 Open Access Article
Open Access ArticleRegioselective Pd-catalyzed hydroamination of substituted dienes
Arnaud
Perrier
,
Michel
Ferreira†
,
Joost N. H.
Reek
and
Jarl Ivar
van der Vlugt
*
Homogeneous & Supramolecular Catalysis, van't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH, Amsterdam, The Netherlands. E-mail: j.i.vandervlugt@uva.nl; Fax: +31 (0)20 525 5604; Tel: +31 (0)20 525 6459
First published on 19th February 2013
Abstract
The regioselective addition of hydrazones and related N-nucleophiles to substituted terpene-based substrates can be steered to either 1,2- or 1,4-addition products, making this a potentially powerful protocol for the preparation of amines from biorelevant feedstocks via C–N bond formation.
Introduction
Amines are very versatile building blocks for a plethora of application areas, including dyes, polymers, pharmaceuticals and agrochemicals. The industrial production of amines still depends, at least to a large extent, on stoichiometric conversion of alkyl halides or alcohols as starting materials, which results in undesirable waste-streams or highly energy-intensive process conditions.1,2 Several catalytic methodologies for the selective formation of C–N bonds based on vinylic or allylic substrates have recently been developed (Scheme 1a).3–8 However, these methodologies generally rely on leaving-group chemistry, multi-step transformations, the inherent generation of by-products or suffer from drawbacks such as limited functional group tolerance.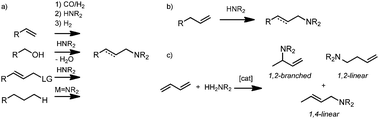 | ||
| Scheme 1 (a) Previously reported catalytic procedures for C–N bond formation; (b) atom-efficient coupling of an alkene with an amine to generate a substituted (allylic) amine; (c) hydroamination of 1,3-diene to yield linear 1,4-, branched 1,2- and linear 1,2-isomers. | ||
The direct, atom-efficient hydroaddition of amines across a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond – hydroamination9 – is a very attractive approach to generate amines from readily available starting materials (Scheme 1b). It would also be of high value to integrate this reaction step with other transformations in a cascade-like sequence. Furthermore, the use of ammonia as starting material, which would lead to valuable primary amines upon addition to alkenes, is a major challenge in this area, despite recent progress in the use of NH3 as a substrate for homogeneous catalysis.10 One particular recent example is the direct amination of allyl alcohols using aqueous ammonia to furnish allylic amines, generating water as a by-product.11 Besides (un)functionalized alkenes, allylic substrates or vinylarenes, dienes have recently emerged as an interesting substrate class for the regioselective hydroamination, potentially giving rise to selective 1,2- or 1,4-addition products (Scheme 1c).12
C double bond – hydroamination9 – is a very attractive approach to generate amines from readily available starting materials (Scheme 1b). It would also be of high value to integrate this reaction step with other transformations in a cascade-like sequence. Furthermore, the use of ammonia as starting material, which would lead to valuable primary amines upon addition to alkenes, is a major challenge in this area, despite recent progress in the use of NH3 as a substrate for homogeneous catalysis.10 One particular recent example is the direct amination of allyl alcohols using aqueous ammonia to furnish allylic amines, generating water as a by-product.11 Besides (un)functionalized alkenes, allylic substrates or vinylarenes, dienes have recently emerged as an interesting substrate class for the regioselective hydroamination, potentially giving rise to selective 1,2- or 1,4-addition products (Scheme 1c).12
Given the recent surge toward the valorization and utilization of biorenewables for the production of high-value, highly functionalized chemicals,13 the exploration of catalytic methodologies for C–N bond formation that are orthogonal to existing functional groups is of prime interest and importance, as this would result in more atom-efficient processes, avoid side-product formation (like water) and retain optimal synthetic diversity in the resulting molecules.
Among the different types of biobased feedstocks available, the class of terpenes appears to be ideally suited to allow easy access to introduction of additional N-containing functionalities. Initial advances concerning interesting terpene-based substrates have recently been reported. Behr and coworkers established the selective Pd-catalyzed 1,4-addition of morpholine to myrcene, resulting in a mixture of (E)- and (Z)-isomers in high yield, using dppb as a ligand.14 This procedure could provide an alternative synthesis route to (−)-menthol, for which the current route, as commercialized by Takasago, includes Li-catalyzed intermolecular hydroamination of diethylamine to myrcene that suffers from low activity.15 The group of Hartwig has investigated the Pd-catalyzed hydroamination of isoprene with a variety of hydrazine or hydroxyl amine derived N-nucleophiles, including hydrazones, and employing large bite angle ligands such as Xantphos.16 Remarkably, selective nucleophilic addition at the 2-position of the diene skeleton was observed, leading to the formation of branched hydrazines.
However, to date no investigations of the use of substituted dienes other than 2,3-dimethyl-1,3-butadiene, isoprene (2-methyl-1,3-butadiene) and 1,3-cyclohexadiene have been reported for the hydroamination with hydrazones (formally hydrohydrazonation) as nucleophiles (Scheme 2). 2-Substituted dienes can potentially deliver a range of interesting N-functionalized products upon regioselective hydroaddition of an amine or derivative thereof, including chiral hydrazines and ultimately branched amines, in the case of selective 1,2-addition.17
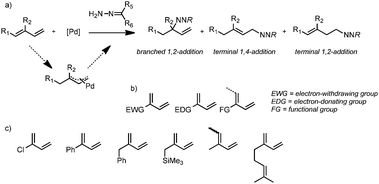 | ||
| Scheme 2 (a) General Pd-catalyzed reaction between dienes and hydrazones via a π-allyl intermediate and the regioisomeric 1,2- and 1,4-products generated. (b) Potential substrate scope and (c) different substrates employed in this work. | ||
Hydrazones may function as protected or masked amines or hydrazines via post-catalysis modification procedures, i.e. N–N bond cleavage or N–C bond hydrolysis, although the former is likely not a trivial conversion. At present, it is unknown if and how the regioselectivity of the hydroaddition reaction can be controlled and to what extent the substitution pattern of the diene plays a governing role in tuning the outcome. We herein report initial studies on the regioselective hydroamination of substituted dienes to explore these issues, unraveling some subtle factors that affect the overall reaction outcome, including the choice of ligand and nucleophile and the character of the diene substituent group.
Results and discussion
The 3-chloro-1-amino-2-butenyl fragment is an often encountered motif in biorelevant organic compounds, but introduction of this functional group, e.g. in a total synthesis procedure, is currently only available via atom-inefficient amination of 1,3-dichloro-2-butene.18 Curiously, the parent primary amine 3-chloro-1-amino-2-butene has so far eluded major attention in the literature. An efficient route to obtain this compound, or a masked version thereof with a protected amine, would be of considerable synthetic interest.Chloroprene as a substrate for hydroamination
We therefore started our investigations of the regioselective hydroamination of functionalized dienes by surveying the reactivity of commercially available 2-chloro-1,3-butadiene (chloroprene) in the Pd-catalyzed hydroamination with various nitrogen nucleophiles. The readily available technical grade chloroprene (50% solution in xylene) can be efficiently upgraded by distillation to obtain pure chloroprene, although we have found that the as-received solution also provides satisfactory results in most cases with no sign of catalyst deactivation.Using benzophenone hydrazone as a nucleophile and an in situ generated Pd catalyst, derived from [Pd(η3-allyl)(μ-Cl)] dimer (1 mol%) and Xantphos (2 mol%) as a ligand (the same system Hartwig reported16), and after a reaction time of 24 hours at r.t., the only compound obtained after work-up was the linear 1,4-addition product, according to NMR spectroscopy, with a Z![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :E isomer ratio of 75:25. This stereoisomeric ratio remained constant with increasing reaction time, which implies that the mechanism of the reaction does not change with conversion, an indication for catalyst stability. We did not observe any diene oligomerization as unwanted side-reaction.
:E isomer ratio of 75:25. This stereoisomeric ratio remained constant with increasing reaction time, which implies that the mechanism of the reaction does not change with conversion, an indication for catalyst stability. We did not observe any diene oligomerization as unwanted side-reaction.
Ligand screening
Strikingly, a screening of diphosphorus ligand systems revealed very strong differences between quite similar frameworks. Well-established diphosphorus ligands with related bite angle dimensions to Xantphos, such as DPEPhos, DBFphos, dppf, BINAP and JosiPhos, did not lead to any activity, even at increased temperature, and only starting materials could be recovered after 24 hours (Table 1). Modification of the xanthene backbone led to ambiguous results: the 4,5-di(tert-butyl) substituted derivative (tBu-Xantphos) showed similar results to parent Xantphos, whereas Nixantphos (–NH instead of C(CH3)2 as the central fragment) showed no activity. Furthermore, the amine nucleophile proved to be of essential influence to steer reactivity, since hydrazine or phenyl hydrazine, benzyl hydroxylamine, urea or phenyl urea, aniline and morpholine resulted in recovery of chloroprene as starting material. Hence, the reactivity and the regioselectivity observed in the hydroamination with chloroprene as substituted diene is markedly different than for seemingly related (unsubstituted) dienes such as isoprene discussed by Hartwig et al.16 or the addition of aliphatic amine nucleophiles to myrcene as disclosed by Behr et al.14 The origin of this dichotomy is not understood so far but might be related to the nature of the 2-substituent and its imposing electronic effect on the diene fragment upon transformation to a π-allyl fragment.
| Nucleophile | Ligand | Conversion (%) | Regioselectivityb |
|---|---|---|---|
|
a [Pd] = 0.02 M; [L] = 0.02 M; [S]:[Pd] = 50:1; Pd = [Pd(η3-allyl)(μ-Cl)]2, 1 mL CH2Cl2, r.t., 24 h.
b Ratio (1,2):(1,4) of 1,2- and 1,4-isomers formed, as determined by NMR spectroscopy.
c BH = benzophenone hydrazone.
d N.R. = no reaction.
|
|||
| BHc | Xantphos | 90 | <1:99 |
| BH | tBu-Xantphos | 88 | <1:99 |
| BH | Nixantphos | N.R.d | — |
| BH | DPEPhos | N.R. | — |
| BH | JosiPhos | N.R. | — |
| BH | BINAP | N.R. | — |
| PhNHNH2 | Xantphos | N.R. | — |
| Urea | Xantphos | N.R. | — |
| Aniline | Xantphos | N.R. | — |
2-Phenyl-1,3-butadiene as a substrate for hydroamination
Intrigued by the regioselectivity observed for chloroprene, we also studied 2-phenyl-1,3-butadiene as electron-poor 2-substituted diene (Scheme 3) using the same in situ catalyst system, i.e. [Pd(η3-allyl)(μ-Cl)]2 and Xantphos, as this combination provided good activity; additional ligand screening was not performed. This compound is synthesized in a straightforward manner from chloroprene via a Ni-catalyzed reaction with phenylmagnesium chloride in good yield after work-up by distillation. To the best of our knowledge, the reactivity of this compound has not been investigated to any significant extent for hydroamination or other hydroaddition reactions to date. Also for this substrate, full conversion with formation of only the 1,4-addition product was observed after 16 hours. In principle, the primary amine should then be accessible via reductive cleavage or hydrolysis of the N–N bond, analogous to published procedures for related masked amine functionalities.19 This amine has previously only been generated via classic and cumbersome synthetic procedures.20 | ||
| Scheme 3 Chemo- and regioselective hydroamination of 2-phenyl-1,3-butadiene with benzophenone hydrazone to yield a mixture of (E)- and (Z)-1,4-addition products. | ||
Other 2-substituted dienes as substrates for hydroamination
Remarkably, we noted very different behaviour for a different set of 2-alkyl-substituted dienes with benzophenone hydrazone, including 2-benzyl-1,3-butadiene and myrcene, which show preferential 1,2-addition of benzophenone hydrazone (Table 2). Use of 2-(trimethylsilyl)methyl-1,3-butadiene as a substrate led to selective desilylation and formation of the same product as was previously reported from isoprene by Hartwig et al.,16 judging from the 1H NMR spectrum of the crude reaction mixture.
| Substrate | Nucleophile | Ligand | Conv.b (%) | b/l ratioc |
|---|---|---|---|---|
|
a [Pd] = 0.02 M; [L] = 0.02 M; [S]:[Pd] = 50:1; Pd = [Pd(η3-allyl)(μ-Cl)]2, 1 mL CH2Cl2, r.t., 24 h.
b Determined by NMR spectroscopy.
c Branched-to-linear ratio.
d BH = benzophenone hydrazone.
|
||||
| 2-Ph | BHd | Xantphos | 88 | 0/100 |
| 2-Bz | BH | Xantphos | 83 | 80/20 |
| 3-Me-C5 | BH | Xantphos | 83 | 80/20 |
| Myrcene | BH | Xantphos | 53 | 95/5 |
| Myrcene | BH | BINAP | 12 | 100/0 |
| Myrcene | Aniline | Xantphos | 100 | 60/40 |
| Myrcene | Morpholine | Xantphos | 100 | 70/30 |
| Myrcene | Piperidine | Xantphos | 100 | 75/25 |
2-Benzyl-1,3-butadiene, which is readily obtained from chloroprene via a Grignard reaction with BzMgBr, was hydroaminated in 83% yield, with a branched to linear (b/l) ratio of 4. However, a pronounced byproduct, formed during reaction, even under ambient conditions, was found to be the isomeric 2-methyl-1-phenylbutadiene, based on comparison of available NMR spectroscopic data for this substituted styrene derivative.21 This byproduct was not observed at the end of the reaction, implying an as-yet unidentified reversible isomerization or subsequent hydroamination of the internal double bond to furnish the same products. The latter would provide a new entry into the application of (biorelevant) trisubstituted dienes as substrates.
Based on this finding, we investigated 3-methyl-1,3-pentadiene as a possible substrate to test the hypothesis that dienes with an internal double bond may also be efficiently hydroaminated to branched products (Scheme 4). Indeed, monitoring the reaction using the same standard conditions, we observed 83% conversion with a b/l ratio (branched 1,2- vs. linear 1,2- and 1,4-products) of 80:20 under the standard catalytic conditions.
 | ||
| Scheme 4 Hydroamination of 3-methyl-1,3-pentadiene with benzophenone hydrazone to generate branched 1,2-addition products (RPh2CN). | ||
Myrcene as a biorenewable diene substrate for hydroamination
We were delighted to see that the bioavailable terpene 7-methyl-3-methylene-1,6-octadiene (myrcene) was also selectively converted to the branched 1,2-addition product upon reaction with benzophenone hydrazone using Xantphos as a ligand (95:5 regioselectivity), with 53% conversion after 24 h at room temperature. Employing BINAP as a ligand led to slower reaction (12% conversion) but with exclusive formation of the 1,2-branched isomer product. Interestingly, also primary and secondary amines, e.g. aniline, piperidine and morpholine, could be used as nucleophiles under mild conditions, but with regioselectivities between 60:40 and 75:25 for the 1,2-:1,4-addition products. The chiral diene nopadiene did not show any reactivity in the same reaction.
Conclusions
In summary, we have reported on subtle differences occurring in the regioselective Pd catalyzed hydroamination of terpene-like dienes under mild catalytic conditions using hydrazone nucleophiles as protected amines. The choice of ligand, nucleophile and substrate were all found to affect the rate and fate of the reaction. Electron-poor 2-substituted butadienes (chloroprene, 2-phenyl-1,3-butadiene) gave rise to sole formation of the linear 1,4-addition product, whereas electron-rich analogues (2-benzyl-1,3-butadiene, 3-methyl-1,3-pentadiene, myrcene) were predominantly converted (60–95%) to the branched 1,2-addition product. The electronic character of the starting diene most likely impacts the hypothesized allylic intermediate, although further investigations are required to elucidate this aspect. We believe that this protocol may lead to novel pathways for the production of chiral amines from readily available biofeedstocks in an atom-efficient manner, if N–N bond cleavage of hydrazones can be achieved using a mild protocol.Experimental section
General methods
All manipulations were carried out under an inert atmosphere using a nitrogen-filled glovebox or standard Schlenk techniques. All glassware was oven or flame dried immediately prior to use. Dichloromethane was of ACS reagent grade, degassed by purging with nitrogen and dried over appropriate drying agents. 2-Phenyl-1,3-butadiene,22 2-benzyl-1,3-butadiene22 and 2-trimethylsilylmethyl-1,3-butadiene23 were prepared following published procedures. All other reagents and solvents were obtained from commercial sources and used without further purification. 1H NMR spectra were obtained at 300 MHz and recorded relative to residual protio-solvent. 13C NMR spectra were obtained at 126 MHz and chemical shifts were recorded relative to the solvent resonance.Procedure for the Pd-catalyzed hydroamination of dienes with benzophenone hydrazone
In a glovebox, [Pd(η3-allyl)(μ-Cl)]2 (3.7 mg, 0.010 mmol), Xantphos (11.6 mg, 0.020 mmol), diene substrate (1.00 mmol) and benzophenone hydrazone (196 mg, 1.00 mmol) were placed into a small vial, dissolved in dichloromethane (1.00 mL), and sealed with a cap containing a PTFE septum. The reaction mixture was stirred at RT for 24 h. The reaction mixture was then purified by alumina flash chromatography (ethyl acetate–hexanes 5:95) and analyzed by NMR spectroscopy. Alternatively, a flame-dried Schlenk tube was charged with a solution of [Pd(η3-allyl)(μ-Cl)]2 (0.020 mmol), Xantphos (0.040 mmol), benzophenone hydrazone (2 mmol) and diene (2 mmol) in dichloromethane (2 mL). The reaction mixture was stirred at appropriate temperature for appropriate time.
1-(3,7-Dimethylocta-1,6-dien-3-yl)-2-(diphenylmethylene)-hydrazine
1H NMR (300 MHz, CDCl3): δ 1.34 (s, 3H), 1.57 (s, 3H), 1.68 (m, 5H), 1.91 (dd, J = 8.1 Hz, 2H), 5.07 (m, 2H), 5.31 (s, 1H), 5.92 (dd, J = 17.7 Hz, 11.1 Hz, 1H), 7.23–7.29 (m, 5H), 7.46–7.51 (m, 5H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 17.5, 22.7, 24.4, 25.6, 39.9, 60.3, 112.4, 124.6, 126.2, 127.4, 128.1, 128.5, 128.8, 129.2, 129.6, 130.1, 131.6, 132.6, 134.0, 139.5, 144.2, 145.0 ppm. HRMS m/z calculated for C22H28N2: 320.2252; found: 320.2260 (M)+.1-(Diphenylmethylene)-2-(2-methyl-1-phenylbut-3-en-2-yl)-hydrazine
1H NMR (300 MHz, CDCl3): δ 1.35 (s, 3H), 3.04 (m, 2H), 5.05 (dd, J = 1.1 Hz, 17.7 Hz, 1H), 5.11 (dd, J = 1.1 Hz, 11.0 Hz, 1H), 6.07 (dd, J = 17.7 Hz, 11.0 Hz, 1H), 7.18–7.38 (m, 10H), 7.47–7.62 (m, 5H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 17.5, 41.6, 60.3, 113.4, 126.8, 127.4, 128.5, 128.9, 130.6, 133.9, 136.2, 138.5, 138.7, 146.2 ppm. HRMS m/z calculated for C24H24N2: 340.1939; found: 340.1899 (M)+.1-(Diphenylmethylene)-2-(3-methylpent-1-en-3-yl)-hydrazine
1H NMR (300 MHz, CDCl3): δ 0.82 (t, J = 7.8 Hz, 3H), 1.32 (s, 3H), 1.70 (m, 2H), 5.06 (m, 2H), 5.94 (dd, J = 17.7 Hz, 10.6 Hz, 1H), 7.22–7.35 (m, 5H), 7.43–7.60 (m, 5H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 7.7, 21.5, 30.6, 62.4, 117.7, 127.5, 127.7, 128.9, 138.5, 138.6, 140.3, 147.1 ppm. HRMS m/z calculated for C19H22N2: 278.1783; found 278.1726 (M)+.1-(3-Chlorobut-2-en-1-yl)-2-(diphenylmethylene)hydrazine
1H NMR (300 MHz, CDCl3): δ 2.03 (s, 3H), 5.95 (s, 1H), 7.21–7.38 (m, 5H), 7.44–7.59 (m, 5H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 21.3, 112.2, 127.5, 128.1, 128.5, 128.8, 138.4, 138.7, 144.8 ppm. HRMS m/z calculated for C16H15ClN2: 270.0924; found: 270.1010 (M)+.1-(Diphenylmethylene)-2-(3-phenylbut-2-en-1-yl)hydrazine
1H NMR (300 MHz, CDCl3): δ 2.12 (s, 3H), 6.81 (s, 1H), 7.17–7.35 (m, 10H), 7.41–7.52 (m, 5H) ppm; 13C{1H} NMR (126 MHz, CDCl3): δ 18.7, 120.2, 123.0, 125.5, 125.6, 128.0, 128.3, 128.5, 128.6, 138.3, 138.9, 141.2, 144.7 ppm. HRMS m/z calculated for C22H20N2: 312.1626; found: 312.1593 (M)+.Acknowledgements
We thank the SmartMix program CatchBio (project 053.70.213) and the European Research Council (ERC Starting Grant EuReCat 279097 to J.I.v.d.V.) for financial support. We acknowledge fruitful discussions with Prof. Dr Johannes G. de Vries, Dr Lavinia Panella, Dr Bernhard Kaptein and Dr Paul Alsters (all from DSM Innovative Synthesis, a unit of DSM Pharma Chemicals) and Dr Gerjan Kemperman (formerly at MSD), partners in the SmartMix consortium. Fatna Ait El Maate is thanked for technical assistance with LC–MS measurements.Notes and references
- Aliphatic amines: K. Eller, E. Henkes, R. Rossbacher and H. Höke, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2000 Search PubMed; Anilines: T. Kahl, K.-W. Schröder, F. R. Lawrence, W. J. Marshall, H. Höke and R. Jäckh, Ullmann's Encyclopedia of Industrial Chemistry, John Wiley & Sons, New York, 2000 Search PubMed.
- A. Ricci, Amino Group Chemistry, Wiley-VCH, Weinheim, 2008 Search PubMed.
- Rh-catalyzed hydroaminomethylation: D. Crozet, M. Urrutigoïty and P. Kalck, ChemCatChem, 2011, 3, 1102 CrossRef CAS; G. T. Whiteker, Top. Catal., 2010, 53, 1025 CrossRef; L. Monnereau, D. Sémeril and D. Matt, Green Chem., 2010, 12, 1670 RSC; B. Hamers, E. Kosciusko-Morizet and D. Vogt, ChemCatChem, 2009, 1, 103 CrossRef; B. Hamers, P. Baüerlein, C. Müller and D. Vogt, Adv. Synth. Catal., 2008, 350, 332 CrossRef; M. L. Clarke and G. J. Roff, Green Chem., 2007, 9, 792 RSC.
- Allylic amination: D. Intrieri, A. Caselli, F. Ragaini, P. Macchi, N. Casati and E. Gallo, Eur. J. Inorg. Chem., 2012, 569 CrossRef CAS; S. M. Paradine and M. C. White, J. Am. Chem. Soc., 2012, 134, 2036 CrossRef; M. E. Harvey, D. G. Musaev and J. Du Bois, J. Am. Chem. Soc., 2011, 133, 17207 CrossRef; M. J. Pouy, L. M. Stanley and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 11312 CrossRef.
- Aziridination: H. Lebel, C. Spitz, O. Leogane, C. Trudel and M. Parmentier, Org. Lett., 2011, 13, 5460 CrossRef CAS; V. Lyaskovskyy, A. I. Olivos Suarez, H. Lu, H. Jiang, X. P. Zhang and B. de Bruin, J. Am. Chem. Soc., 2011, 133, 12264 CrossRef; S. A. Cramer and D. M. Jenkins, J. Am. Chem. Soc., 2011, 133, 19342 CrossRef; I. D. G. Watson, L. Yu and A. K. Yudin, Acc. Chem. Res., 2006, 39, 194 CrossRef.
- Amination/amidation: T. W. Liwosz and S. R. Chemler, J. Am. Chem. Soc., 2012, 134, 2020 CrossRef CAS; P. B. White and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 18594 CrossRef; J. D. Neukom, N. S. Perch and J. P. Wolfe, J. Am. Chem. Soc., 2010, 132, 6276 CrossRef; A. Iglesias, E. G. Pérez and K. Muñiz, Angew. Chem., Int. Ed., 2010, 49, 8109 CrossRef; F. Cardona and A. Goti, Nat. Chem., 2009, 1, 269 CrossRef.
- C–H amination: J. F. Berry, Dalton Trans., 2012, 41, 700 RSC; E. R. King, E. T. Hennessy and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 4917 CrossRef CAS; H. Lu and X. P. Zhang, Chem. Soc. Rev., 2011, 40, 1899 RSC; H. Lu, H. Jiang, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2010, 49, 10192 CrossRef; F. Collet, R. H. Dodd and P. Dauban, Chem. Commun., 2009, 5061 RSC; S. Fantauzzi, A. Caselli and E. Gallo, Dalton Trans., 2009, 5434 RSC.
- Reductive amination: J. Norinder and A. Börner, ChemCatChem, 2011, 3, 1407 CrossRef CAS; N. Andrushko, V. Andrushko, P. Roose, K. Moonen and A. Börner, ChemCatChem, 2010, 2, 640 CrossRef; C. Wang, A. Pettman, J. Bacsa and J. Xiao, Angew. Chem., Int. Ed., 2010, 49, 7548 CrossRef . Alternative methods: C. Gunanathan and D. Milstein, Science, 2007, 317, 790 CrossRef; T. Zweifel, J. V. Naubron and H. Grützmacher, Angew. Chem., Int. Ed., 2009, 48, 559 CrossRef.
- T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef.
- J. I. van der Vlugt, Chem. Soc. Rev., 2010, 39, 2302 RSC.
- K. Das, R. Shibuya, Y. Nakahara, N. Germain, T. Ohshima and K. Mashima, Angew. Chem., Int. Ed., 2012, 51, 150 CrossRef CAS; C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588 CrossRef; D. Pingen, C. Müller and D. Vogt, Angew. Chem., Int. Ed., 2010, 49, 8130 CrossRef; S. Imm, S. Bähn, L. Neubert, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8126 CrossRef.
- U. M. Dzhemilev, G. A. Tolstikov and R. I. Khusnutdinov, Russ. J. Org. Chem., 2009, 45, 957 CrossRef CAS.
- K. A. D. Swift, Top. Catal., 2004, 27, 143 CrossRef CAS; J. L. F. Monteiro and C. O. Veloso, Top. Catal., 2004, 27, 169 CrossRef; A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef; A. Behr and L. Johnen, ChemSusChem, 2009, 2, 1072 CrossRef; A. J. D. Silvestre and A. Gandini, in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, Amsterdam, 2009, pp. 17–38 Search PubMed.
- A. Behr, L. Johnen and N. Rentmeister, Adv. Synth. Catal., 2010, 352, 2062 CrossRef CAS.
- K. Takabe, T. Katagiri, J. Tanaka, T. Fujita, S. Watanabe and K. Suga, Org. Synth., 1989, 67, 44 CAS.
- A. M. Johns, Z. Lu and J. F. Hartwig, Angew. Chem., Int. Ed., 2007, 46, 7259 CrossRef CAS.
- 2-Phenyl-1,3-butadiene is prepared via Ni-catalyzed coupling of PhMgCl to chloroprene: C. Sahlberg, A. Quader and A. Claesson, Tetrahedron Lett., 1983, 24, 5137 CrossRef CAS . 2-Benzyl-1,3-butadiene is prepared in a similar fashion. Alternative syntheses: S. Nunomoto, Y. Kawakami and Y. Yamashita, Bull. Chem. Soc. Jpn., 1981, 54, 2831 CrossRef; R. R. Pidaparthi, M. E. Welker, C. S. Day and M. W. Wright, Org. Lett., 2007, 9, 1623 CrossRef.
- L. F. Tietze, F. Behrendt, F. Major, B. Krewer and J. M. von Hof, Eur. J. Org. Chem., 2010, 6909 CrossRef CAS; L. F. Tietze, F. Major, I. Schuberth, D. A. Spiegl, B. Krewer, K. Maksimenka, G. Bringmann and J. Magull, Chem.–Eur. J., 2007, 13, 4396 CrossRef; L. F. Tietze, T. Herzig, T. Feuerstein and I. Schuberth, Eur. J. Org. Chem., 2002, 1634 CrossRef; L. F. Tietze, T. Herzig, A. Fecher, F. Haunert and I. Schuberth, ChemBioChem, 2001, 2, 758 CrossRef.
- See ref. 16 and: R. E. Johnson, J. T. Hane, D. C. Schlegel, R. B. Perni, J. L. Herrmann Jr., C. J. Opalka, P. M. Carabateas, J. H. Ackerman and J. Swestock, J. Org. Chem., 1991, 56, 5218 CrossRef CAS.
- R. C. Mansfield and C. J. Schmidle, J. Org. Chem., 1956, 21, 699 CrossRef CAS; A. Buschauer, A. Friese-Kimmel, G. Baumann and W. Schunack, Eur. J. Med. Chem., 1992, 27, 321 CrossRef.
- A. Padwa, T. Caruso, S. Nahm and A. Rodriguez, J. Am. Chem. Soc., 1982, 104, 2865 CrossRef CAS . The precise mechanism for the observed isomerization is not known to date, but the intermediacy of a Pd-allylic species is envisaged as a likely candidate.
- D. Djahanbini, B. Cazes, J. Gore and F. Gobert, Tetrahedron, 1985, 41, 867 CrossRef CAS; I. Ojima, M. Yatabe and T. Fuchikami, J. Org. Chem., 1982, 47, 2051 CrossRef; A. Hosomi, M. Saito and H. Sakurai, Tetrahedron Lett., 1979, 20, 429 CrossRef; M. E. Jung and C. A. McCombs, Tetrahedron Lett., 1976, 17, 2935 CrossRef.
- The selective 1,2-hydroamination of anilines to 1,1-dimethylallene furnishes similar compounds: (a) J. F. Beck and J. A. R. Schmidt, RSC Adv., 2012, 2, 128 RSC See also: ; (b) K. L. Butler, M. Tragni and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2012, 51, 5175 CrossRef CAS.
Footnote |
| † Present address: Université Artois, Unité de Catalyse et de Chimie du Solide (UCCS – UMR CNRS 8181) F-62307 Lens, France. |
| This journal is © The Royal Society of Chemistry 2013 |