 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
To aggregate, or not to aggregate? considerations in the design and application of polymeric thermally-responsive nanoparticles
Matthew I.
Gibson
* and
Rachel K.
O'Reilly
*
Department of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, UK CV4 7AL. E-mail: m.i.gibson@warwick.ac.uk; rachel.oreilly@warwick.ac.uk; Fax: +44 (0)2476 524112
First published on 9th April 2013
Abstract
The aim of this review is to highlight some of the challenges in designing thermally responsive nanoparticles, where the responsivity is endowed by a responsive polymeric corona. A review of the literature reveals many contradictory observations upon heating these particles through their transition temperature. Indeed, both an increase in size due to aggregation and particle shrinkage have been reported for apparently similar materials. Furthermore, careful review of the literature shows that responsive nanoparticles do not have the same transition temperature or properties as their constituent polymers. These observations raise serious questions as to how to achieve the rational design of a responsive particle with a predictable and reproducible response. Here we highlight specific cases where conflicting results have been observed for spherical particles and put these results into the context of flat-surface grafted polymer brushes to explain the behaviour in terms of grafting density, curvature, chain end effects and the role of the underlying substrate. A better understanding of these observations should lead to the improved design of nanoparticles with real function and applications.
 Matthew I. Gibson | Dr Matthew I. Gibson is an Assistant Professor (2012–) and Science-City Research Fellow (2010–) in the Department of Chemistry at the University of Warwick, UK. Prior to this he received his PhD (2007, with Neil Cameron, Durham) and spent a post-doctoral period with Harm-Anton Klok (EPFL, Switzerland). Matthew now leads a diverse research group, with backgrounds in chemistry, biochemistry and biology, working on the global theme of interfacing materials with biology for healthcare applications. Current research includes developing biochemically responsive polymers, probing carbohydrate–protein interactions for pathogen inhibition/detection, and also the design of antifreeze-protein mimetics for cellular cryopreservation and regenerative medicine. Matthew was awarded the 2012 MacroGroup UK Young Researchers Medal for his contribution to polymer science. |
 Rachel K. O'Reilly | Professor Rachel O'Reilly is an EPSRC career acceleration fellow in the Chemistry Department at the University of Warwick. She graduated from the University of Cambridge in 1999 and completed her PhD at Imperial College, London in 2003. She then moved to the US under the joint direction of Professors Craig J. Hawker and Karen L. Wooley. In 2004 she was awarded a research fellowship from the Royal Commission for the Exhibition for 1851 and in 2005 took up a Royal Society Dorothy Hodgkin Fellowship at the University of Cambridge. In 2009 she moved to her current position and was promoted to a full professorship in 2012. She was the 2012 recipient of the IUPAC-Samsung young polymer scientist award and the 2013 Hermann Mark young scholar award from the ACS. Her research focuses on bridging the interface between creative synthetic, polymer and catalysis chemistry, to allow for the development of materials that are of significant importance in medical, materials and nanoscience applications. |
Introduction
Inspired by the responsive and adaptable nature of biological macromolecules and assemblies, it has long been the goal of chemists to create synthetic (macro)molecules which can match Nature's performance. Modern controlled and highly tolerant polymerisation methodologies coupled with highly-orthogonal modification chemistries now allow access to macromolecules bearing virtually any functionality with a range of topologies and architectures, and even offers some hope of sequence control. Responsive polymers which are able to undergo large structural changes in response to an external stimulus have attracted much attention working towards achieving the goal of generating ‘smart’ materials capable of giving a measurable output in response to environmental changes. In pursuit of this, materials which respond to electrical currents, electromagnetic fields, light, pH, enzymes, salts, biomarkers and more have been synthesised and are the subject of many reviews.1–3 One of the most promising and most studied stimuli is heat. Many synthetic (and natural) polymers display lower critical solution temperature (LCST) or upper critical solution temperature (UCST) behaviour promoting reversible desolvation or solvation, respectively, when heated above a critical transition temperature, depicted in Fig. 1. Some common LCST thermoresponsive polymers are shown in Table 1. The temperature where phase separation occurs is termed the cloud point, which is the measurable macroscopic effect. The actual LCST/UCST is defined as being the intersection of the spinodal and coexistence curves – this difference is crucial in the discussion below.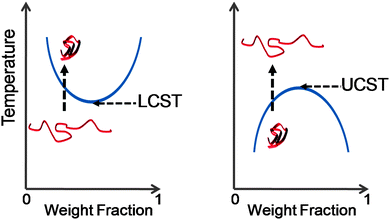 | ||
| Fig. 1 Schematic showing phase transition associated with LCST (lower critical solution temperature) and UCST (upper critical solution temperature) behaviour. Blue line represents the phase separation boundary, which produces a cloud point in solution. | ||
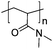
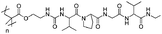
A further advantage of modern polymerisation methods is that they allow for the incorporation (typically at chain ends) of discrete functionality to enable immobilisation onto surfaces. In particular, immobilisation of polymers onto nanoparticles to form a responsive corona offers potential in a wide range of technological, biological and medical applications where the interfacial properties are being modulated. This includes reducing non-specific protein binding, improving biocompatibility, preventing coagulation/aggregation, enabling sensing capabilities or modulating solubility.
The aim of this review is to introduce the reader to some of the design criteria which must be considered when synthesising thermoresponsive (specifically LCST) polymer-coated nanoparticles. Particular focus will be placed on the influence of immobilisation on the observed phase transitions and the measurement techniques necessary to observe these. This will be put into context by comparison with the (significant) literature of flat-surface grafted polymer brushes and also compared to related solution studies of responsive assemblies. This is not intended to be a complete and comprehensive review on thermally responsive polymers, but rather to set the scene for the discussion of the individual factors which contribute to their properties and the challenges faced in ensuring these particles have predictable, desirable, and appropriate thermal responses in their targeted application.
Synthesis of responsive nanoparticles
There are 3 general methods used, in the context of this article, to access responsive nanoparticles with polymeric coronas; grafting from premade nanoparticles, grafting to premade nanoparticles and solution self-assembly of amphiphilic block copolymers; these are summarised in Fig. 2. Synthetic methodology is not the focus of this review and readers are directed to the individual references and also a recent review.4 However, in order to place into context the concepts described later it is necessary to highlight some of the key methodologies used to obtain responsive particles discussed herein.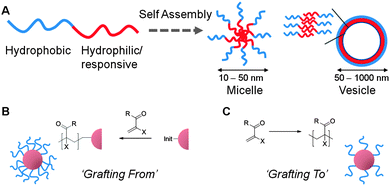 | ||
| Fig. 2 Synthetic concepts used to obtain naoparticles with a thermo-responsive polymer corona. (A) Self-assembly of block copolymeramphiphiles; (B) grafting of polymers from an initiator-functionalised nanoparticle core; (C) grafting of polymer chains onto pre-formed nanoparticle cores. | ||
Block copolymers where (at least) one block is hydrophobic and the other is hydrophilic (or in this case responsive) spontaneously self-assemble in aqueous solution to reduce the unfavourable interactions between water and the hydrophobic block. Depending on the ratio of each of the block lengths, either micelles (high curvature) or vesicles with a bilayer structure (low curvature) are formed, Fig. 2A. Micelles have hydrophobic interiors whereas vesicles have a discrete hydrophobic bilayer and hydrophilic interior domain. This bottom up assembly approach has been demonstrated to afford well-defined and responsive polymeric nanostructures. An alternative route to core–shell nanostructures is to use pre-formed nanoparticles, typically based on inorganic substrates such as silica, gold, iron oxide or even polymer colloids, which offers the opportunity to predictably and precise control the particle size and shape. These pre-formed substrates can subsequently be modified by one of two methods, Fig. 2B and C. The first method is the ‘grafting to’ approach, where an already-synthesised polymer which has a reactive ‘handle’ capable of binding to the particle surface is immobilised. This approach has the advantage of being able to fully characterise each component prior to nanoparticleassembly allowing for excellent size and property control in the resultant hybrid particle. The second method the ‘grafting from’ approach requires a polymerisation initiator (e.g. for radical, ring opening or metathesis) to be first attached to the surface of the particle and polymer chains are subsequently grown from this attachment point. This method tends to give denser and longer coronal polymer chains and the consequences of this are discussed in later sections.
Applications of corona-responsive particles
The potential applications of any corona-responsive particle lie in their fundamental ability to change a physical parameter (be it solubility, size, aggregation, charge or more) upon application of an external stimuli, or upon encountering a different environment containing a desired concentration of a particular stimulant. In this section, the potential emerging, applications of responsive nanostructures, which may be either as polymers or particles, are summarised.An early application of thermoresponsive polymers was to modulate enzyme function.5 Hoffman and co-workers conjugated PNIPAM onto β-glucosidase to enable facile isolation, and to modulate its activity. While most applications of the LCST focus on a soluble to insoluble transition, this has also been exploited to trigger a change in lipophilicity to promote phase transfer from aqueous to organic solutions.6,7 This solubility switch results in a change of partition coefficient, which is a key characteristic in the design of ‘drug like’ molecules which are able to cross cellular membranes. In particular, this switching may be useful for hyperthermic targeting of disease sites, such as tumours which are typically 1–2 °C warmer than surrounding tissues. PNIPAM was shown to preferentially accumulate in a murine tumour model8 and in vitro studies have shown that heating (40 °C) PNIPAM-coated gold nanoparticles above their LCST (37 °C) results in cellular internalisation.9 Alternatively, thermoresponsive polymers have been used to reversibly mask tumour-targeting ligands, so that upon applying a local temperature gradient, the ligands are exposed, promoting receptor-mediated endocytosis.10 A further example of this uses gold nano-rods which undergo a localised heating effect when exposed to near-infrared (NIR) light (650–900 nm). This was exploited to trigger a phase transition in surface-grafted PNIPAM.11 Selective accumulation of the thermoresponsive nano-rods in murine kidneys was achieved by targeted exposure of the NIR light. Polyionic complexes comprising of DNA and poly(ethyleneimine) with a PNIPAM corona were used to deliver green fluorescent protein plasmid into B16F10 cells by switching the temperature from 25 to 37 °C which is above the LCST of the PNIPAM component.12Fig. 3 shows an example of thermally-triggered internalisation of P(NIPAM-co-DMAAm)-block-poly(lactic acid) micelles into endothelial cells upon heating ∼3 °C above their LCST.13 In addition to using the LCST to trigger cellular uptake or aggregation, it has also been exploited for the controlled release of encapsulated drug molecules. Okano et al. developed micelles with a PNIPAM corona, containing the hydrophilic drugAdriamycin. Upon heating, the corona collapsed, destabilising the micelle and causing Adriamycin release.14 As this review is focused on nanoparticles with responsive coronas, we do not cover the assembly and encapsulation of drugs into particles with responsive cores, or the use of a thermoresponsive polymer to drive self-assembly of polymer–protein hybrids,15 but these are covered in several recent reviews.16–18
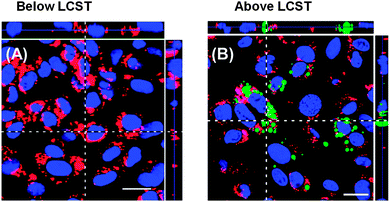 | ||
| Fig. 3 Laser scanning confocal images showing thermally-triggered uptake of P(NIPAM-co-DMAAm)-block-poly(lactic acid) micelles into endothelial cells. Cell nuclei are shown in blue, micelles in green. Red stains are organelle-specific stains. Reprinted (adapted) with permission from Mol. Pharmaceutics 2010, 7, 926. Copyright (2013) American Chemical Society.13 | ||
Covalent incorporation of a fluorescent dye sensitive to the hydration of the polymer chain provides a fluorometric output for LCST phase transitions, and can easily be incorporated into nanoparticle assemblies.19 By heating above the LCST, expulsion of water molecules changes the dye’s hydration state and hence fluorescence behaviour. Gota et al. used thermoresponsive PNIPAM nanogels to probe the intracellular temperature within a fibroblast-like cell line with a sensitivity of ∼0.5 °C in the range 27–33 °C, shown in Fig. 4.20 Similarly, Qiao et al. synthesised block copolymers of PNIPAM and polystyrene which formed micellar thermoresponsive nanoparticles with thermally-tuneable emission.21
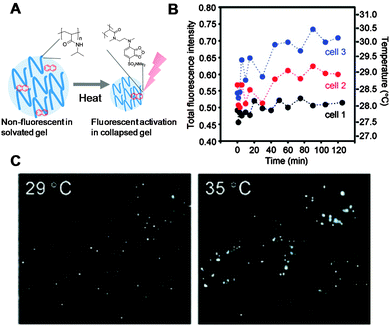 | ||
| Fig. 4 Intracellular thermometer based on solvochromic dye labelled PNIPAM nanogels. (A) Schematic of a thermoresponsive nanogel containing a solvochromic dye. Increasing temperature above LCST induces fluorescence emission due to an increase in hydrophobicity; (B) intracellular fluorescence measurements of individual cells exposed to Camptothecin, which leads to intracellular temperature changes; (C) fluorescence microscope images of cells incubated with nanoparticles at different temperatures demonstrating increased fluorescence at high temperatures. Figures B and C reprinted with permission from J. Am. Chem. Soc., 2009, 131, 2766–2767. Copyright (2013) American Chemical Society.20 | ||
It is also worth highlighting the concept of isothermal LCST transitions, whereby the desirable transition of a thermoresponsive polymer/particle is triggered by application of a secondary stimulus, which effectively shifts the phase diagram of the polymer to trigger the transition. Examples of these triggers include pH-triggered disassembly, addition of salts,22 bacteria23 or intracellular glutathione.24
The concept of phase transitions has been exploited extensively within amphiphilic block copolymer assemblies for triggering changes in the self-assembly processes which can be utilised in controlled release or morphology switching applications, Fig. 5. Interestingly, using polymers such as PNIPAM it has been reported that temperatures well above its expected LCST (∼32 °C) are required to induce the morphology transition. These transitions were observed to be relatively slow when using PNIPAM which was attributed to its high glass transition temperature and strong interchain H-bonding which prevents fast and reversible switching. Indeed, further work by Grubbs demonstrated that poly(ethylene oxide)-co-poly(butyleneoxide) was a more effective responsive block and allowed for fast and effective morphology switching.25 More recently Chen and Jiang have demonstrated that through design of a lightly associated permanently hydrophobic domain, fast and reversible micelle–vesicle transitions could be observed at temperatures close to the LCST of PNIPAM.26 They accounted for this through the reduced restriction of conformational changes in the responsive block in these nanostructures.
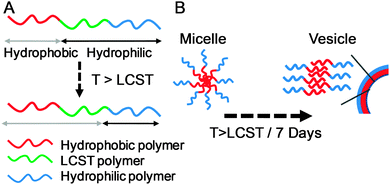 | ||
| Fig. 5 Concept of morphology-switching self-assembled block-copolymernanoparticles. (A) Triblock copolymer motif with defined hydrophobic, responsive and hydrophilic segments; (B) micelle to vesicle (bilayer) transition upon heating the responsive block above its LCST. | ||
Responsive polymer brushes on flat substrates
In order to understand and discuss the influence of immobilisation of polymers during the synthesis of responsive nanoparticles, it is necessary to briefly discuss the significant progress which has been made in the study of responsive polymers on flat surfaces. Such systems have been probed by several complimentary techniques including contact angle, atomic force microscopy, elipsometry, quartz-crystal microbalance, infrared and neutron reflectivity. Polymer brushes, where one chain-end of a polymer is immobilised onto a solid substrate, have attracted wide interest as ultra thin (10's–100's of nanometre) functional coatings. The two general methods to obtain these brushes are the same as for coating pre-formed nanoparticles with polymers (i.e. the ‘grafting from’ and ‘grafting to’ approaches). Generally speaking, ‘grafting from’ gives rise to high polymer brush densities and thicker brushes than the ‘grafting to’ method. The synthetic methods to obtain these are outside of the scope of this review, but readers are pointed to some detailed review articles.27,28 Since the original work by Alexander and de Gennes29,30 on the conformations of surface-tethered brushes there has been a huge research effort to understand their structure. Original theories made the key assumption that the density of the polymer chains was constant throughout the brush and hence the chain ends are all at the same distance from the surface29,30 (Fig. 6A). However, the more recent mean field theory of polymer brushes suggests that the structure shown in Fig. 6B is a more accurate representation, with a higher density of monomer units at the substrate compared to the surface.31 This distinction of the two independent regions in polymer brushes is crucial in their applications, especially in responsive systems where the bulk transitions (such as in a nano-actuator), or the surface (such as reversible cell adhesion) are critical.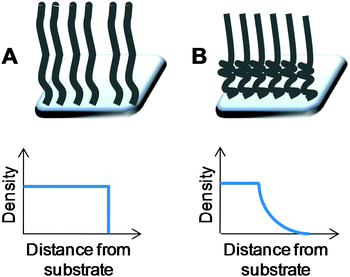 | ||
| Fig. 6 Generalised depictions of density (monomer unit concentration) profiles in polymer brushes as a function of distance from the surface to which the chains are grafted. (A) Constant density with chain ends at equal distances; (B) variable density brushes with density decreasing above a critical distance. | ||
Huck and co-workers have studied the effects of confinement of thermally responsive, nanopatterned, poly(2-(2-methoxethoxy)ethyl methacrylate) brush by atomic force microscopy (AFM).32 Feature sizes of 35 nm which contained only ∼300 individual polymer chains were compared to those which were essentially infinitely large in one dimension. Upon heating–cooling cycles the 35 nm features (whose polymer chains can explore more space) showed larger swelling ratios compared to the laterally infinite brushes. The smaller brush features also displayed significantly broader collapse transitions occurring over the range of 20–40 °C, as opposed to the sharp transition between 25–32 °C observed for the infinitely large brush domain. This was rationalised as in small feature sizes each individual chain can explore more space, as they are less sterically confined (i.e. larger volume/brush is available) which accounts for the observed broadness of the transition. Such a comparison is similar to a curved nanoparticle surface, which is discussed in the next section. Whilst AFM provides detailed structural information it probes the bulk changes, rather than surface properties. To address this, Lalyaux et al. used a combination of quartz crystal microbalance (QCM) and contact angle (CA) measurements to simultaneously probe the collapse of polymer chains and the change in CA as a function of temperature.33 Between 30 and 40 °C the thermally triggered collapse (shrinkage) of PNIPAM was observed due to de-solvation of the polymer. However, there was no increase in the CA until the temperature passed 40 °C, suggesting significant polymer brush solvation even above the predicted LCST. This has profound implications for the design of responsive particles as an increase in hydrophobicity (or surface energy) triggers aggregation, but this result indicates that the LCST transition has already occurred prior to any observed surface changes. Using elipsometry, Vansco and co-workers separated the collapse of each segment of the polymer brush.34 Low grafting density brushes demonstrated a smooth, broad phase transition compared to the higher density brushes which had a sharp phase transition which may represent the different levels of hydration of the different segments. Modelling the elipsometric data suggested that the flexible surface segments began to collapse prior to any changes in the densely packed layers. Neutron reflectivity studies35 and QCM studies of electrolyte-induced transitions36 have also confirmed this non-linear collapse of PNIPAM brushes. Low density PNIPAM brushes (<0.1 chains per nm2) displayed no appreciable change in contact angle or thickness upon heating up to 40 °C, in contrast to higher densities which became more hydrophobic upon heating. This observation strongly influenced protein–cell binding within these polymer brushes.37,38
Taken together and placed in the context of nanoparticles, it can be seen that surface and bulk properties of polymer films must be considered when designing thermoresponsive nanoparticles. In particular, the transition temperatures and measurable macroscopic properties are not necessarily the same as those of their linear, soluble polymer counterparts.
Thermally responsive nanoparticles
The remainder of this review will discuss thermally responsive polymer-coated nanoparticles with a particular focus on those displaying lower critical solution temperature behaviour. By focusing on LCST polymeric materials, comparison to flat-surfaces ‘polymer brushes’ will be made and observed trends highlighted.Transitions and macroscopic outcomes
When studying the phase transitions of responsive polymers the most commonly measured macroscopic property is the onset of aggregation via turbidimetry – monitoring the decrease in transmitted light once the polymer has been heated above its cloud point. Although this has the advantage of being an experimental observation, it is actually measuring the outcome of the transition not the transition itself; these are not necessarily the same. Complementary techniques such as microcalorimetry and 1H NMR spectroscopy have revealed that the actual transitions begin at temperatures below that of the cloud point, and highlights a problem in using macroscopic events to characterise a molecular process.39 A consequence of this is that is not clear how the thermoresponsive properties (onset, cloud point, transition width) of the polymers will translate when incorporated onto the surface of nanoparticles compared to behaviour when free in solution. Furthermore, the intermediate states during the transition, which include contraction and knotting before final collapse, make this a rather complex process. This will not be discussed in this review, but readers are pointed to the work of Wu and co-workers on single-polymer chain collapse using light scattering techniques.40,41Gibson et al. observed that poly[oligo(ethyleneglycol)methacrylate] coated gold nanoparticles displayed significantly lower cloud points than the responsive polymer alone in solution, highlighting this point.42 The free POEGMA had a cloud point of >95 °C whereas a 50 nm diameter gold nanoparticle functionalised with the same polymer showed a cloud point of 75 °C. Such a dramatic decrease had not been previously reported and represents a serious challenge in the predictable design of responsive nanoparticles with controlled transitions. This size-dependant decrease was further shown to occur for a wide range of POEGMAs with different LCSTs, and most interestingly also for self-assembled polymeric micelles.43 Indeed, with poly(N-vinylcaprolactam) as the responsive corona and poly(vinyl acetate) as the core, 50 nm block copolymer micelles had transition temperatures 30 °C lower than the homopolymer alone. Although these decreases have not been explicitly defined before, a review of the literature reveals several examples where reported cloud points for responsive nanoparticles are lower than that of the responsive segment alone. An early example from Li and co-workers44 demonstrated that a PNIPAM coated 13 nm gold nanoparticle had a cloud point ∼29 °C compared to ∼35 °C for the free polymer in solution. Similarly, examples of poly(N-vinyl lactam),45 PNIPAM46 on gold nanoparticles and PNIPAM,47 poly(N-vinyl lactam)48 micelles, POEGMA on silica49 and PNIPAM absorbed onto poly(styrene) colloids all resulted in depressed thermal transitions/cloud points.50 Tenhu and co-workers have investigated the influence of gold nanoparticle core size (in the range of 2–4.1 nm) with PNIPAM oligomer coatings (5–28 repeat units) on their thermal responses. Using microcalorimetry, a depression of the transition temperature from 13 to 8.7 °C was measured compared to the free oligomer. Interestingly, the reported transitions are far below that of what is typically reported for PNIPAM (∼32 °C). This appears to be due to significant end-group effects for oligomers, which has been shown for several systems.24,51,52
In order to explain this particle size dependent trend on transition temperature, which is not universally observed, comparison to the previously discussed cases of flat substrates with grafted polymer brushes is required. Restricting polymer chain flexibility in a confined environment would be expected to lead to dramatic changes in both the conformation and the transitions of thermally responsive polymers. When we consider a spherical surface the free space per polymer chain at equal grafting densities, will be strongly dependent on the degree of curvature, shown schematically in Fig. 7A. Fig. 7B shows how the average volume per chain for surface grafted polymers on spherical nanoparticles varies as a function of particle radius. Although this is an oversimplified model it highlights how dramatic effects should be seen for nanoparticles with diameters in the range 1–50 nm, where small changes in size have dramatic influence on curvature, which fits with the observations of Gibson et al.42,43 Zhulina et al.53 predicted in 1991 that spherical polymer brushes would undergo temperature-triggered collapse from the outer layer due to the effective lower density compared to the interior of the brush and this has since been shown experimentally on flat substrates.34 As particle size changes, there is also an effective change in local functional unit concentration. This presents a feasible explanation for changes observed in cloud point, as this property is intrinsically concentration dependant in solution. The final factor to consider is the actual grafting density of the polymer onto the surface; the relative volume per chain will vary between high and low grafting densities resulting in differences in local monomer unit concentrations and hence may influence the thermal transition temperatures. To the best of our knowledge the collapse transition temperature as a function of grafting density has not yet been studied in detail, although some observations have been made (vide infra).54
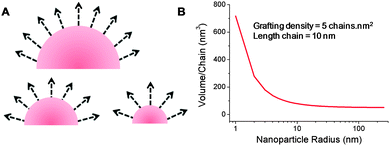 | ||
| Fig. 7 Variation of free volume per polymer chain as a function of nanoparticle curvature, assuming constant grafting density and chain elongation. (A) Schematic of nanoparticle surface with polymer chains represented as vectors; (B) calculated volume/chain as a function of nanoparticle radius. | ||
This interplay between grafting density, curvature and elongation of the polymer chains is complicated when considering particles, particularly in terms of the macroscopic consequences of the polymers passing through their transitions. As discussed above for polymer brushes, a transition (in terms of partial collapse of the polymer chains) can occur without significant changes in the contact angle, implying there is still hydration–solvation. In the case of nanoparticles, one can imagine the situation where the polymer brush collapses without giving rise to nanoparticle aggregation, or delaying aggregation until further heating is applied. This subtle distinction is absolutely crucial for the properties/applications: for example a change in surface chemistry might be the desirable outcome (e.g. for protein binding), or aggregation (e.g. for sensing) however this may only be observed at a temperature above the actual transition temperature.
Most reports of responsive nanoparticles in solution report the change in optical transmission (aggregation) as an indicator of a thermal response, specific changes in particle size are also observed. Kim et al. synthesised 17 nm gold nanoparticles with a PNIPAM corona by surface-initiated ATRP with a relatively thick polymer layer.55 Upon heating above 30 °C, the nanoparticle diameter was observed by DLS to contract from 190 to 107 nm which is almost identical to what is observed in solid-supported, densely packed polymer brushes. This is in stark contrast to the observed effects for PNIPAM-coated 13 nm gold particles made by an in situ ‘grafting to approach’, via RAFT.44 Upon heating above 27 °C these particles readily aggregated with an observed size increase from ∼100 to 800 nm. This contrasting behaviour for two similarly sized nanoparticles (shown in Fig. 8) with identical chemical structures serves as an example to highlight two contrasting macroscopic outcomes of nanoparticle-tethered polymer phase transitions and highlights the challenge of designing a particle with a particular response for a specific application.
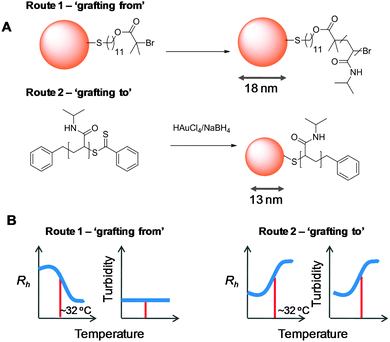 | ||
| Fig. 8 Effect of synthetic method on macroscopic properties upon heating through the LCST. (A) Synthesis of two PNIPAM@Au nanoparticles from the literature with similar diameters by the ‘grafting to’44 and ‘grafting from’56 methods. (B) Summary of observed properties. Rh is hydrodynamic radius (nm) of particles determined by dynamic light scattering. Approximate LCST is indicated on the graphs by a red bar. | ||
Although not a complete review of the (very significant) literature of responsive nanoparticles, a summary of polymer-coated responsive nanoparticles made by both ‘grafting to’ and from approaches is shown in Table 2. This only includes examples where the hydrodynamic diameters of the nanoparticles were explicitly measured by dynamic light scattering, to allow for comparison before and after the thermal transition.
| Particle | Polymer/method | Size transition | Ref. |
|---|---|---|---|
| a Hollow PNIPAM particles obtained via an inorganic template. | |||
| Hollowa | PNIPAM in situpolymerisation | Decrease 400–250 nm | 57 |
| Au | PNIPAM grafting from | Decrease 200–100 nm | 55 |
| Dextran | PNIPAM grafting from | Decrease 250 to 150 μm | 56 |
| Silica | PNIPAM grafting from | Decrease 350–260 nm | 58 |
| Silica | POEGMA grafting from | Decrease 200–190 nm | 49 |
| Micelle | PNIPAM self-assembly | Increase 50–350 nm | 59 |
| Gold | PNIPAM grafting to | Increase 80–110 nm | 60 |
| Gold | POEGMA grafting to | Increase 15–200 nm | 61 |
| Gold | POEGMA grafting to | Increase 20–250 nm | 6 |
The most obvious trend in Table 2 is that responsive nanoparticles with thick, densely packed, brushes (i.e. those prepared by the ‘grafting from’ method) tend to show shrinkage rather than colloidal aggregation upon heating through their LCST, and that those with less densely packed brushes (‘grafting to’) tend to colloidally aggregate following transition. Wu and co-workers observed that 600 nm polystyrene particles with low densities of PNIPAM grafted did not show significant size changes upon heating from 20 to 35 °C, but as the grafting density increased, the contraction upon heating became more pronounced.54 These observations can be speculated to be due to 2 different components; (i) accessibility of chains ends and (ii) reduced grafting densities, leading to an increased contribution from the underlying surface as a consequence of mushroom/brush regime differences. Solution studies of thermo-responsive polymers, particularly by Rimmer et al., have shown that the hydrophilic chains ends of branched PNIPAM can stabilise polymer aggregates above their cloud point to give stable colloids.62 This implies that the chain ends impart significant stability (hydrophilicity) above the cloud point. Low density brushes would be expected to have less significant chain-end effects than high density brushes. A distinct transition exists between very lightly grafted polymers in the ‘mushroom’ regime compared to the densely packed ‘brush’ regime. The brush regime is typically defined as being when the distance between grafting sites of the polymers is greater than twice the radius of gyration (Rg) of the individual polymers. ‘Grafting to’ is intrinsically limited by the excluded volume effect, preventing high densities, unlike ‘grafting from’ approaches, shown schematically in Fig. 9. Brittain and Minko have reviewed the definitions of polymer brushes highlighting this feature which is pertinent in this review.63 In the mushroom regime, the polymer chains do not present sufficient surface coverage to ensure that only polymer–solvent interactions are present, and indeed solvent–substrate interactions are also present. A direct consequence of this is that the hydrophilicity/hydrophobicity of the polymer corona is therefore influenced by the nature of the underlying substrate. This effect is well known from colloid science for steric stabilisation. Using density gradients of poly(acrylamide), Genzer and coworkers64 identified the transition between mushroom and brush by exploring changes in both film thickness and contact angle. Within each individual regime, the responsive behaviour of the polymer and resulting macroscopic properties (such as wetting or aggregation) will therefore be significantly different and the contribution of the underlying substrate will also vary.65
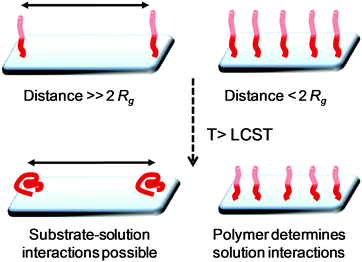 | ||
| Fig. 9 Effect of polymer brush morphology on conformation transitions above their LCST. Rg is the radius of gyration of an individual polymer chain. | ||
Experimental evidence for the surface exposure was obtained by Mastratto et al. PNIPAM-coated AuNPs were synthesised by the grafting to approach (to give polymer chains in the mushroom regime) with hydrophilic cell targeting ligands also included at the surface.10 Below the LCST of the PNIPAM the polymer effectively shielded the underlying surface by steric effects, but above the LCST the polymers collapsed, exposing the ligands which triggered cellular recognition/internalisation, Fig. 10. The lack of aggregation above the LCST (which would be expected based on the previous ‘grafting to’ AuNP studies) and the specific receptor–cell interactions provided evidence that at low grafting densities the underlying substrate can control macroscopic events and in particular phase transitions.
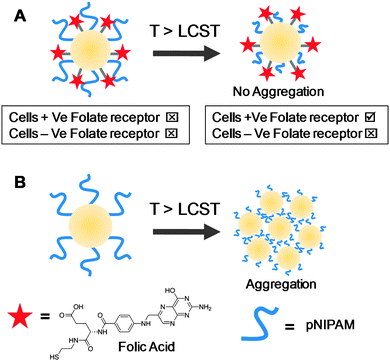 | ||
| Fig. 10 Influence of underlying surface chemistry on macroscopic properties of nanoparticles functionalised with thermoresponsive polymers. (A) Au nanoparticles with both PNIPAM and folic acid on surface. Upon heating above the LCST folate receptors are exposed. Tick indicates successful uptake into specified cells, and cross no uptake. No aggregation is observed above the LCST suggesting the hydrophilic folic acid provides colloidal stability; (B) Au nanoparticle prepared in same method as (A), but without folic acid. Upon heading above the LCST particles aggregate. Images related to data presented in ref. 10 (A) and ref. 44 (B). | ||
As introduced in the above sections, self-assembled block copolymer spherical micelles with a thermoresponsive block in the corona can undergo a morphology switch to form vesicular or cylindrical nanostructures. Reports on this process, using PNIPAM as the central (responsive) block do not report any increase in turbidity nor significant shrinkage upon heating.66,67 While this may appear to conflict with the observations outlined in Table 2, these systems contained a permanently hydrophilic component in the corona, hence they maintain their water-solubility throughout the transition and do not show an increase in diameter until the morphology transformation occurs, Fig. 11. The most surprising observation of these systems is the amount of time required for the morphological transitions to occur, typically taking several days; in the initial stages there is a small decrease in hydrodynamic diameter of the spherical micelles, followed by a relatively abrupt increase in size and dispersity upon formation of vesicles. It is also observed that the systems must be heated well above the LCST of the constituent polymers (ca. 10 °C) implying some confinement effects, as observed with surface grafted brushes. 1H NMR spectroscopy also confirmed the observed increased transition temperature in the nanostructures.66 Complementary studies of self-assembled oligomers containing responsive oligoethyleneglycol (OEG) units with precise numbers of the responsive branches have been undertaken by Thayumanavan and coworkers.68 Micelles constructed from oligomers with more OEG branches showed narrower transitions, at lower temperatures, compared to micelles constructed from oligomers with fewer OEG branches. During transition from micelle to vesicle, the curvature and density of the responsive chains will vary and hence will dramatically change the actual transition temperature during the morphology switch. This, combined with interfacial/surface energy considerations, no doubt contribute to the complex behaviour observed for these systems.
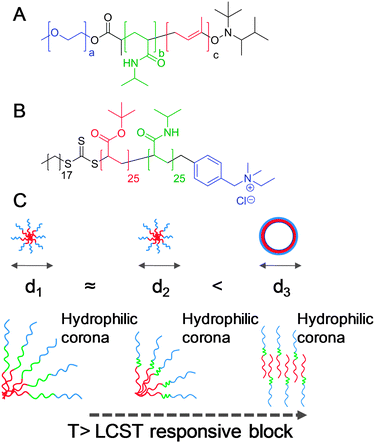 | ||
| Fig. 11 Morphology switching thermally responsive, self-assembled polymer micelles containing a permanently hydrophobic segment (red), permanently hydrophilic segment (blue) and a thermoresponsive block (green). (A) Poly(ethyleneoxide)-block-poly(NIPAM)-block-poly(butadiene); (B) poly(t-butylacrylate)-block-PNIPAM with quaternary ammonium end group; (C) sequence of events leading to morphology collapse, starting with a micelle with responsive corona: heating above the LCST leading to collapse of the responsive block and shrinkage (and shift in hydrophilic/hydrophobic ratio) followed by rearrangement to lower curvature vesicular morphology. | ||
Direct experimental evidence of the conformational regime assumed by nanoparticle-grafted polymer chains has been observed by Gauthier and co-workers, using proteins as monodisperse substrates, Fig. 12A.69 More than 100 discrete polymer–protein conjugates were synthesised by the surface initiated ATRP of oligo(ethyleneglycol)methacrylate from chymotrypsin decorated with varying densities of ATRP initiating sites. This methodology provided precise and quantifiable control over grafting density, number average degree of polymerisation and PEG side-chain length (which influences the LCST). Using 1H NMR spectroscopy as a probe for polymer flexibility, the transition between mushroom and brush regimes as the grafting density increased from 2–12 polymers/protein was clearly observed. Furthermore, this transition correlated with a decrease in the observed cloud points of the conjugates, Fig. 12B. This supports both hypotheses that increasing steric crowding or reducing exposure of the hydrophilic protein surface can lower the responsive transition temperature relative to the free polymers in solution.
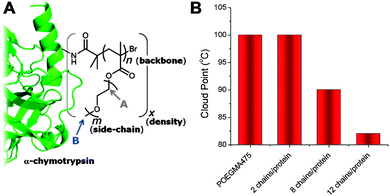 | ||
| Fig. 12 Thermoresponsive polymers grafted to the surface of a protein. (A) synthetic strategy and chemical space explored; (B) observed cloud point of chymotrypsin-graft PPEGMA475 as a function of grafting density. Modified from ref. 69. | ||
Outlook
Here we have summarised the current state of the art in the design of thermally responsive nanoparticles. The key question which is raised here is “what output is desired when assembling a responsive polymer onto a responsive nanoparticle?” While apparently simple, this review has shown that translating the thermally-responsive properties of polymers into nanoformulations is not necessarily straightforward and can give rise to unexpected results.The literature was surveyed to reveal that apparently similar, responsive nanoparticles give rise to distinctly different responses: either aggregation or shrinkage. Here, this is linked to the structure of the grafted polymer layer, particularly the influence of the grafting density. By relating the surface of nanoparticles to the well-studied flat-surface tethered polymers (‘brushes’) it was possible to hypothesise that the differences are due to the different conformation regimes which the polymers can assume; brushes (higher density) or ‘mushrooms’ (lower density). In the mushroom regime, above the collapse (LCST) temperature of polymers, the chains are sufficiently spaced for the underlying substrate to dominate interfacial properties and hence drive aggregation (or ligand expression). In the brush regime, the densely packed brushes cover the surface and hence shrinkage, due to expulsion of water, is observed but the chain ends maintain solubility/mobility preventing aggregation in many cases.
These observations highlight the need to use appropriate spectroscopic tools when monitoring the properties of responsive polymer-coated particles and to ensure that distinction is made between the polymer thermal collapse and the macroscopic aggregation/shrinkage and also careful monitoring of the actual transition temperatures, in addition to clouding points. If responsive particles are to be translated into real-world applications, consistent terminology, testing and appropriate consideration of underlying physical phenomena must be discussed and disclosed. The need to continue investigating these materials, also offers hope for the development of new, yet unknown, functional and responsive nanoparticles with many potential applications.
Acknowledgements
MIG is a Birmingham Science City Interdisciplinary Research Fellow funded by the Higher Education Funding Council for England (HEFCE). ROR holds an EPSRC Career Acceleration Fellowship.Notes and references
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449 RSC.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2009, 5, 2513–2533 RSC.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef.
- O. J. Cayre, N. Chagneux and S. Biggs, Soft Matter, 2011, 7, 2211–2234 RSC.
- G. Chen and A. S. Hoffman, Bioconjugate Chem., 1993, 4, 509–514 CrossRef CAS.
- E. W. Edwards, M. Chanana, D. Wang and H. Möhwald, Angew. Chem., Int. Ed., 2008, 47, 320–323 CrossRef CAS.
- Y. Saaka, R. C. Deller, A. Rodger and M. I. Gibson, Macromol. Rapid Commun., 2012, 33, 779–784 CrossRef CAS.
- D. E. Meyer, B. C. Shin, G. A. Kong, M. W. Dewhirst and A. Chilkoti, J. Controlled Release, 2001, 74, 213–224 CrossRef CAS.
- S. Salmaso, P. Caliceti, V. Amendola, M. Meneghetti, J. P. Magnusson, G. Pasparakis and C. Alexander, J. Mater. Chem., 2009, 19, 1608–1615 RSC.
- F. Mastrotto, P. Caliceti, V. Amendola, S. Bersani, J. P. Magnusson, M. Meneghetti, G. Mantovani, C. Alexander and S. Salmaso, Chem. Commun., 2011, 47, 9846–9848 RSC.
- T. Kawano, Y. Niidome, T. Mori, Y. Katayama and T. Niidome, Bioconjugate Chem., 2009, 20, 209–212 CrossRef CAS.
- H. S. Bisht, D. S. Manickam, Y. You and D. Oupicky, Biomacromolecules, 2006, 7, 1169–1178 CrossRef CAS.
- J. Akimoto, M. Nakayama, K. Sakai and T. Okano, Mol. Pharmaceutics, 2010, 7, 926–935 CrossRef CAS.
- J. E. Chung, M. Yokoyama, M. Yamato, T. Aoyagi, Y. Sakurai and T. Okano, J. Controlled Release, 1999, 62, 115–127 CrossRef CAS.
- C. Lavigueur, J. G. Garcia, L. Hendriks, R. Hoogenboom, J. J. L. M. Cornelissen and R. J. M. Nolte, Polym. Chem., 2011, 2, 333–340 RSC.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173–1222 CrossRef CAS.
- S. R. Abulateefeh, S. G. Spain, J. W. Aylott, W. C. Chan, M. C. Garnett and C. Alexander, Macromol. Biosci., 2011, 11, 1722–1734 CrossRef CAS.
- C. Li and S. Liu, Chem. Commun., 2012, 48, 3262–3278 RSC.
- C. Gota, K. Okabe, T. Funatsu, Y. Harada and S. Uchiyama, J. Am. Chem. Soc., 2009, 131, 2766–2767 CrossRef CAS.
- J. Qiao, L. Qi, Y. Shen, L. Zhao, C. Qi, D. Shangguan, L. Mao and Y. Chen, J. Mater. Chem., 2012, 22, 11543–11549 RSC.
- J. P. Magnusson, A. Khan, G. Pasparakis, A. O. Saeed, W. Wang and C. Alexander, J. Am. Chem. Soc., 2008, 130, 10852–10853 CrossRef CAS.
- J. Shepherd, P. Sarker, K. Swindells, I. Douglas, S. MacNeil, L. Swanson and S. Rimmer, J. Am. Chem. Soc., 2010, 132, 1736–1737 CrossRef CAS.
- M. J. Summers, D. J. Phillips and M. I. Gibson, Chem. Commun, 2013 10.1039/c2cc34236g.
- Y. Cai, K. B. Aubrecht and R. B. Grubbs, J. Am. Chem. Soc., 2010, 133, 1058–1065 CrossRef.
- K. Wei, L. Su, G. Chen and M. Jiang, Polymer, 2011, 52, 3647–3654 CrossRef CAS.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS.
- S. Edmondson, V. L. Osborne and W. T. S. Huck, Chem. Soc. Rev., 2004, 33, 14–22 RSC.
- S. Alexander, J. Phys., 1977, 38, 983–987 CAS.
- P. G. de Gennes, Macromolecules, 1980, 13, 1069–1075 CrossRef CAS.
- S. T. Milner, Science, 1991, 251, 905–914 CrossRef CAS.
- A. M. Jonas, Z. Hu, K. Glinel and W. T. S. Huck, Nano Lett., 2008, 8, 3819–3824 CrossRef CAS.
- X. Lalyaux, B. Mathy, B. Nysten and A. M. Jonas, Langmuir, 2010, 26, 838–847 CrossRef.
- E. S. Kooij, X. Sui, H. J. W. Zandvliet and G. J. Vansco, J. Phys. Chem. B, 2012, 116, 9261–9268 CrossRef CAS.
- H. Yim, M. S. Kent, S. Satija, S. Mendez, S. S. Balamurugan, S. Balamurugan and G. P. Lopez, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 72, 051801 CrossRef CAS.
- Y. K. Jhon, R. R. Bhat, C. Jeong, O. J. Rojas, I. Szleifer and J. Genzer, Macromol. Rapid Commun., 2006, 27, 697–701 CrossRef CAS.
- C. Xue, N. Yonet-Tanyeri, N. Brouette, M. Sferrazza, P. V. Braun and D. E. Leckband, Langmuir, 2011, 27, 8810–8818 CrossRef CAS.
- D. Cunliffe, C. de las Heras Alarcón, V. Peters, J. R. Smith and C. Alexander, Langmuir, 2003, 19, 2888–2899 CrossRef CAS.
- J. Deng, Y. Shi, W. Jiang, Y. Peng, L. Lu and Y. Cai, Macromolecules, 2008, 41, 3007–3014 CrossRef CAS.
- C. Wu and S. Zhou, Macromolecules, 1995, 28, 5388–5390 CrossRef CAS.
- X. Wang and C. Wu, Macromolecules, 1999, 32, 4299–4301 CrossRef CAS.
- M. I. Gibson, D. Paripovic and H.-A. Klok, Adv. Mater., 2010, 22, 4721–4725 CrossRef CAS.
- N. S. Ieong, K. Brebis, L. E. Daniel, R. K. O'Reilly and M. I. Gibson, Chem. Commun., 2011, 47, 11627–11629 RSC.
- M.-Q. Zhu, L.-Q. Want, G. J. Exarhos and A. D. Q. Li, J. Am. Chem. Soc., 2004, 126, 2656–2657 CrossRef CAS.
- M. Beija, J.-D. Marty and M. Destarac, Chem. Commun., 2011, 47, 2826–2828 RSC.
- J. Shan, Y. Zhao, N. Granqvist and H. Tenhu, Macromolecules, 2009, 42, 2696–2701 CrossRef CAS.
- R.-S. Lee and K.-P. Wu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3163–3173 CrossRef CAS.
- N. S. Ieong, M. Redhead, C. Bosquillon, C. Alexander, M. Kelland and R. K. O’Reilly, Macromolecules, 2011, 44, 886–893 CrossRef CAS.
- D. Li, G. L. Jones, J. R. Dunlap, F. Hua and B. Zhao, Langmuir, 2006, 22, 3344–3351 CrossRef CAS.
- T. Hu, J. Gao and C. Wu, J. Macromol. Sci. Phys., 2000, B39, 407–414 CrossRef CAS.
- J. Du, H. Willcock, J. P. Patterson, I. Portman and R. K. O'Reilly, Small, 2011, 7, 2070–2080 CrossRef CAS.
- S. Jana, S. P. Rannard and A. I. Cooper, Chem. Commun., 2007, 2962–2964 RSC.
- E. B. Zhulina, O. V. Borisov, V. A. Prymitsyn and T. M. Birshtein, Macromolecules, 1991, 24, 140–149 CrossRef CAS.
- M.-Q. Chen, T. Serizawa, M. Li, C. Wu and M. Akashi, Polym. J., 2003, 35, 901–910 CrossRef CAS.
- D. J. Kim, S. M. Kang, B. Kong, W.-J. Kim, H.-j. Paik, H. Choi and I. S. Choi, Macromol. Chem. Phys., 2005, 206, 1941–1946 CrossRef CAS.
- D. J. Kim, J.-y. Heo, K. S. Kim and I. S. Choi, Macromol. Rapid Commun., 2003, 24, 517–521 CrossRef CAS.
- H. Gao, W. Yang, K. Min, L. Zha, C. Wang and S. Fu, Polymer, 2005, 46, 1087–1093 CrossRef CAS.
- K. Zhang, J. Ma, B. Zhang, S. Zhao, Y. Li, Y. Xu, W. Yu and J. Wang, Mater. Lett., 2007, 61, 949–952 CrossRef CAS.
- M. Yang, Y. Ding, L. Zhang, X. Qian, X. Jiang and B. Liu, J. Biomed. Mater. Res., Part A, 2007, 81, 847–857 CrossRef.
- J. Raula, J. Shan, M. Nuopponen, A. Niskanen, H. Jiang, E. I. Kauppinen and H. Tenhu, Langmuir, 2003, 19, 3499–3504 CrossRef CAS.
- C. Boyer, M. R. Whittaker, M. Luzon and T. P. Davis, Macromolecules, 2009, 42, 6917–6926 CrossRef CAS.
- S. Rimmer, S. Carter, R. Rutkaite, J. W. Haycock and L. Swanson, Soft Matter, 2007, 3, 971–973 RSC.
- W. J. Brittain and S. Minko, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 3505–3512 CrossRef CAS.
- T. Wu, K. Efimenko and J. Genzer, J. Am. Chem. Soc., 2002, 124, 9394–9395 CrossRef CAS.
- I. Luzinov, S. Minko and V. V. Tsukruk, Soft Matter, 2008, 4, 714–725 RSC.
- A. Sundararaman, T. Stephan and R. B. Grubbs, J. Am. Chem. Soc., 2008, 130, 12264–12265 CrossRef CAS.
- A. O. Moughton and R. K. O'Reilly, Chem. Commun., 2010, 46, 1091–1093 RSC.
- F. Wang, A. Klaikherd and S. Thayumanavan, J. Am. Chem. Soc., 2011, 133, 13496–13503 CrossRef CAS.
- M. Liu, P. Tirino, M. Radivojevic, D. J. Phillips, M. I. Gibson, J.-C. Leroux and M. A. Gauthier, Adv. Funct. Mater., 2013 DOI:10.1002/adfm.201202227.
| This journal is © The Royal Society of Chemistry 2013 |