an apparent non-ASF Fischer–Tropsch product distribution.
Gradients in FT process conditions and resulting chain growth probability and product distributions were modeled on the particle scale and the reactor scale. For both cases, a process-condition-dependent α-model proposed by Vervloet et al.14 was used, which captured the experimental trends well:
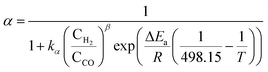 | (1) |
To demonstrate the effect of reactant concentration profiles at the particle scale, a steady state reaction-diffusion model was applied for a spherical catalyst particle. This model, described in detail by Vervloet et al.,14 is based on dimensionless steady state reaction-diffusion mass balances in a catalyst particle. The relevant process variables considered in the particle model are the particle diameter dcat, the syngas ratio H2/CO, the temperature T and the catalyst activity multiplication factor FYS, relative to the Yates & Satterfield kinetics.18
To describe the behavior at the reactor scale, a simple 1-D reactor model was set up for a single tube. Since the FT process is strongly exothermal, often axial temperature gradients will be found. Such gradients can also lead to a deviation from a single α product distribution. We illustrated this by imposing a simple linear temperature profile which can also be encountered in industrial practice.17 In this steady state reactor model, mass transfer limitations and radial gradients were assumed to be absent and the syngas ratio was kept constant at 2. The gas phase, which was modeled to only include H2 and CO, was therefore in instant equilibrium with the liquid phase according to Henry's law. The linear temperature increase was from T = 210 to 240 °C. Other process conditions and assumptions can be found in Appendix A.
At both the particle and the reactor scale, the reaction rate was assumed to follow Langmuir–Hinshelwood kinetics, proposed by Yates and Satterfield,18 where the catalyst activity multiplication factor FYS accounts for the increase in catalyst activity since the publication of the original values. The reaction rate and adsorption constant reported by Maretto and Krishna19 are used. Finally, the overall Fischer–Tropsch product distribution was calculated in two different ways: firstly, using a single average α, based on the reaction rate weighted integrated α over the catalyst or reactor volume; and secondly, by summating local (ASF) product distributions weighed with the reaction rate and incremental volume.
At the particle scale, we demonstrated the effect of diffusion limitations on the chain growth probability, product distribution and C5+ selectivity of the FT reaction. A system with a moderate diffusion length, representative of packed bed reactors, was simulated. This example is illustrated in Fig. 1, where the reactant concentrations, reaction rate, α gradients and the resulting product distributions are shown. In this diffusion-limited catalyst particle, the calculation of the product distribution from the average α gave a straight distribution with a C5+ selectivity of 54%, while the weighted summation of local distributions gave a curved plot with a positive deviation in the higher carbon numbers and a C5+ selectivity of 79%. There are thus large deviations in the shape of the product distribution and in the C5+ selectivity between these two calculation methods.
The cause of the curved product distribution is the occurrence of gradients in the local syngas ratio in the catalyst particle due to diffusion limitations. Towards the centre of the particle, CO becomes depleted, causing an increase in the H2/CO ratio, resulting in a decrease in the local α. Correspondingly, the local productivities of the components are different, resulting in a curved total product distribution when summated. In contrast to the chain growth probability, a maximum reaction rate can occur inside the particle at lower CO concentrations due to the kinetics.
The prominence of this CO depletion depends strongly on the diffusion length, temperature, syngas ratio and catalyst activity. At H2/CO = 2, a common choice for experimental conditions since it is close to the overall stoichiometric consumption of the reactants, even catalyst particles as small as 200 μm can become depleted in CO at enhanced catalyst activity. This potentially leads to non-linear ASF product distributions when the concentration dependency of α is accounted for. A high temperature and/or catalyst activity amplifies this effect, because CO is depleted more rapidly at higher reaction rates.
Different conditions and diffusion lengths were simulated to represent various reactor systems (i.e. short diffusion lengths to represent the slurry reactor and thin coated reactor internals, and moderate and large diffusion lengths to represent packed bed reactors). We modeled a catalyst with a conservative activity (FYS = 2, representing a factor 2 improvement in catalyst activity from the original values18), and a high catalyst activity (FYS = 10, representing the best state-of-the-art catalysts). The product distribution was calculated in two different ways: using the average chain growth probability and by summating local product distributions. In strongly diffusion-limited systems a significant difference between these two distributions was found (Table 1).
| H2/CO | d cat = 50 μm | d cat = 1 mm | d cat = 2 mm | ||||
|---|---|---|---|---|---|---|---|
| 180 °C | 230 °C | 180 °C | 230 °C | 180 °C | 230 °C | ||
| F YS = 2 | 0.5 | = | = | = | = | = | = |
| 1 | = | = | = | = | = | = | |
| 2 | = | = | = | ! | ! | ! | |
| F YS = 10 | 0.5 | = | = | = | = | = | = |
| 1 | = | = | = | = | = | = | |
| 2 | = | = | ! | ! | ! | ! | |
To compare this modeling with experimental results, we used the reaction-diffusion model to simulate the process conditions used by Visconti et al.6 (p = 20 bar, T = 230 °C, H2/CO = 2.1, dcat = 100 μm). For this purpose, the catalyst dependent parameters in the variable-α model (kα, β and ΔEa, see eqn (1)) were tuned to fit the data. The resulting product distribution is compared to the experimental data in Fig. 2. The modeled product distribution captures the experimental data well. It is especially noteworthy that the CO conversion level is reproduced, as well as the curved product distribution, including the value for C1. This result implies that diffusion limitations in the particle could cause a non-ASF product distribution such as that found experimentally.
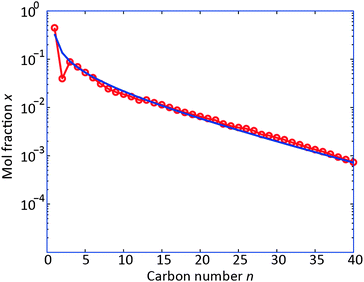 | ||
| Fig. 2 Product distribution at T = 503 K, H2/COin = 2.1, dcat = 100 μm, P = 20 bar. Red circles: experimental data, from ref. 6; blue line: reaction-diffusion model results with variable-α model14 with FYS = 10 and model parameters ΔEa = 120.4 × 103 J mol−1, β = 3.8 and kα = 2.77 × 10−3. | ||
At the reactor scale, the effect of temperature gradients on the product distribution is demonstrated by imposing a linear temperature increase of 30 K over the length of the reactor (see Fig. 3). Generally, and captured in eqn (1), α decreases with increasing temperature along the reactor. The effect of this α-gradient is a non-linear ASF product distribution, similar to the shape found in diffusion-limited catalyst particles.
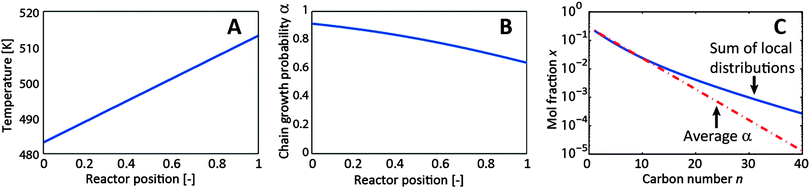 | ||
| Fig. 3 Results of the 1-D reactor with the imposed linear temperature gradient. (A) Temperature profile in the reactor, (B) gradient in chain growth probability, (C) resulting product distributions calculated from local concentrations and from average α. | ||
It was found that the effect of the imposed temperature profile on the apparent chain growth, and thus on the product distribution, is smaller than the effect of the reactant ratio at the catalyst particle scale. This is due to the parameters in the variable-α model. Although the used reactor model is a gross simplification of a real FT reactor, this example clearly shows that typical axial temperature gradients encountered in FT reactors can cause a non-ASF product distribution too.
Conclusions
Non-ASF product distributions in Fischer–Tropsch synthesis can occur when there is a gradient in process variables (temperature, concentration) leading to a gradient in the chain growth probability α. The origin of this effect can be found on the particle scale, i.e. concentration gradients caused by diffusion limitations in the catalyst, or on the reactor scale, i.e. gradients in local conditions such as temperature. When such gradients are modeled, using the average chain growth probability to predict the product composition can lead to erroneous results. The correct product distribution should be obtained by volume based summation of local product distributions, weighted with the local reaction rate.Appendix A
Parameter values used in models (Tables 2 and 3).| Feed rate gas | 0.15 m s−1 |
| Feed rate liquid | 0.01 m s−1 |
| Volume fraction solid | 0.6 |
| Volume fraction gas | 0.2 |
| Henry coefficient CO | 339.3 bar |
| Henry coefficient H2 | 428.0 bar |
| Pressure | 30 bar |
| Pressure drop | Negligible |
| Reactor tube diameter | 25 mm |
| H2/CO feed ratio | 2 |
| Reactor length | 2 m |
| Catalyst activity multiplication factor | 2 |
| Intrinsic catalyst density | 2500 kg m−3 |
| Catalyst porosity | 0.50 |
| Catalyst pore tortuosity | 1.5 |
| CO diffusivity in product medium | 5.584 × 10−7 m2 s−1 |
| CO diffusivity activation energy | 14.85 × 103 J mol−1 |
| H2 diffusivity in product medium | 1.085 × 10−6 m2 s−1 |
| H2 diffusivity activation energy | 13.51 × 103 J mol−1 |
Notes and references
- R. B. Anderson, R. A. Friedel and H. H. Storch, J. Chem. Phys., 1951, 19, 313–319 CrossRef CAS.
- G. P. Van Der Laan and A. A. C. M. Beenackers, Catal. Rev., 1999, 41, 255–318 CAS.
- L. S. Glebov and G. A. Kliger, Usp. Khim., 1994, 63, 192–202 CAS.
- R. J. Madon, S. C. Reyes and E. Iglesia, J. Phys. Chem., 1991, 95, 7795–7804 CrossRef CAS.
- Y. Y. Ji, H. W. Xiang, J. L. Yang, Y. Y. Xu, Y. W. Li and B. Zhong, Appl. Catal., A, 2001, 214, 77–86 CrossRef CAS.
- C. G. Visconti, E. Tronconi, L. Lietti, P. Forzatti, S. Rossini and R. Zennaro, Top. Catal., 2011, 54, 786–800 CrossRef CAS.
- C. M. Masuku, D. Hildebrandt and D. Glasser, Chem. Eng. Sci., 2011, 66, 6254–6263 CrossRef CAS.
- I. Puskas and R. S. Hurlbut, Catal. Today, 2003, 84, 99–109 CrossRef CAS.
- A. P. Raje and B. H. Davis, Energy Fuels, 1996, 10, 552–560 CrossRef CAS.
- J. Gao, B. Wu, L. Zhou, Y. Yang, X. Hao, J. Xu, Y. Xu and Y. Li, Ind. Eng. Chem. Res., 2012, 51, 11618–11628 CrossRef CAS.
- B. W. Wojciechowski, Catal. Rev.: Sci. Eng., 1988, 30, 629–702 CAS.
- G. A. Huff and C. N. Satterfield, J. Catal., 1984, 85, 370–379 CrossRef CAS.
- H. Schulz and M. Claeys, Appl. Catal., A, 1999, 186, 91–107 CrossRef CAS.
- D. Vervloet, F. Kapteijn, J. Nijenhuis and J. R. van Ommen, Catal. Sci. Technol., 2012, 2, 1221–1233 CAS.
- M. E. Dry, Appl. Catal., A, 1996, 138, 319–344 CrossRef CAS.
- G. Lozano-Blanco, K. Surla, J. W. Thybaut and G. B. Marin, Oil Gas Sci. Technol., 2010, 66, 423–435 CrossRef.
- N. Hooshyar, D. Vervloet, F. Kapteijn, P. J. Hamersma, R. F. Mudde and J. R. van Ommen, Chem. Eng. J., 2012, 207–208, 865–870 CrossRef CAS.
- I. C. Yates and C. N. Satterfield, Energy Fuels, 1991, 5, 168–173 CrossRef CAS.
- C. Maretto and R. Krishna, Catal. Today, 1999, 52, 279–289 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |