Open Access Article
Open Access Article
DOI:
10.1039/C3CS60032G
(Review Article)
Chem. Soc. Rev., 2013,
42, 5809-5832
Synthesis of functional ‘polyolefins’: state of the art and remaining challenges
Received
28th January 2013
First published on 19th April 2013
Abstract
Functional polyolefins (i.e., polyethene or polypropene bearing functional groups) are highly desired materials, due to their beneficial surface properties. Many different pathways exist for the synthesis of these materials, each with its own advantages and drawbacks. This review focuses on those synthetic pathways that build up a polymer chain from ethene/propene and functionalised polar vinyl monomers. Despite many recent advances in the various fields of olefin polymerisation, it still remains a challenge to synthesise high molecular-weight copolymers with tuneable amounts of functional groups, preferably with consecutive insertions of polar monomers occurring in a stereoselective way. To overcome some of these challenges, polymerisation of alternative functionalised monomers is explored as well.

Nicole M. G. Franssen
| Nicole Franssen (1985) studied chemistry at the Utrecht University, where she did an internship with Prof. Klein Gebbink followed by an internship with Prof. Vollhardt at the University of California, Berkeley (USA). After obtaining her MSc degree in 2008, she moved to the University of Amsterdam (UvA, The Netherlands) where she did her PhD research under the supervision of Prof. Reek and Prof. de Bruin. Her research focused on exploring the scope of homogeneously catalysed carbene polymerisation reactions and she obtained her PhD degree in 2012. |

Joost N. H. Reek
| Joost Reek (1967) did his PhD in Nijmegen (Prof. Nolte). After a postdoctoral fellowship in Sydney, Australia, with Prof. Crossley, he joined the group of Prof. van Leeuwen at the University of Amsterdam (UvA, The Netherlands) as an assistant professor (1998), with research activities focusing on transition metal catalysis. In 2006 he became a full professor. His current research activities include transition metal catalysis, supramolecular catalysis and catalysis for green energy applications. He further started a spin-off company (InCatT) to explore the commercial potential of new supramolecular strategies for combinatorial approaches in (asymmetric) catalysis. |

Bas de Bruin
| Bas de Bruin (1971) did his PhD research at the RUN in Nijmegen (Prof. A. W. Gal, 1999). He did his postdoc at the MPI für Bio-Anorganische Chemie in Mülheim a/d Ruhr (Prof. K. Wieghardt), after which he returned to the RUN as an assistant professor. In 2005 he moved to the University of Amsterdam (UvA, The Netherlands), where he was promoted to full professor in 2013. He is involved in various research activities, including radical organometallic chemistry, EPR spectroscopy, olefin oxygenation, polymer synthesis, mechanistic studies and computational catalysis, with a focus on the development of new (bio-inspired) catalytic transformations. |
Polymers bearing polar functionalised side groups are highly desired materials, due to their unique and rapidly expanding range of material properties. Compared to their non-functionalised analogues they exhibit beneficial properties with respect to adhesion, toughness, print/paintability, miscibility and rheological properties.1 The wide variety of existing polar monomers and their ability to be copolymerised in various ratios with non-functionalised monomers such as ethene and propene allows the synthesis of a nearly infinite amount of different polymer formulations, each with completely different material properties. Nowadays, most functional polymers are synthesised on demand, i.e. the polymer composition and microstructure are tuned in such a way that they match the desired material properties for specialty applications (Fig. 1).2
 |
| | Fig. 1 Examples of application areas of functional polymers. | |
2 Functionalised polyolefins
Due to the outstanding material properties of non-functionalised polyolefins (i.e. polyethene and polypropene), obtaining functional polymers based on polyolefins is of growing interest.3 Introducing small amounts of polar functionalities into either polyethene or polypropene has a tremendous effect on the surface properties of the polymer.2,4 In this way it is possible for polyolefins to enter a completely new area of applications, in which the material properties of polyolefins are combined with new beneficial surface properties imposed by the polar functionalities of the functionalised monomers.
Functionalised polyolefins can be classified into four categories, based on their structures (Fig. 2): randomly functionalised copolymers that exist as either branched (a) or linear structures (b), end-functionalised copolymers (c), block copolymers (d), and graft copolymers (e).3,5 In the following sections, each class of functional polyolefins will be discussed separately. Although many polar comonomers have been used, giving rise to polymers with a wide range of different properties, we focus on copolymers of polyolefins and polar vinyl monomers (i.e. monomers in which the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond is directly substituted by the polar functionality), thereby excluding copolymers based on e.g. polyetheneglycol and poly-tetrahydrofuran (poly-THF). Most of the emphasis will lie on copolymers of acrylates, although some attention is given to co-monomers bearing other functionalities. Amounts of polar monomers incorporated will be listed in mol% if available, unless stated otherwise. These types of copolymers have been widely covered in the patent literature of the last few decades. However, since we want many readers to have access to the examples covered in this review, we mainly focused on articles published in scientific journals. Where appropriate, a reference to the corresponding patent literature is given as suggestion for further reading. Scientific publications that appeared in the literature in the last decade are the main focus of this review, although appropriate older examples are mentioned as well to give a complete overview.
C double bond is directly substituted by the polar functionality), thereby excluding copolymers based on e.g. polyetheneglycol and poly-tetrahydrofuran (poly-THF). Most of the emphasis will lie on copolymers of acrylates, although some attention is given to co-monomers bearing other functionalities. Amounts of polar monomers incorporated will be listed in mol% if available, unless stated otherwise. These types of copolymers have been widely covered in the patent literature of the last few decades. However, since we want many readers to have access to the examples covered in this review, we mainly focused on articles published in scientific journals. Where appropriate, a reference to the corresponding patent literature is given as suggestion for further reading. Scientific publications that appeared in the literature in the last decade are the main focus of this review, although appropriate older examples are mentioned as well to give a complete overview.
3 Randomly functionalised copolymers
Randomly functionalised polyolefins (Fig. 2a and b) offer a way to drastically alter the properties of the corresponding homo-polyolefins by applying a minimum of changes. The beneficial effect of randomly placed polar functionalities stems from breaking-up the crystal packing of the otherwise non-functionalised polyolefins, thereby broadening their application window. The resulting copolymers exhibit completely different properties compared to more ordered structures (Fig. 2c–e) due to the random distribution of monomers along the backbone. Randomly functionalised polyolefins exist in many variations, differing not only in the type of functional groups but also in the amount of functionalisation and the polymer microstructure, i.e. the placement of functionalities along the non-functionalised chain (cf.Fig. 2a vs. b). Minor changes in the polymer structure often lead to major changes in the copolymer properties, making this class of copolymers an excellent starting point for the development of new materials. Functionalised polyolefins exhibiting linear structures as depicted in Fig. 2b are of particular interest since they can be regarded as a functionalised form of HDPE, thus benefiting from the highly developed HDPE chemistry, including properties and applications.5,6
Random copolymers can be synthesised in a variety of ways as depicted in Scheme 1, and each pathway will be discussed separately in the following sections.
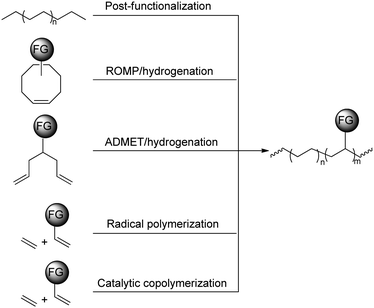 |
| | Scheme 1 Different synthesis pathways for the synthesis of randomly functionalised copolymers. | |
3.1 Polymer post-functionalisation
Polymer post-functionalisation is currently the most-used approach for the commercial synthesis of functionalised polyolefins, since this technique benefits most from the well-developed synthesis of non-functionalised polyolefins (for a review on the most recent advances in catalytic polyolefin synthesis, see Guan7 and Qiao8). Polymer post-functionalisation of existing polymeric structures can be carried out in the bulk as well as on the surface, thereby offering ample opportunities for obtaining desired material properties.2 The amount of functionalisation can be tuned by changes in stoichiometry and in principle any modification that can be carried out on a small molecule is useful for polymer modifications.9 One of the most utilized approaches comprises reaction of free radicals with the C–H bonds of the polymer backbone, leading to H abstraction and the formation of a polymeric radical. These free radicals can be generated in various ways, with peroxide initiation being most common. In most cases maleic anhydride is added to this polymeric radical, giving rise to anhydride-functionalised polymer chains. These anhydride functionalities can in turn be converted to several other groups upon reaction with nucleophiles such as amines. A different post-functionalisation approach deals with insertions of carbenes or nitrenes into the C–H bonds of the polymer backbone (Scheme 2). This method allows for somewhat more selectivity and control over the reaction in contrast to the above-described free radical approach. From an industrial perspective, surface modification techniques based on plasma, corona and flame treatment are relevant as well.6 However, since non-functionalised polyolefins contain no reactive groups, chemical modifications of the inert C–H bonds generally require harsh conditions (for an example, see Scheme 2).6,9 Many undesirable side reactions (e.g. cross-linking or chain scission; for more details see ref. 6 or 9) may occur under these conditions, thereby severely altering the properties of the polymer, also in an undesired manner.10 Aside from this, polymer post-functionalisation is often hampered by solubility problems and cumbersome purification steps. Although catalytic methodologies are being developed, this technique is still far from ideal due to the above-mentioned drawbacks.9
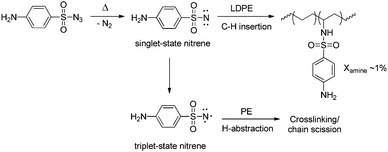 |
| | Scheme 2 Functionalisation of polyethene via post-functionalisation: C−H insertion of a nitrene generated from a tosylazide. The bottom pathway emphasises the undesirable side reactions. | |
3.2 Ring-opening metathesis polymerisation
Ring-opening metathesis polymerisation (ROMP) of functionalised cyclic alkenes and subsequent hydrogenation of the resulting polyalkenamers is a viable alternative for obtaining functionalised polyolefins, although this technique requires specific monomers and occurs in a multi-step fashion. The production of linear, acyclic, functionalised polyolefins by homopolymerisation of functionalised cyclooctenes (Scheme 1) is of particular interest. However, the number of reports describing ROMP of cyclooctenes followed by hydrogenation is rather limited, since they exhibit lower ring-strain and are therefore more reluctant towards polymerisation than e.g. norbornene analogues. The presence of functional groups in cyclooctenes also hinders polymerisation.11 Due to the development of more efficient and polar-tolerant catalysts (mainly based on ruthenium)12,13 the field is rapidly emerging and polymerisations of (functionalised) cyclooctenes are of growing importance.
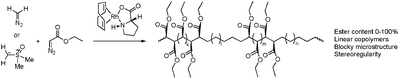
Homopolymerisation of cyclooctenes functionalised in the 5-position gave rise to polymers that resemble copolymers of non-functionalised and functionalised olefins, theoretically containing 25% of the corresponding polar vinyl monomers separated by either 6, 7 or 8 methylene units.14–21 In this way, functional polyolefins bearing acrylate and ketone functionalities have been prepared (Scheme 3). Similar strategies have been applied to the synthesis of copolymers with highly regio- and stereoregular alcohol and amine functionalities (Scheme 4).22–27 Due to their way of synthesis, these copolymers have precise sequence distributions that have a major effect on polymer crystallinity and physical properties.28 More randomly distributed copolymers with lower contents of polar monomers (<10%) are accessible via ROMP copolymerisation of functionalised and non-functionalised cyclic monomers (Scheme 5).11,28 The whole field of ROMP has recently been reviewed and summarised by Buchmeiser29 and Slugovc.30
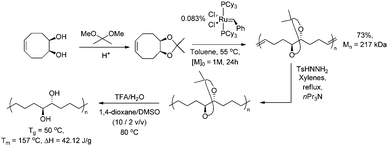 |
| | Scheme 4 Highly stereo- and regioregular analogues of hydroxy-functionalised PE obtained by ROMP of protected dihydroxy-functionalised cyclooctene. | |
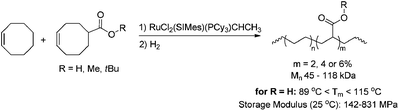 |
| | Scheme 5 Synthesis of analogues of ethene-acrylate copolymers obtained by ROMP copolymerisation of functionalised and non-functionalised cyclooctenes containing theoretically 2, 4 or 6% incorporation of polar vinyl monomers. | |
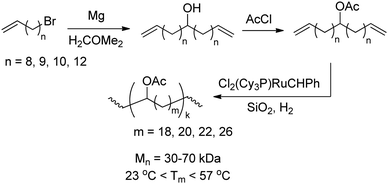 |
| | Scheme 6 Synthesis of acetate-functionalised monomers and subsequent ADMET polymerisation. | |
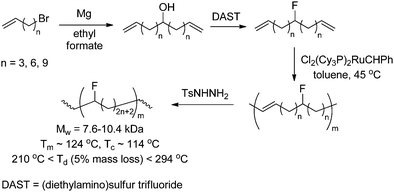 |
| | Scheme 7 Fluorinated polyethene obtained via ADMET polymerisation of fluor-containing monomers. | |
3.4 Direct copolymerisation of polar and non-polar vinyl monomers
Despite some advantages of the above-mentioned techniques for obtaining functionalised polyolefins, direct copolymerisation of polar vinyl monomers and non-polar olefins remains of particular interest.46 Obviously, direct copolymerisation circumvents the need for functionalisation via often cumbersome polymer post-functionalisation steps. By tuning the insertion efficiency of the functionalised monomer vs. the non-polar olefin, direct copolymerisation allows quantitative control over the amount of functionalisation. This difference in insertion efficiency, in combination with changes in the feed ratio of both monomers, also allows variations in copolymer architecture (i.e., changing the arrangement of the monomers along the chain).1 These features offer a broad window for tuning the copolymer properties, making direct copolymerisation a powerful method for the development of a wide variety of new materials. However, despite being the most logical approach, direct copolymerisation of polar and non-polar olefins is not that easy to achieve in a controlled way as is illustrated by the examples below. The two main copolymerisation pathways to obtain such copolymers are based on radical polymerisation or involve chain growth via catalytic processes, and each method will be discussed separately in the following sections.
3.4.1 Radical pathways.
Radical polymerisation of olefins and polar vinyl monomers (e.g. acrylates, vinyl acetate and acrylic acid) is widely applied on a commercial scale, and the resulting copolymers find many applications in daily life.47 The incorporated acetate or acid functionalities can be converted into their respective salts, giving rise to the formation of cross-linked ionomers, which find applications in areas that require extreme toughness (e.g. golf ball coatings) or enhanced sealing properties (e.g. bacon packaging).48
However, radical polymerisation of non-polar olefins and polar vinyl monomers is not easily achieved, since non-polar olefins are poorly reactive monomers in radical polymerisation. Due to the lack of stabilizing conjugation in non-polar olefins the stability of the generated radicals is too low to allow smooth polymerisation, and to be successful quite harsh polymerisation conditions (high temperatures and pressures) are required, resulting in the formation of highly branched polyolefins.47,49 Since the reactivity of polar vinyl monomers towards radical polymerisation is much higher, the resulting copolymers contain large amounts of polar functionalities.5 Reports dealing with the synthesis of such copolymers under milder and more controlled conditions are rather scarce.
Earlier attempts deal with the use of boron-protected polar vinyl monomers, leading to the formation of highly electron-deficient monomers. In this way, alternating copolymers of ethene/propene and ethyl acrylate could be obtained by free-radical polymerisations under relatively mild conditions (25–50 °C, 6–20 atm. ethene), with considerable amounts of tacticity (Scheme 9).50–52 At low ethene/propene pressures the acrylate content in the copolymers is higher than 50%.
Copolymerisations using non-protected polar vinyl monomers have been developed as well, based on atom-transfer radical polymerisation (ATRP) catalysed by copper complexes (Scheme 10).49,53 Copolymers of both ethene and propene with methyl acrylate were obtained containing up to 14 and 21% of non-functionalised olefin respectively (PDI < 1.5). Similar results were obtained in copolymerisations initiated by 2,2-azobis(isobutyronitrile) (AIBN), although those copolymers exhibited much broader molecular-weight distributions (PDI = 9 for ethene).54 Consecutive insertions of non-polar olefins were not observed, due to the reactivity difference between both monomers. The incorporation of olefin could be slightly tuned by varying the feed ratio of both monomers. A drawback of this metal-catalysed process is that the copolymers often contain traces of metal impurities and are therefore colored.54 Metal-free routes towards these copolymers have been developed based on nitroxide-mediated radical polymerisation (NMRP).55 This allowed the synthesis of copolymers of methyl acrylate and ethene/propene containing ∼15% of non-functionalised olefin.
Reversible addition-fragmentation chain-transfer polymerisation (RAFT) or degenerative-transfer (DT) polymerisation of ethene and methylacrylate yields fully alternating copolymers, with a relatively narrow molecular-weight distribution (Scheme 11).56
3.4.2 Catalytic routes.
Transition-metal (TM) catalysed routes towards random copolymers with a linear structure are highly desired, and potentially allow the synthesis of functionalised copolymers with control over the amount of polar functionalities and their distribution along the polymer backbone. In that respect, TM catalysed processes are advantageous over radical polymerisation reactions.57 However, despite many years of research and recent advances in the field of coordination/insertion polymerisation (i.e. migratory insertion of an olefin into a metal–alkyl bond) (Scheme 12), direct copolymerisation of olefins with polar vinyl monomers still remains a challenge. In fact, achieving control over polymer composition and architecture, and development of catalysts that allow incorporation of a variety of functionalities during olefin polymerisation are two of the main goals set by the Council for Chemical Research (CCR) to be achieved in 2020, driven by the future technology needs of the chemical industry in the area of catalysis.54 Early transition-metal (ETM) catalysts are most active in the homopolymerisation of ethene and propene, but they are easily poisoned by the functional groups in the co-monomers due to their high oxophilicity, and thus electronic or steric protecting strategies are required to prevent coordination to the metal center.1,5 Recently, direct copolymerisation has been successfully achieved by applying late transition-metal (LTM) catalysts (mainly based on palladium), since they are more tolerant to the presence of functional groups and allow their incorporation without the need for protection. However, due to the presence of the electron-withdrawing or -donating functionalities directly attached to the double bond, the electronic environment around the reactive C–C bonds is drastically changed compared to the non-functionalised analogues. This has a major effect on the relative rates of incorporation of both monomers, often leading to the formation of copolymers containing only a few per cent of polar monomers.5 In principle, the effect of the polar functionality on the reactivity can be minimised by implementing a methylene spacer between the double bond and the functionality, but these monomers are less abundant and therefore more expensive.1 Another problem that can hamper TM catalysed copolymerisation is β-heteroatom elimination of the functional group, which has been observed for vinyl acetate, vinyl halides and vinyl ethers.1,5 Aside from this, after insertion of the polar monomer, the functional groups can coordinate to the metal centre leading to the formation of stable chelates, breaking-up of which significantly slows down further insertions. Nevertheless, despite these possible problems, successful examples of direct copolymerisation have been reported and the number of publications has rapidly grown in the past few years. The field of transition-metal catalysed copolymerisations has been the subject of several reviews.1,5,6,48,57–59 The following sections will give an overview of the current state-of-the-art with respect to controlled synthesis of functional copolymers via direct copolymerisation of non-functionalised olefins and polar vinyl monomers.
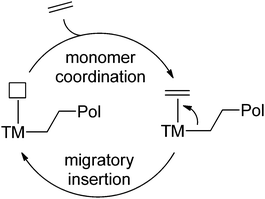 |
| | Scheme 12 Schematic representation of TM-catalysed coordination/insertion polymerisation. | |
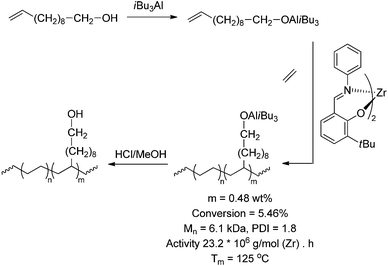 |
| | Scheme 14 Synthesis of polyethene containing alcohol functionalities via direct copolymerisation with aluminium-protected OH-functionalised monomers. | |
Amine functionalities have also been protected with trimethylsilyl (TMS) groups (Scheme 15),79,80 since monomers containing silyl functionalities can easily be copolymerised with ethene or propene using ETM catalysts (Scheme 16).81–86 Different protection strategies have also been applied for copolymerisation with acrylates, since acrylate monomers are the most difficult to be copolymerised with non-polar olefins due to the formation of inactive O-enolate complexes or π-allyl-η3-complexes after insertion of the acrylate monomer into the M–C bond.1,5 Formation of these inactive species can be circumvented by the use of acrylate salts as monomers, formed by complexation of the acrylate anions to metallocene cations (Scheme 17).87 In this way, the synthesis of aminofunctional linear low density polyethene (LLDPE) could be realised.
 |
| | Scheme 16 Silyl-functionalised polyethene synthesised via direct copolymerisation catalysed by ETM complexes. | |
Another approach that has been applied in combination with ETM catalysts is functionalisation via borane monomers. Borane functionalities do not hamper ETM catalysed polymerisation, and since they are easily modified they form an excellent starting point for the introduction of various functionalities. Chung et al. reported formation of random copolymers of ethene and propene with vinyl monomers containing 9-borabicyclononane (9-BBN) functionalities.88–91 These pendant borane functionalities could be converted to a variety of functional groups (Scheme 18), including alcohols, amines, halogens and radical species that can serve as initiators for the synthesis of graft copolymers (vide infra, Section 5.2).
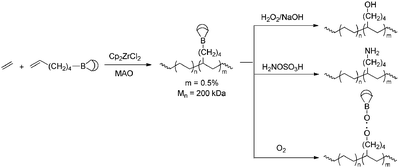 |
| | Scheme 18 Functionalisation of polyethene via ETM-catalysed direct copolymerisation with borane-containing monomers, followed by subsequent functionalisation steps. | |
However, all the above-described examples deal with polar monomers containing several methylene groups as spacers between the CC double bond and the polar group. Very few examples of direct copolymerisation of polar vinyl monomers without this additional spacer have been reported for ETM catalysis, thereby limiting this approach to a selected class of monomers. Aside from this, the incorporation of polar monomers is rather low (in general less than 5%) due to diminished reactivity of the polar monomers compared to the non-functionalised analogues (similar to the lower reactivities of α-olefins), and the distribution along the polymer backbone is difficult to tune.
3.4.2.2 Late transition-metal catalysts.
Late transition-metal (LTM) catalysts are able to incorporate polar vinyl monomers that bear the polar functionality at the vinylic position, directly attached to the CC double bond, via direct copolymerisation with non-polar olefins, without the need for protection. The most successful examples have been reported for nickel and palladium catalysts.5,92
Palladium catalysts bearing α-diimine ligands are highly active in ethene homopolymerisation and the scope of this reaction has been extended to copolymerisations of ethene/propene with acrylates,93–95 acrylic acid,48 methyl vinyl ketone,93 and monomers bearing acetyl functionalities (Scheme 19).96 These systems yield highly branched copolymers (typically 100 branches per 1000 carbon atoms) due to rapid β-hydride elimination followed by reinsertion (i.e. chain walking), and the polar monomers are mainly located at the end of the branches. Incorporation of polar monomers up to 12% was achieved by varying the monomer feed ratio, although the polymerisation efficiency decreased significantly upon incorporation of the polar monomers. The incorporation of polar monomers could be increased to more than 20% by slightly modifying the catalyst, i.e. by blocking the axial positions.95 For none of these systems consecutive insertions of polar monomers were observed, and the system proved to be inactive towards homopolymerisation of polar vinyl monomers. Copolymerisation of olefins with methyl methacrylate could not be achieved with these Pd-diimine catalysts and this feature was ascribed to the very low insertion rate of methacrylates.97,98 Acrylonitrile and other nitrogen-containing monomers also hampered copolymerisation, most likely via N-coordination of the monomers to the catalyst.48 The catalyst system is more tolerant to functional groups if additional methylene spacers are present between the CC double bond and the functionality similar to the ETM systems discussed above.99 Although this approach is viable, it is not a unique feature of these Pd-diimine catalysts and therefore of minor importance.
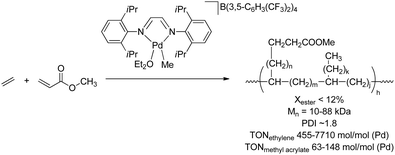 |
| | Scheme 19 Synthesis of highly branched ethene-acrylate copolymers via coordination/insertion polymerisation catalysed by a Pd-diimine complex. | |
The corresponding nickel-diimine catalysts showed far less activity in the copolymerisation of olefins and polar vinyl monomers, due to strong coordination of the oxygen functionalities in the polar monomers to the metal centre, thereby poisoning the catalyst.100 This problem can be overcome by applying quite harsh polymerisation conditions (high temperatures (120 °C) and pressures up to 340 bar), and in this way copolymers of ethene and methyl acrylate were obtained with around 1% incorporation of the polar monomers (Scheme 20).101,102 In contrast to the above-mentioned Pd systems, these Ni catalysts give rise to copolymers with a wider range of microstructures, from moderately linear (i.e. 30 branches per 1000 carbon atoms) to highly branched, and the incorporation of acrylates takes place mainly in-chain. These Ni systems and their behaviour in copolymerisation reactions with polar monomers have mainly been discussed in the patent literature (for examples, see e.g. Nozaki5 and Ittel48).
To overcome the problem of O-coordination to the oxophilic Ni center, several anionic ligands have been used to achieve copolymerisation of ethene and methyl acrylate, mainly based on [P–O], [P–N] or [N–O] coordination to the metal centre.
Application of these catalysts in combination with Lewis acids as cocatalysts resulted in the formation of moderately linear low-Mw copolymers (Mn ∼ 1500 Da) with rather low amounts of polar functionalities (<1%), although the methyl acrylate units are incorporated in the polymer backbone rather than at the branches (Scheme 21).103–105 The main advantage of these systems is that they allow copolymerisation of olefins with methyl methacrylate, and copolymers with a MMA content up to 17% could be obtained (Scheme 22).106–109 Papers dealing with MMA contents of 80% have been reported as well, but these high acrylate contents point in the direction of radical polymerisation rather than sole coordination/insertion polymerisation.110
Along these lines, palladium complexes bearing phosphine–sulfonate [P–O] ligands have been applied in copolymerisation reactions of ethene and several acrylates, giving rise to linear copolymers with acrylate incorporation up to 17%.57,111–114 The degree of branching in the copolymers is very low (1 branch per 1000 carbon atoms), giving rise to truly linear copolymers with the polar monomers positioned along the polymer backbone. No consecutive acrylate insertions were observed for these systems Recently it was found that by slightly modifying the catalysts, the acrylate incorporation can be increased up to 52% giving rise to consecutive acrylate insertions (albeit up to 3 or 4) (Scheme 23).115 Unfortunately, the Mw of the corresponding copolymers decreased drastically (to as low as 2 kDa) upon incorporation of acrylate. This approach has been extended to the copolymerisation of ethene and a variety of co-monomers, including acrylonitrile,116 vinyl ether,117 vinyl acetate,118 acryl amides (Scheme 24),119 acrylic acid,120,121 vinyl sulfones122 and allylic monomers containing functional groups.123,124
The above-described examples emphasise the potential of this approach and the number of publications has rapidly grown in the past few years. The tolerance of these catalysts towards a large variety of functional groups makes them excellent candidates for the synthesis of copolymers of olefins and polar vinyl monomers. Up to now, Pd[P–O] systems have only been applied for copolymerisations with ethene. Another drawback is the limited occurrence of consecutive polar monomer insertions, which limits the potential of this approach for block copolymer synthesis. However, since the research in this field is still ongoing, future improvements of the catalysts and polymerisation conditions are expected to broaden the scope of these catalyst systems further.
4 Chain-end functionalised copolymers
Chain-end functionalisation is a key step in polyolefin synthesis, since this is the starting point for constructing more complex macromolecular architectures such as block copolymers and graft copolymers from the otherwise unreactive polyolefin chains (vide infra). Polyolefins bearing a terminal functional group at either one end or both ends (i.e. telechelic polymers) are prepared by three general methods: controlled end-capping of living TM-catalysed polymerisation (a), in situ chain transfer reactions during TM catalysed coordination polymerisation (b), or modification of preformed unsaturated chain ends (c).3,6,125,126 These approaches are discussed in the following paragraphs, with emphasis on those examples that have been exploited for the synthesis of more complex macrostructures.
4.1 End-capping of living polymerisations
Although end-capping of living polymerisation is the classical approach for introducing terminal functionalities, this approach is less suitable for end-functionalisation of polyolefins with polar groups.3 Aside from being metal-consuming and thus of limited practical value, this approach requires the use of very specific catalysts under certain polymerisation conditions and, in fact, not many examples have been reported.127 Living coordination polymerisation of polypropene was achieved using a vanadium catalyst at a very low temperature (−65 °C) and syndiotactic polypropene end-capped with iodine was obtained by addition of I2 at the end of the polymerisation.128–131 Aldehyde-functionalised polypropene was prepared in a similar process by reaction with carbon monoxide (Scheme 25).132,133
 |
| | Scheme 25 Synthesis of aldehyde-functionalised polypropene by end-capping of living vanadium-catalysed polymerisation by carbon monoxide. | |
End-capped polyethene was obtained by using a cobalt catalyst bearing para-substituted phenyl groups.134 These phenyl groups are able to initiate living coordination polymerisation of ethene, thereby introducing the terminal functionality in the initiation step. More recently, this approach has been extended to the synthesis of acrylate-functionalised polyethenes via living coordination polymerisation at selected palladium-diimine catalysts.135,136 Pd complexes chelated by one acrylate unit were able to initiate living polymerisation of ethene, thus allowing end-functionalisation of polyethene in the initiation step. This approach has also been used for the synthesis of telechelic polyethenes, in which the second end functionality was introduced by insertion of an acrylate unit after the polymerisation reaction (Scheme 26).136
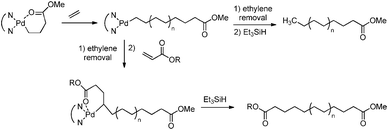 |
| | Scheme 26 Synthesis of polyethene functionalised with an acrylate unit at either one or both chain ends via end-capping of living polymerisation catalysed by palladium. | |
Introduction of functional groups during the initiation step is not limited to the use of LTM catalysts. ETM catalysts bearing phenoxyimine (FI) ligands were used to obtain both polyethene and polypropene end-capped with aluminium or silyl functionalities, which can be converted to hydroxyl end groups (Scheme 27).137 Similar to the LTM systems above, this method can be extended to the synthesis of telechelic polymers by insertion of functionalised monomers after polymerisation of the non-functionalised monomers.
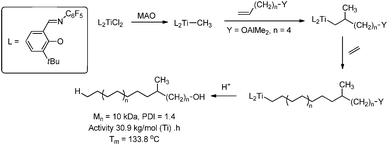 |
| | Scheme 27 Introduction of functional groups during the initiation of polyethene growth using a titanium catalyst bearing phenoxyimine (FI) ligands. | |
4.2 Chain-transfer reactions
End-functionalisation via chain transfer is the most efficient approach for obtaining polyolefins bearing a terminal functionality. The process is very metal-efficient, since each catalytically active species produces multiple polymer chains, all bearing the desired end functionality. The overall polymerisation efficiency is therefore retained, while the molecular weight of the resulting polymers can be tuned by varying the chain-transfer agent (CTA) to monomer ratio ([CTA]/[monomer]). Different varieties of chain-transfer reactions have been applied for this purpose and a general overview is given in Scheme 28. A well-known chain transfer reaction in polyolefin synthesis (often the cause of formation of low-Mw materials) is β-hydride elimination, yielding polyolefin chains with vinylic end groups that can be further modified through chemical transformation reactions. Chain transfer to a (main-group) metal, either catalytic or irreversible, is a different approach that is of growing importance due to the well-understood behaviour of ETM/metal alkyl catalyst systems in polyolefin synthesis. By taking advantage of the reactivity of the newly formed metal–carbon bonds different functional groups can be introduced and examples of this approach will be provided in the following paragraphs. Another chain-transfer approach is direct introduction of functionalities using suitable (functionalised) chain-transfer agents (CTA) that terminate the growing polymer chain with a functional group while the catalyst is simultaneously re-activated for the formation of a new polyolefin chain.3,6 The field of chain-transfer reactions in polyolefin synthesis has recently been reviewed by Marks.138
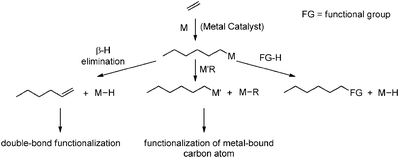 |
| | Scheme 28 General overview of chain-transfer reactions that can be used for end-functionalisation of polyolefins. | |
Chain transfer of the growing polymer chain to a second (main-group) metal present in the polymerisation mixture has already been known for a long time139 and is mainly observed for ETM catalysts that operate in cooperation with zinc and main-group metal alkyl compounds. Under certain conditions this chain transfer occurs in a catalytic fashion, giving rise to a process called chain-shuttling or catalytic chain-transfer polymerisation (CCTP).140 This approach is very suitable for obtaining polyolefin chains that are fully end-capped with a (main-group) metal, although the non-catalytic version of this chain-transfer process has also been successfully used for end-functionalisation of polyolefins. Chain transfer from ETM catalysts to zinc yielding Zn-alkyl terminated polypropene was first utilised to synthesise polypropenes with a variety of functional groups (hydroxyl,141,142 carbonyl,143 halogen,143 vinyl144,145 or amine groups146), making use of the high reactivity of Zn–C bonds towards many reagents (Scheme 29). Catalytic chain transfer from ETM catalysts to zinc has also been applied and led to the synthesis of polyolefins bearing alkyne or azide functionalities.147 For a more general overview of the potential of catalytic chain transfer to zinc in polymerisation, see e.g. Gibson,148 Arriola149 and Sita150 and references therein. Transfer to zinc has also been used in combination with LTM catalysts, yielding hydroxyl-terminated polyethene.151
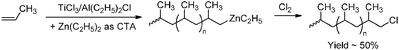 |
| | Scheme 29 Polypropene end-functionalised with halogen functionalities via intermediate chain transfer to zinc. | |
Similar results are obtained when ETM catalysed polymerisations are conducted in the presence of aluminium alkyls, leading to polyolefins bearing hydroxyl groups (Scheme 30).152–158
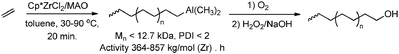 |
| | Scheme 30 Synthesis of hydroxy-functionalised polyethene via ETM catalysis in combination with chain transfer to aluminium. | |
Transfer to magnesium followed by subsequent functionalisation has been exploited for lanthanide catalysts, giving rise to polymers with various functionalities,159 such as152 alkoxyamine-functionalised polymers,160,161 polymers bearing thiocarbonyl functionalities162 and azide-functionalised polyethene (Scheme 31).163
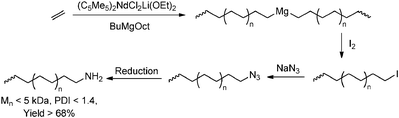 |
| | Scheme 31 End-functionalisation of polyethene via lanthanide catalysis in combination with catalytic chain transfer to magnesium alkyls. | |
In the presence of various silanes, chain transfer of the growing polymer chain to the silyl functionality can occur from both ETM and lanthanide catalysts (Scheme 32).164–167 This process is versatile, and both polyethene and polypropene bearing a wide range of silyl functionalities have been prepared. Although this should in principle be a good starting point for the synthesis of polyolefins with a variety of other functionalities,168 additional modifications of the silyl functionalities were not reported.
 |
| | Scheme 32 Chain transfer to silanes as effective method for the synthesis of silyl end-capped polypropene. | |
Chain-transfer to organoboranes leads to the formation of polymers with a terminal borane functionality and a corresponding metal-hydride species, which is still active towards polymerisation. Chung and Xu reported formation of polyethene terminated with a 9-BBN moiety, obtained via chain-transfer from ETM catalysts to the 9-BBN dimer.169,170 The borane functionalities can be oxidised to yield polyolefins with a terminal hydroxyl group,169 but more interesting is their ability to be used as initiators in free radical polymerisation via controlled oxidation (Scheme 33). This allows the formation of block copolymers via coordination/insertion polymerisation in combination with subsequent free radical polymerisation (vide infra).
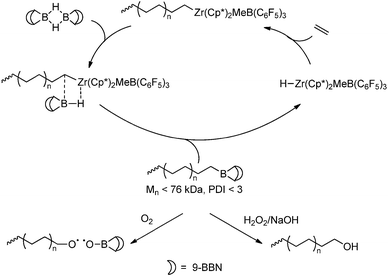 |
| | Scheme 33 Functionalisation of polyethene with a terminal borane group and subsequent transformations into a hydroxyl functionality and a suitable initiator for radical polymerisation. | |
Another chain-transfer reaction that is of interest is the transfer to styrene derivatives (in particular p-methylstyrene) that in turn can function as initiators for the growth of polar polymer segments (vide infra, Section 4.3). These styrene functionalities can be introduced by first inserting the styrene-derivative via a coordination/insertion mechanism, followed by chain transfer to hydrogen, yielding the styrene-terminated polymer chain and a metal-hydride catalyst (Scheme 34).171–173 These systems benefit from the low activity of the ETM catalysts towards the incorporation of styrenes and, in fact, the system becomes inactive after insertion of the styrene-derivative. Chain-transfer to hydrogen is necessary to regenerate catalyst and allow the formation of additional polymer chains. This approach works for both polyethene and polypropene and several functionalised styrene-derivatives have been used, including styrenes bearing chloride and amine groups.172
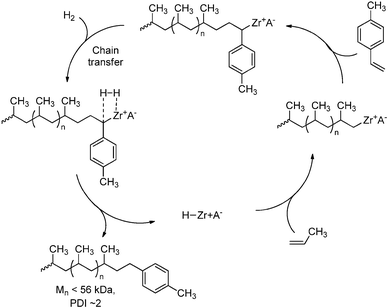 |
| | Scheme 34 Chain transfer of a growing polypropene chain to p-methylstyrene. | |
4.3 Functionalisation of unsaturated chain ends
Functionalisation of polyolefins by chemical modification of preformed unsaturated chain-ends has been applied for the introduction of several functionalities, mainly via hydroboration, hydrosilylation and hydroalumination reactions.3,126 However, this approach has several disadvantages, which limits its potential for the synthesis of end-functionalised polyolefins. The chain-end unsaturation of the preformed polyolefins needs to be nearly quantitative to ensure complete functionalisation, although several ETM systems exist to date that mainly terminate via β-hydride elimination (e.g. Et(Ind)2ZrCl2 that has been applied by Chung174). The subsequent chemical transformations are hampered by the very low concentration of the double bonds in the polymer mixture and, even more importantly, by the poor solubility of these polymers in the reaction medium/solvent associated with the use of these reagents.6,125
Hydroalumination and subsequent oxidation have been used to functionalise polypropene with a hydroxyl group.157,175 Later, this approach was used for hydroxy-functionalisation of ethene–propene copolymers (EPR), followed by further conversion of the hydroxyl group to a methacryloyl functionality suitable for copolymerisation reactions (Scheme 35).175 Introduction of halogen functionalities at the chain end was also achieved via this method.176
Hydrosilylation reactions have been applied for converting the unsaturated chain ends of polypropene into a suitable initiator for radical polymerisation (Scheme 36),177 in order to synthesise block copolymers (vide infra).
Hydroboration is by far the most successful transformation reaction, which has been attributed to the advantageous reactivity and solubility of dialkylborane reagents in hydrocarbon media.6 Polypropene or polyethene-co-hexadiene bearing an unsaturated chain end was transformed into borane-functionalised polypropene by a hydroboration reaction with alkylboranes.174,178 Subsequent transformations of these newly formed polypropenes led to the formation of polyolefins end-functionalised with hydroxyl,174,178 amine,174 silyl174 or maleic anhydride functionalities179,180 (see Scheme 37).
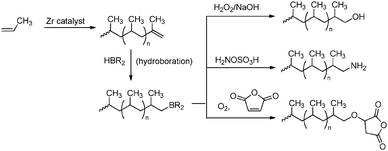 |
| | Scheme 37 End-functionalisation of polypropene via hydroboration reactions converting the unsaturated chain ends. | |
 |
| | Fig. 3 TEM pictures of polypropene-polymethylmethacrylate block-copolymers (PP-b-PMMA) showing phase separation at the microscopic level, with different morphologies for a PMMA content of 60% (a) and 75% (b).181 (Reproduced with permission; Copyright 2003 Wiley) | |
 |
| | Fig. 4 Mechanistic transformations in living/controlled polymerisation aimed at the synthesis of block copolymers. Solid lines indicate pathways suitable for the synthesis of functional polyolefins, while pathways indicated with dashed lines are not. | |
 |
| | Scheme 38 Schematic representation of copolymer synthesis via mechanistic transformation reactions. | |
The two main transformation pathways for the synthesis of functional block copolymers based on vinyl monomers are cross-over reactions between catalytic coordination/insertion polymerisations and either radical polymerisations or conjugate addition mechanisms.6,49,58,125 Cross-over to radical mechanisms is mainly described for block copolymers of ethene/propene and acrylates. The non-polar block is formed by catalytic coordination polymerisation at either early151,169,170,177,181,191–196or late TM catalysts,151,197–199 or lanthanide complexes,161,162 followed by subsequent transformation of the chain end into a radical initiator suitable for acrylate polymerisation (Scheme 39 (ref. 200) and Scheme 40). Several possibilities for transforming the chain end into a suitable initiator have been discussed above (Section 4.2). A drawback of this approach is the rather limited control over the polymerisation in terms of stereoselectivity due to the use of radical polymerisation.
Very recently, Monteil and coworkers developed a pathway for the synthesis of multiblock copolymers from ethene and acrylates, based on a reversible shuttling process, going back and forth from catalytic insertion polymerisation to radical polymerisation (Scheme 41), occurring at the same metal (Ni) center.201,202 This so-called dual radical/catalytic polymerisation allows the synthesis of multiblock copolymers of various compositions that can be tuned by varying the feed ratio of both monomers and the absolute ethene pressure. As stated above, the stereoselectivity of the consecutive polar monomer insertions is moderate as a result of the underlying radical mechanism.
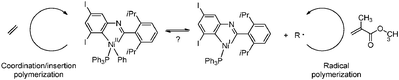 |
| | Scheme 41 Reversible shuttling process between a NiII species responsible for coordination/insertion polymerisation of ethene and a NiI species active in controlled radical polymerisation of acrylates. Multiple shuttling cycles result in the formation of ethene-acrylate multiblock copolymers. | |
Transformation of coordination/insertion polymerisation to addition polymerisation of acrylates (i.e. metal-catalysed anionic polymerisation) is limited to the use of lanthanide (metallocene)203–209 or early-TM metallocene210,211 catalysts for the synthesis of the non-polar block (for an example, see Scheme 42). Polymerisations of the polar monomers, to some extent, take place stereoselectively, allowing the synthesis of isotactic and syndiotactic (rich) blocks.210,212 However, this approach is only unidirectional: due to mechanistic incompatibilities block copolymers can only be obtained starting with the non-polar block. Copolymerisation attempts starting with acrylate polymerisation only lead to the formation of homopolymers.58 This also implies that this cross-over approach is not suitable for the synthesis of statistical, random copolymers, which is a major drawback.
A different approach to the synthesis of block copolymers is linking two preformed polymer segments together via a coupling reaction. Despite the fact that coupling reactions of any kind are widely available and can nowadays be applied very efficiently for coupling of polymers (for a review see e.g. Schubert et al.213), this approach is not often used for linking non-polar polyolefin segments and segments functionalised with polar groups. This is due to the very limited abundance of polyolefin segments terminated with a functionality suitable for coupling reactions, since the field of end-functionalisation of polyolefins has only recently started to bloom (see Section 4.2). Nevertheless, this approach will likely gain importance in the near future and the first successful example of coupling polyethene segments to polyacrylates via (hetero) Diels–Alder chemistry has been reported (Scheme 43).214
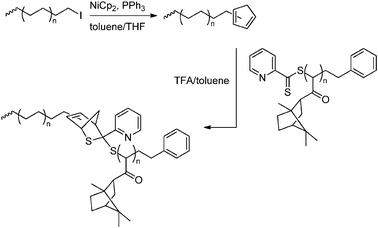 |
| | Scheme 43 Formation of a block copolymer by coupling a preformed non-polar and a preformed polar segment via a hetero Diels–Alder reaction. | |
The above-mentioned examples clearly emphasise the need for the development of alternative synthesis pathways for polar/non-polar block copolymers that allow more control over the synthesis and preferably do not require multiple consecutive reaction steps. Despite recent progress there is clearly room for optimisation in block copolymer synthesis.
For similar reasons as discussed above for the synthesis of block copolymers, graft copolymers are generally prepared via transformation reactions involving two mechanistically distinct polymerisation mechanisms. Whereas this is rather straight-forward in the case of block copolymers (i.e. involving modification of one chain end), the synthesis of graft copolymers is more challenging since it requires modifications on multiple sites along the polymer backbone. Nevertheless, graft copolymers based on polyolefins have been described in the literature and the section below gives an overview of their synthesis pathways.125
Three general methods exist for the synthesis of graft copolymers: grafting onto (a), grafting from (b) and grafting through (c) approaches (Fig. 5) (for a more general review on graft copolymers see e.g. Hadjichristidis215 and Huang216).
5.2.1 Grafting onto.
The “grafting onto” approach requires the synthesis of a polymer backbone with randomly placed functionalities, as well as the formation of end-functionalised polymers bearing functional groups with a complementary reactivity to those of the main chain. Coupling of the two functionalities in a separate step leads to the synthesis of graft copolymers in which the end-functionalised polymers become branches of the main chain. In principle, this technique is very suitable for obtaining graft copolymers based on polyolefins, since it allows the synthesis of the polymer backbone and the branches in separate steps (and thus by different polymerisation mechanisms). Although this approach has been used to couple polyolefins to several other polymer segments (e.g. Li et al.217), to our knowledge, no examples of this approach exist in the literature covering the synthesis of polyolefin-graft-poly(polar vinyl monomer) copolymers.216
5.2.2 Grafting from.
The second approach, “grafting from”, involves the synthesis of a polymer backbone bearing randomly placed functionalities, which can serve as an initiator for the polymerisation of a second monomer via a different mechanism. Similar to block copolymer synthesis, the most successful transformations are those based on catalytic coordination/insertion polymerisation of the polyolefin backbone, combined with radical polymerisation to form the functionalised branches.3,125 The polymer backbone is synthesised via ETM-catalysed insertion polymerisation and the functionalities are incorporated via direct copolymerisation of selected functional monomers with either ethene or propene (or both). The nature of the co-monomer depends highly on the desired follow-up polymerisation reaction to form the polymeric branches. Common functionalities are p-methylaryl (for an example, see Chung et al.218) 9-borabicyclononane (9-BBN, see Scheme 44),89,91 alkoxyamine,219 hydroxyl (Scheme 45)220,221 and vinylic groups,174,222 some of which require additional transformations in order to serve as initiator in subsequent radical polymerisation of the polar monomers. Although some of these examples are formally outside the scope of this review, they contribute to a better understanding of the possible ways to functionalise polyolefins and are therefore included in this section. Various forms of radical polymerisation have been applied, including peroxyborane-initiated radical polymerisation (Scheme 44),90,223 nitroxide-mediated radical polymerisation (NMRP)219 and atom-transfer radical polymerisation (ATRP) (Scheme 45),220–222 leading to the formation of polyolefin-graft-polyacrylate copolymers.
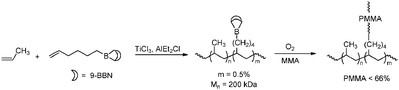 |
| | Scheme 44 Synthesis of PP-g-PMMA copolymers via ETM catalysed coordination/insertion polymerisation followed by peroxyborane-initiated radical polymerisation. The borane initiation sites were incorporated in the polymer backbone via direct copolymerisation of borane-functionalised monomers with propene. | |
The polar segments can also be introduced by cross-over to anionic polymerisation from e.g. p-methylstyrene functionalities,218 although examples in this field are scarce due to the special conditions required for incorporation of polar monomers.224 Although the above-mentioned examples indicate that the “grafting from” approach has proven to be successful for the synthesis of graft copolymers, a major drawback of this method is that it requires multiple reaction steps.
5.2.3 Grafting through.
In the “grafting through” or macro-monomer approach a polymer–oligomer chain with a polymerisable end group is formed, that can be copolymerised with other monomers to yield graft copolymers. The polyolefin macro-monomer is synthesised via coordination polymerisation using either ETM (Scheme 46)151,175 or LTM135 catalysts, and the end group is transformed into a methacryloyl functionality that can be copolymerised with other acrylate monomers via ATRP. The polyolefin segment can also be prepared via catalytic chain shuttling polymerisation using lanthanide/magnesium catalysis.163 After introduction of a suitable functionality via “click” chemistry, these macro-monomers were copolymerised with other acrylates via radical polymerisation (Scheme 47).
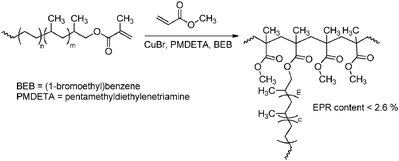 |
| | Scheme 46 PMMA-g-PE/PP copolymers obtained by copolymerisation of MMA with PE/PP macro-monomers via the “grafting through” approach. For the synthesis of the macro-monomer, see Scheme 35. | |
The “grafting through” approach yields polyacrylate-graft-polyolefin copolymers, thus being complementary to the graft copolymers obtained via the “grafting from” method. The length of the grafts and the grafting density can easily be tuned by changing the degree of polymerisation of the side chains and the polymer backbone, respectively, allowing the synthesis of graft copolymers in a controlled way.216 However, this approach has several drawbacks. Due to the incompatibility of the monomers and the high viscosity of the polymerisation medium the conversions of the ATRP reaction are generally low, leading to the presence of unreacted macro-monomers in the copolymer mixture that need to be removed in additional steps. Similar to the systems discussed above, the “grafting through” process occurs in a multi-step fashion, although the sensitivity of the catalytic systems to the functionalities necessary for ATRP (i.e. halogens) often requires several additional transformations, making this approach somewhat more cumbersome.125
6 Alternative polar monomers
The above-described examples illustrate that the most successful routes towards functionalised polyolefins involve, at some point, polymerisation via a coordination–insertion mechanism, catalysed by transition metals. Transition-metal catalysis offers the advantages of more controlled polymerisations, e.g. in terms of polymer microstructure and stereoregularity. However, attempts to obtain functionalised polyolefins solely by transition-metal catalysed coordination–insertion processes have met only limited success (vide supra). This has to do with the large differences of non-polar olefins and polar vinyl monomers in their reactivity towards different metals. Therefore, the development of alternative pathways to introduce functionalities directly into the growing polymer chain via a transition metal catalysed process, based on monomers other than polar vinyl compounds is of growing importance. One such approach is the use of so-called C1 monomers that build up a polymer chain with one backbone-carbon unit at a time, as an alternative to traditional olefinic monomers or C2 monomers that extend the growing polymer chain by two carbon units at each insertion step.
The most widely used C1 monomer is carbon monoxide (CO) and ample examples exist in the literature describing co- and ter-polymerisations of CO and olefins formed via migratory-insertion processes, leading to successful introduction of ketone functionalities in the polymer chain (Scheme 48) (for a review on this topic see e.g. Drent,225 Bianchini,226 Sen227 and Nozaki5). However, this approach is obviously not suitable for the direct synthesis of a wider variety of functionalised polymers without the need for any further modifications.
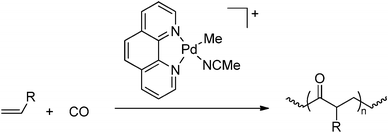 |
| | Scheme 48 Synthesis of alternating CO-olefin copolymers via a coordination/insertion mechanism catalysed by Pd catalysts. | |
Carbenes (i.e. CRR' units) can be regarded as a more versatile class of C1 monomers since they allow incorporation of a wider variety of functionalities by varying the substituents. Carbenes are readily generated from diazo compounds228 and other carbene precursors (e.g. ylides of sulfoxides229 and α,α-dihalocarbons). Diazo compounds are excellent and widely applied carbene precursors due to their ease of preparation and their relative ‘green’ character: the only byproduct upon carbene generation is dinitrogen, while other carbene precursors give rise to formation of byproducts that are not so easily removed from the reaction mixture (e.g. dmso).230,231 The large scale applicability of carbene precursors is somewhat limited in substrate scope due to some potential safety hazards associated with some diazo compounds. Many diazo compounds are toxic, and some of them are even potentially explosive under certain conditions.232 Especially aliphatic diazoalkanes are very unstable and require handling at low temperatures and low concentrations. Diazocarbonyl compounds (diazoketones or diazoesters), on the other hand, are much more stable and quite safe to work with. Hence they are frequently applied as carbene precursors in several organic transformations, even in large scale industrial reactions. They are excellent substrates for the introduction of highly-functionalised carbon units, since a wide variety of substituted diazocarbonyl compounds is readily available via well-defined synthesis routes developed in the past decades.233 Because the handling of aliphatic carbene precursors is more problematic, much recent effort has been put into the development of safer alternative methods that allow either in situ generation of the inherently unstable aliphatic diazo compounds (for a review see Fulton et al.234) or avoid the use of diazo compounds entirely (for a review see e.g. Müller235). The availability of these alternative methods to generate (aliphatic) carbenes will certainly stimulate further research into the use of carbenes in (catalytic) organic transformations in general.
In the presence of transition metals, carbenes can undergo migratory insertion into TM–C bonds, similar to insertions of alkenes and CO236,237 (Scheme 49) and this elementary step has received quite some recent interest as a tool for the introduction of functionalities. While some main-group metals and several Lewis acids are suitable for the generation of carbenes and catalysis of the subsequent migratory insertion reactions, the most successful results were obtained with late transition-metal (LTM) catalysts and at these metal centres migratory insertions occur both in a stoichiometric and in a catalytic fashion.238
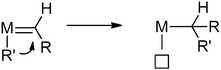 |
| | Scheme 49 Migratory insertion of carbenes. | |
6.1 Carbene polymerisation
If the reactions are carried out using an excess of carbene precursor with respect to the metal catalyst, multiple carbene insertions can occur resulting in the formation of oligomers or polymers (provided that the rate constant for carbene insertion is larger than that for elimination–termination processes or follow-up reactions). As such, metal-catalysed insertions of carbenes provide a viable pathway for the synthesis of densely functionalised polymers that (so far) cannot be synthesised in any other way. The following sections give an overview of the development of TM catalysed coordination/insertion polymerisation based on carbenes. The whole field of so-called ‘C1 polymerisation’ including catalysis by non-LTM systems has recently been reviewed by de Bruin et al.239 and Ihara.240
6.1.1 Polymers from diazo-alkanes and phenyl diazomethane.
Reports dealing with the polymerisation of diazoalkanes date back to the early 1900's and although many different metals have been attempted as catalysts for this reaction, most of the earlier reports deal with the activity of copper241,242 and gold243–245 (Scheme 50). With both metals high molecular-weight polymers were obtained (i.e. Mw up to 50 kDa for polymers of diazomethane) and this topic has been reviewed in the 1970's.242,246 The activity of other metals is less extensively studied, but there are some reports mentioning the activity of nickel,247 iridium248 and palladium249 towards polymerisation of diazoalkanes. The behaviour of the nickel system is rather unique, since it produces exclusively high-Mw polymers without the formation of byproducts (i.e. dimers and oligomers) and the catalyst remains active after all monomers have been converted.247 The use of palladium catalysts, on the other hand, leads to formation of small amounts of low-Mw oligomers containing large amounts of azo-groups, indicating that the reaction is not fully selective towards carbene insertion (Scheme 51).
Aside from the above-described homogeneous catalysts also some heterogeneous metal surfaces catalyse the polymerisation of carbenes (Au, Cu, Ti, Fe, Mg, W, Ni, V, Mn, Ta, Pt, Co, Zn, Cd, Cr, Al, Mo; listed in order of decreasing yield), with thin films of gold being the most active.243,250–252 In a way, these polymerisation reactions, involving multiple migratory carbene insertion steps, also model key steps of hydrocarbon formation in Fischer–Tropsch synthesis.253,254
6.1.2 Polymers from carbenes bearing polar functionalities.
C1 polymerisation of carbenes bearing polar functionalities is much more challenging due to the increased stability of the corresponding carbene precursors and most metal catalysts form only the corresponding dimers from these precursors.247,255,256
From 2003 on Ihara and co-workers investigated the polymerisation of diazo compounds bearing polar functionalities catalysed by a variety of PdII catalysts. They successfully subjected diazoesters,249,256–259 diazoketones249,260–262 and diazoacetamides263 to so-called ‘poly(substituted methylene) synthesis’, which allowed them to obtain various highly-functionalised oligomers/polymers in either homo- or co-polymerisation processes, yielding low Mw atactic material. These results indicate that the catalyst system is not sensitive to the presence of functional groups in the monomer, emphasizing the wide range of applicability of this technique towards the synthesis of functional materials. Recently the authors have shown that higher-Mw polymers of diazoesters (up to 24 kDa) could be obtained in reasonable yields by applying a Pd0(NHC)/BPh4− system as a catalyst.258,259 This approach has been extended to the synthesis of poly(ether ketone)s264 and poly(ester ether ketone)s265 by reaction of diazoketones with aromatic diols/cyclic ethers and diacids/cyclic ethers respectively (Fig. 6).
Stereoselective polymerisation of diazoesters was achieved by applying rhodium(diene) catalysis, giving rise to high-Mw, highly syndiotactic (co)polymers in high yields (up to 95%) (Scheme 52). The remaining products are atactic oligomers (Mw ∼ 1200 Da) and dimers.266–270 Using this approach, a variety of functionalised carbene precursors could be polymerised in a stereoselective way, such as n-butyl diazoacetate, 3-butenyl diazoacetate,270 benzyl diazoacetate and tBu-diazoacetate, and the reaction allows the synthesis of random and [homo-A]-[random-B > A]-type block copolymers (Scheme 53).268 The resulting syndiotactic polymers are highly crystalline materials that reveal both thermotropic and lyotropic liquid crystalline properties.268,270 The Mw of the polymers can be tuned by varying the Rh catalyst and the polymerisation conditions.268 Recently, it was shown that these Rh catalysts are also able to copolymerise functionalised carbenes with non-functionalised carbenes generated from either diazomethane271 or sulfoxonium ylides272 (Scheme 54). The resulting copolymers exhibit a blocky microstructure and the functional-group content can be tuned in a large window by varying the feed ratio of the two monomers. Copolymerisation of functionalised carbenes with ethene is also possible, albeit leading to quite inhomogeneous polymer mixtures.273 These results emphasise the potential of this new technique for the development of new materials that are up to now inaccessible via traditional olefin polymerisation.
This Rh-mediated carbene polymerisation has the advantage of giving stereoregular (syndiotactic) polymers with high molecular weights from polar C1 monomers,239 whereas other C1 polymerisation techniques mainly yield atactic and/or low-Mw material. This approach seems to work exclusively with Rh catalysts. Most of the corresponding iridium analogues did not show polymerisation activity towards carbenes, with only one exception.266,274 No other metals able to polymerise carbenes in a stereoselective manner have been reported. The stereoregularity of the polymers obtained by Rh catalysis is, according to DFT calculations, most likely a result of chain-end control during the propagation steps, since the Rh catalysts used in these reactions are non-chiral.266 The reaction proceeds via a migratory insertion mechanism (Scheme 55)266 and no or little chain transfer seems to occur.275–278 Most recently, detailed mechanistic studies revealed that the active species responsible for this polymerisation is a RhIII(allyl)(alkyl) species (Fig. 7) that can be formed by modifications of rather simple RhI(diene) precursors (e.g. 1,5-cyclooctadiene (cod) derivatives) under the applied reaction conditions.267,277,279 These findings are expected to lead to future improvements of this approach in terms of yields, initiation efficiencies of the catalysts and in the synthesis of stereoregular polymers with different tacticities. This clearly indicates that this new reaction has not yet been exploited at its full potential.
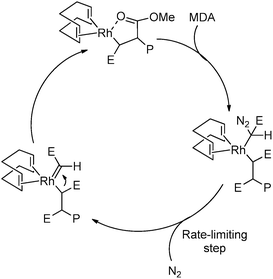 |
| | Scheme 55 Proposed mechanism for the Rh-mediated polymerisation of polar functionalised carbenes, proceeding via a migratory insertion pathway (P = growing polymer chain, E = COOMe). | |
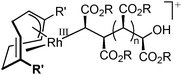 |
| | Fig. 7 RhIII(allyl)(alkyl) species proposed as active species for the Rh-catalysed stereoselective polymerisation of diazoesters. | |
7 Concluding remarks
The research area reviewed here has seen a remarkable amount of activity over the past decade, resulting in the development of a variety of improved methods that allow the synthesis of functional polyolefins in a more controlled way. Each pathway described in this review has its own advantages and limitations and its potential for the synthesis of functional polyolefins has been described in detail.
Despite many recent advances in this field, the synthesis of functional polyolefins in a controlled way with tuneable amount of functionalities still remains a challenge. Randomly functionalised copolymers are still the most difficult to prepare in a controlled way, although advances in the field of late transition-metal catalysed polymerisation allow nowadays the synthesis of these materials via a direct copolymerisation reaction of non-functionalised olefins and functional polar monomers. This approach is somewhat limited with respect to incorporation of large amounts of polar functionalities, since consecutive insertions of polar monomers take place only up to three or four times. Attempts to improve the catalysts used in this process are still ongoing, so we expect this technique to gain more potential in the future. Recent advances in the field of metathesis polymerisation allowed the synthesis of analogues of these randomly functionalised polyolefins with very precise control over the distribution of functionalities along the polymer backbone. The resulting polymers have shown to be excellent structures for studying the effect of functional-group distributions on the crystal packing and physical properties. Chain-end functionalised polyolefins are readily available via a variety of pathways, although this approach benefits most from the recent advances in the field of chain-transfer reactions, which allow almost quantitative chain-end functionalisation. The resulting materials are excellent starting points for the synthesis of block and graft copolymers in which the functional fragments are synthesised in a sequential reaction step via a different reaction mechanism. So far, these materials are not available via a single reaction mechanism due to the intrinsic reactivity differences between polar vinyl monomers and non-functionalised olefins. However, as stated above, advances in the field of late transition-metal catalysis might lead to the development of pathways for the synthesis of block and graft copolymers via a coordination/insertion mechanism in the (near) future.
To overcome some of these challenges, alternative pathways that lead to analogues of functional polyolefins are continuously being developed. One such promising alternative is the use of functionalised carbenes (C1 monomers) instead of the traditional polar vinyl monomers for the insertion of the polar functionalities. These carbenes can be polymerised to high molecular weight, highly stereoregular and densely functionalised polymers in a transition-metal catalysed process.
Acknowledgements
This work is part of the research programme of the Dutch Polymer Institute DPI, PO Box 902, 5600 AX Eindhoven, The Netherlands, project #646/647. We further kindly thank the European Research Council (ERC, EU, 7th framework program, grant agreement 202886-CatCIR) and the University of Amsterdam (UvA) for financial support.
Notes and references
- L. S. Boffa and B. M. Novak, Chem. Rev., 2000, 100, 1479 CrossRef CAS
 .
.
- A. O. Patil, Chem. Innovation, 2000, 30, 19 CAS .
- J.-Y. Dong and Y. Hu, Coord. Chem. Rev., 2006, 250, 47 CrossRef CAS .
- I. Novák, E. Borsig, L. Hrčková, A. Fiedlerová, A. Kleinová and V. Pollák, Polym. Eng. Sci., 2007, 47, 1207 Search PubMed .
- A. Nakamura, S. Ito and K. Nozaki, Chem. Rev., 2009, 109, 5215 CrossRef CAS .
-
T. C. Chung, Functionalisation of polyolefins, Academic Press, London, 2002 Search PubMed .
-
Metal Catalysts in Olefin Polymerisation, ed. Z. Guan, Springer, 2009, vol. 26, p. 255 Search PubMed .
- J. Qiao, M. Guo, L. Wang, D. Liu, X. Zhang, L. Yu, W. Song and Y. Liu, Polym. Chem., 2011, 2, 1611 RSC .
- N. K. Boaen and M. A. Hillmeyer, Chem. Soc. Rev., 2005, 34, 267 RSC .
- A. Peacock, J. Macromol. Sci., Part C: Polym. Rev., 2001, 41, 285 CrossRef .
- J. Alonso-Villanueva, J. L. Vilas, I. Moreno, J. M. Laza, M. Rodríguez and L. M. León, J. Macromol. Sci., Part A: Pure Appl. Chem., 2011, 48, 211 CrossRef CAS .
- M. R. Buchmeiser, Chem. Rev., 2000, 100, 1565 CrossRef CAS .
- C. W. Bielawski and R. H. Grubbs, Prog. Polym. Sci., 2007, 32, 1 CrossRef CAS .
- M. A. Hillmyer, W. R. Laredo and R. H. Grubbs, Macromolecules, 1995, 28, 6311 CrossRef CAS .
- S. J. McLain, E. F. McCord, S. D. Arthur, E. Hauptman, J. Feldman and W. A. Nugent, Polym. Mater.: Sci. Eng., 1997, 76, 246 CAS .
-
D. A. Bansleben, T. C. Huynh-Tran, R. L. Blanski, P. A. Hughes, W. P. Roberts, R. H. Grubbs and G. R. Hartfield, U.S. Pat. 6,203,293, 2001 Search PubMed .
-
D. A. Bansleben, T. C. Huynh-Tran, R. L. Blanski, P. A. Hughes, W. P. Roberts, R. H. Grubbs and G. R. Hartfield, PCT Int. Pat. Appl. WO00/18579, 2000 Search PubMed .
-
S. Y. Cho and U. H. Cho, Korean Pat. Appl. KR349626, 2002 Search PubMed .
-
S. Y. Cho and U. H. Cho, Korean Pat. Appl. KR2001036073A, 2001 Search PubMed .
- H. Yang, M. Islam, C. Budde and A. E. Rowan, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2107 CrossRef CAS .
- C. H. Stephens, H. Yang, M. Islam, S. P. Chum and S. J. Rowan, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 2062 CrossRef CAS .
-
D. A. Bansleben, T. C. Huynh-Tran, R. L. Blanski, P. A. Hughes, W. P. Roberts, R. H. Grubbs and G. R. Hartfield, PCT Int. Pat. Appl. WO99/50331, 1999 Search PubMed .
-
D. A. Bansleben, T. C. Huynh-Tran, R. L. Blanski, P. A. Hughes, W. P. Roberts, R. H. Grubbs and G. R. Hartfield, U.S. Pat., 6,153,714, 2000 Search PubMed .
-
D. A. Bansleben, T. C. Huynh-Tran, R. L. Blanski, P. A. Hughes, W. P. Roberts, R. H. Grubbs and G. R. Hartfield, U.S. Pat., 6,506,860, 2003 Search PubMed .
- O. A. Scherman, R. Walker and R. H. Grubbs, Macromolecules, 2002, 35, 5366 CrossRef CAS .
- O. A. Scherman, R. Walker and R. H. Grubbs, Macromolecules, 2005, 38, 9009 CrossRef CAS .
- J. P. Jordan, O. A. Scherman and R. H. Grubbs, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2003, 44, 841 CrossRef CAS .
- S. E. Lehman Jr., K. B. Wagener, L. Saunders Baugh, S. P. Rucker, D. N. Schulz, M. Varma-Nair and E. Berluche, Macromolecules, 2007, 40, 2643 CrossRef .
-
M. R. Buchmeiser, in Handbook of Ring-Opening Polymerisation, ed. P. Dubois, O. Coulembier and J.-M. Raquez, Wiley-VCH Verlag GMBH, 2009, p. 197 Search PubMed .
- A. Leitgeb, J. Wappel and C. Slugovc, Polymer, 2010, 51, 2927 CrossRef CAS .
- T. W. Baughman, C. D. Chan, K. I. Winey and K. B. Wagener, Macromolecules, 2007, 40, 6564 CrossRef CAS .
- K. L. Opper, B. Fassbender, G. Brunklaus, H. W. Spiess and K. B. Wagener, Macromolecules, 2009, 42, 4407 CrossRef CAS .
- K. L. Opper, D. Markova, M. Klapper, K. Müllen and K. B. Wagener, Macromolecules, 2010, 43, 3690 CrossRef CAS .
- D. J. Valenti and K. B. Wagener, Macromolecules, 1998, 31, 2764 CrossRef CAS .
- M. D. Watson and K. B. Wagener, Macromolecules, 2000, 33, 5411 CrossRef CAS .
- T. W. Baughman and E. van der Aa, Macromolecules, 2005, 38, 2550 CrossRef CAS .
- T. W. Baughman, E. van der Aa and K. B. Wagener, Macromolecules, 2006, 39, 7015 CrossRef CAS .
- M. D. Watson and K. B. Wagener, Macromolecules, 2000, 33, 8963 CrossRef CAS .
- K. L. Opper and K. B. Wagener, Ma, 2009, 30, 915 CAS .
- J. K. Leonard, Y. Wei and K. B. Wagener, Macromolecules, 2012, 45, 671 CrossRef CAS .
- E. Boz, A. J. Nemeth, K. B. Wagener, K. Jeon, R. Smith, F. Nazirov and M. R. Bockstaller, Macromolecules, 2008, 41, 1647 CrossRef CAS .
- E. Boz, A. J. Nemeth, I. Ghiviriga, K. Jeon, R. G. Alamo and K. B. Wagener, Macromolecules, 2007, 40, 6545 CrossRef CAS .
- E. Boz, A. J. Nemeth, R. G. Alamo and K. B. Wagener, Adv. Synth. Catal., 2007, 349, 137 CrossRef CAS .
- H. Mutlu, L. Montero de Espinosa and M. Meier, Chem. Soc. Rev., 2011, 40, 1404 RSC .
- K. L. Opper and K. B. Wagener, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 821 CrossRef CAS .
- A. R. Padwa, Prog. Polym. Sci., 1989, 14, 811 CrossRef CAS .
-
Encyclopedia of Polymer Science and Engineering, ed. H. F. Mark, N. M. Bikales, C. G. Overberger and G. Mendes, Wiley, New York, 1986, vol. 13 Search PubMed .
- S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169 CrossRef CAS .
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963 CrossRef CAS .
- A. L. Logothetis and J. M. McKenna, J. Polym. Sci., Part A: Polym. Chem., 1977, 15, 1441 CrossRef CAS .
- A. L. Logothetis and J. M. McKenna, J. Polym. Sci., Part A: Polym. Chem., 1977, 15, 1431 CrossRef CAS .
- A. L. Logothetis and J. M. McKenna, J. Polym. Sci., Part A: Polym. Chem., 1978, 16, 2797 CrossRef CAS .
- S. Liu, S. Elyashiv and A. Sen, J. Am. Chem. Soc., 2001, 123, 12738 CrossRef CAS .
- S. Liu and A. Sen, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 6175 CrossRef CAS .
- B. Gu, S. Liu, J. D. Leber and A. Sen, Macromolecules, 2004, 37, 5142 CrossRef CAS .
- S. Liu, B. Gu, H. A. Rowlands and A. Sen, Macromolecules, 2004, 37, 7924 CrossRef CAS .
- A. Berkefeld and S. Mecking, Angew. Chem., Int. Ed., 2008, 47, 2538 CrossRef CAS .
- E. Y.-X. Chen, Chem. Rev., 2009, 109, 5157 CrossRef CAS .
- D. Takeuchi, Dalton Trans., 2010, 39, 311 RSC .
-
A. W. Langer and R. R. Haynes, U.S. Pat., 3,775,279, 1973 Search PubMed .
- C.-E. Wilén, M. Auer and J. H. Näsman, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 1163 CrossRef .
- H. Hagihara, T. Ishihara, H. T. Ban and T. Shiono, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1738 CrossRef CAS .
- M. D. Purgett and O. Vogl, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2051 CrossRef CAS .
- K. Hakala, B. Löfgren and T. Helaja, Eur. Polym. J., 1998, 34, 1093 CrossRef CAS .
- P. Aaltonen and B. Löfgren, Eur. Polym. J., 1997, 33, 1187 CrossRef CAS .
- M. D. Purgett and O. Vogl, J. Macromol. Sci., Part A: Pure Appl. Chem., 1987, 24, 1465 CrossRef .
- W. Zuo, M. Zhang and W.-H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 357 CrossRef CAS .
- H. Terao, S. Ishii, M. Mitani, H. Tanaka and T. Fujita, J. Am. Chem. Soc., 2008, 130, 17636 CrossRef CAS .
- M. Fernandes and W. Kaminsky, Macromol. Chem. Phys., 2009, 210, 585 CrossRef CAS .
- K. Hakala, T. Helaja and B. Löfgren, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 1966 CrossRef CAS .
- X. Zhang, S. Chen, H. Li, Z. Zhang, Y. Lu, C. Wu and Y. Hu, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5944 CrossRef CAS .
- H. Hagihara, K. Tsuchihara, J. Sugiyama, K. Takeuchi and T. Shiono, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5600 CrossRef CAS .
- M. M. Marques, S. G. Correira, J. R. Ascenso, A. F. G. Ribeiro, P. T. Gomes, A. R. Dias, P. Foster, M. D. Rausch and J. C. W. Chien, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2457 CrossRef CAS .
- X. Zhang, S. Chen, H. Li, Z. Zhang, Y. Lu, C. Wu and Y. Hu, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 59 CrossRef CAS .
- P. Aaltonen and B. Löfgren, Macromolecules, 1995, 28, 5333 CrossRef .
- P. Aaltonen, G. Fink, B. Löfgren and J. Seppälä, Macromolecules, 1996, 29, 5255 CrossRef CAS .
- N. Kawahara, S. Kojoh, S. Matsuo, H. Kaneko, T. Matsugi and N. Kashiwa, J. Mol. Catal. A: Chem., 2005, 241, 156 CrossRef CAS .
- Y. Huang, K. Yang and J.-Y. Dong, Macromol. Rapid Commun., 2006, 27, 1278 CrossRef CAS .
- M. J. Schneider, R. Schäfer and R. Mülhaupt, Polymer, 1997, 38, 2455 CrossRef CAS .
- V. Kotzabasakis, N. Petzetakis, M. Pitsikalis and N. Hadjichristidis, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 876 CrossRef CAS .
- S. A. Amin and T. J. Marks, J. Am. Chem. Soc., 2006, 128, 4506 CrossRef CAS .
- S. A. Amin and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 2938 CrossRef CAS .
- J. Liu and K. Nomura, Macromolecules, 2008, 41, 1070 CrossRef CAS .
- K. Nomura, K. Kakinuki, M. Fujiki and K. Itagaki, Macromolecules, 2008, 41, 8974 CrossRef CAS .
- D.-J. Byun, S.-M. Chin, C. J. Han and S. Y. Kim, Polym. Bull., 1999, 43, 333 CrossRef CAS .
- S. H. Lipponen and J. Seppälä, Organometallics, 2011, 30, 528 CrossRef CAS .
- B. M. Novak and H. Tanaka, Polym. Mater.: Sci. Eng., 1999, 80, 45 CAS .
- S. Ramakrishnan, E. Berluche and T. C. Chung, Macromolecules, 1990, 23, 378 CrossRef CAS .
- T. C. Chung and D. Rhubright, Macromolecules, 1991, 24, 970 CrossRef CAS .
- T. C. Chung and D. Rhubright, Macromolecules, 1993, 26, 3467 CrossRef CAS .
- T. C. Chung, L. Lu and C. L. Li, Polym. Int., 1995, 37, 197 CrossRef CAS .
- S. Ito and K. Nozaki, Chem. Rec., 2010, 10, 315 CrossRef CAS .
- L. K. Johnson, S. Mecking and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 267 CrossRef CAS .
- S. Mecking, L. K. Johnson, L. Wang and M. Brookhart, J. Am. Chem. Soc., 1998, 120, 888 CrossRef CAS .
- C. S. Popeney, D. Camacho and Z. Guan, J. Am. Chem. Soc., 2007, 129, 10062 CrossRef CAS .
- W. Li, X. Zhang, A. Meetsma and B. Hessen, J. Am. Chem. Soc., 2004, 126, 12246 CrossRef CAS .
- A. Sen and S. Borkar, J. Organomet. Chem., 2007, 692, 3291 CrossRef CAS .
- S. Borkar, H. Yennawar and A. Sen, Organometallics, 2007, 26, 4711 CrossRef CAS .
- G. Chen, X. S. Ma and Z. Guan, J. Am. Chem. Soc., 2003, 125, 6697 CrossRef CAS .
- A. Michalak and T. Ziegler, Organometallics, 2003, 22, 2660 CrossRef CAS .
- S. J. McLain, K. J. Sweetman and L. K. Johnson, PMSE prepr., 2002, 86, 320 CAS .
- L. K. Johnson, A. Bennet, K. Dobbs, E. Hauptman, A. Ionkin, S. D. Ittel, E. F. McCord, S. J. McLain, C. Radzewich, Z. Yin, L. Wang, Y. Wang and M. Brookhart, PMSE prepr., 2002, 86, 319 CAS .
- M. Brasse, J. Cámpora, P. Palma, E. Álvarez, V. Cruz, J. Ramos and M. L. Reyes, Organometallics, 2008, 27, 4711 CrossRef CAS .
-
L. Wang, E. Hauptman, L. K. Johnson, E. F. McCord, Y. Wang and S. D. Ittel, Pat., WO 0,192,342, 2001 Search PubMed .
-
L. Wang, L. K. Johnson and A. S. Ionkin, Pat., WO 0,192,348, 2001 Search PubMed .
- C. Carlini, V. de Luise, M. Martinelli, A. M. R. Galetti and G. Sbrana, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 620 CrossRef CAS .
- A. M. R. Galetti, C. Carlini, S. Giaiacopi, M. Martinelli and G. Sbrana, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1134 CrossRef .
- V. C. Gibson and A. Tomov, Chem. Commun., 2001, 1964 RSC .
- X.-F. Li, Y.-G. Li, Y.-S. Li, Y.-X. Chen and N.-H. Hu, Organometallics, 2005, 24, 2502 CrossRef CAS .
- C. Carlini, M. Martinelli, A. M. R. Galetti and G. Sbrana, Macromol. Chem. Phys., 2002, 303, 1606 CrossRef .
- E. Drent, R. van Dijk, R. van Ginkel, B. van Oort and R. I. Pugh, Chem. Commun., 2002, 744 RSC .
- T. Kochi, K. Yoshimura and K. Nozaki, Dalton Trans., 2006, 25 RSC .
- K. M. Skupov, P. R. Marella, M. Simard, G. P. A. Yap, N. Allen, D. Conner, B. L. Goodall and J. P. Claverie, Macromol. Rapid. Commun., 2007, 28, 2033 CrossRef CAS .
- D. Guironnet, L. Caporaso, B. Neuwald and I. Göttker-Schnetmann, J. Am. Chem. Soc., 2010, 132, 4418 CrossRef CAS .
- D. Guironnet, P. Roesle, T. Rünzi, I. Göttker-Schnetmann and S. Mecking, J. Am. Chem. Soc., 2009, 131, 422 CrossRef CAS .
- T. Kochi, S. Noda, K. Yoshimura and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 8948 CrossRef CAS .
- S. Luo, J. Vela, G. R. Lief and R. F. Jordan, J. Am. Chem. Soc., 2007, 129, 8946 CrossRef CAS .
- S. Ito, K. Munakata, A. Nakamura and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14606 CrossRef CAS .
- T. Friedberger, P. Wucher and S. Mecking, J. Am. Chem. Soc., 2012, 134, 1010 CrossRef CAS .
- J. C. Daigle, L. Piche and J. P. Claverie, Macromolecules, 2011, 44, 1760 CrossRef CAS .
- T. Rünzi, D. Fröhlich and S. Mecking, J. Am. Chem. Soc., 2010, 132, 17690 CrossRef .
- C. Bouilhac, T. Rünzi and S. Mecking, Macromolecules, 2010, 43, 3583 CrossRef .
- J.-C. Daigle, L. Piche, A. Arnold and J. P. Claverie, ACS Macro Lett., 2012, 1, 343 CrossRef CAS .
- S. Ito, M. Kanazawa, K. Munakata, N. Yoshikuni, J.-I. Kuroda, Y. Okimura and K. Nozaki, J. Am. Chem. Soc., 2011, 133, 1232 CrossRef CAS .
- R. G. Lopez, F. D'Agosto and C. Boisson, Prog. Polym. Sci., 2007, 32, 419 CrossRef .
- M. J. Yanjarappa and M. Sivaram, Prog. Polym. Sci., 2002, 27, 1347 CrossRef CAS .
- G. J. Domski, J. M. Rose, G. W. Coates and A. D. Bolig, Prog. Polym. Sci., 2007, 32, 30 CrossRef CAS .
- Y. Doi, S. Ueki and K. Soga, Makromol. Chem., 1979, 180, 1359 CrossRef CAS .
- Y. Doi, S. Ueki and K. Soga, Macromolecules, 1979, 12, 814 CrossRef CAS .
- Y. Doi, S. Suzuki and K. Soga, Macromolecules, 1986, 19, 2896 CrossRef CAS .
- Y. Doi and T. Keii, Adv. Polym. Sci., 1986, 73/74, 201 CrossRef .
- Y. Doi and M. Murata, Makromol. Chem., Rapid Commun., 1984, 5, 811 CrossRef CAS .
- Y. Fukui and M. Murata, Macromol. Chem. Phys., 2001, 202, 1430 CrossRef CAS .
- M. Brookhart, J. M. DeSimone, B. E. Grant and M. J. Tanner, Macromolecules, 1995, 28, 5378 CrossRef CAS .
- S. C. Hong, S. Jia, M. Teodorescu, T. Kowalewski, K. Matyaszewsi, A. C. Gottfried and M. Brookhart, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 2736 CrossRef CAS .
- A. C. Gottfried and M. Brookhart, Macromolecules, 2003, 36, 3085 CrossRef .
- H. Makio and T. Fujita, Macromol. Rapid. Commun., 2007, 28, 698 CrossRef CAS .
- S. B. Amin and T. J. Marks, Angew. Chem., Int. Ed., 2008, 47, 2006 CrossRef CAS .
- K. Ziegler, E. Holzkamp and H. Breil, Angew. Chem., 1955, 67, 541 CrossRef CAS .
- J.-F. Pelletier, A. Mortreux and X. Olonde, Angew. Chem., Int. Ed., 1996, 35, 1854 CrossRef CAS .
- T. Shiono, K. Yoshida and K. Soga, Makromol. Chem., Rapid Commun., 1990, 11, 285 CrossRef .
- D. R. Burfield, Polymer, 1984, 25, 1817 CrossRef CAS .
- T. Shiono, H. Kurosawa and K. Soga, Makromol. Chem., 1992, 193, 2751 CrossRef CAS .
- T. Shiono, H. Kurosawa and K. Soga, Macromolecules, 1995, 28, 437 CrossRef CAS .
- H. Kurosawa, T. Shiono and K. Soga, Macromol. Chem. Phys., 1994, 195, 1381 CrossRef CAS .
- T. Shiono, H. Kurosawa and K. Soga, Macromolecules, 1994, 27, 2635 CrossRef CAS .
- C. Zhang, H. Niu and J.-Y. Dong, Appl. Organomet. Chem., 2011, 25, 632 CrossRef CAS .
- G. J. P. Britovsek, S. A. Cohen, V. C. Gibson and M. van Meurs, J. Am. Chem. Soc., 2004, 126, 10701 CrossRef CAS .
- D. J. Arriola, E. M. Carnahan, P. D. Hustad, R. L. Kuhlman and T. T. Wenzel, Science, 2006, 312, 714 CrossRef CAS .
- L. Sita, Angew. Chem., Int. Ed., 2009, 48, 2464 CrossRef CAS .
- H. Kaneyoshi, K. Inoue and K. Matyaszewski, Macromolecules, 2005, 38, 5425 CrossRef CAS .
- D.-J. Byun and S. Y. Kim, Macromolecules, 2000, 33, 1921 CrossRef CAS .
- C. J. Han, M. S. Lee, D.-J. Byun and S. Y. Kim, Macromolecules, 2002, 35, 8923 CrossRef CAS .
- J. Imuta, N. Kashiwa and Y. Toda, J. Am. Chem. Soc., 2002, 124, 1176 CrossRef CAS .
- S. Tsubaki, J. Jin, T. Sano, T. Uozumi and K. Soga, Macromol. Chem. Phys., 2001, 202, 1757 CrossRef CAS .
- S. Kojoh, T. Tsutsui, M. Kioka and N. Kashiwa, Polym. J., 1999, 31, 332 CrossRef CAS .
- K. K. Kang, T. Shiono and T. Ikeda, Macromolecules, 1997, 30, 1231 CrossRef CAS .
- T. Shiono, K. K. Kang, H. Hagihara and T. Ikeda, Macromolecules, 1997, 30, 5997 CrossRef CAS .
- J. Mazzolini, E. Espinosa, F. D'Agosto and C. Boisson, Polym. Chem., 2010, 1, 793 RSC .
- R. Godoy Lopez, C. Boisson, F. D'Agosto, R. Spitz, F. Boisson and D. Bertin, Macromolecules, 2004, 37, 3540 CrossRef .
- R. Godoy Lopez, C. Boisson, F. D'Agosto, R. Spits, F. Boisson, D. Gigmes and D. Bertin, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 2705 CrossRef .
- R. Godoy Lopez, C. Boisson, F. D'Agosto, R. Spitz, F. Boisson, D. Gigmes and D. Bertin, Macromol. Rapid. Commun., 2006, 27, 173 CrossRef .
- R. Briquel, J. Mazzolini, T. Le Bris, O. Boyron, F. Boisson, F. Delolme, F. D'Agosto, C. Boisson and R. Spitz, Angew. Chem., Int. Ed., 2008, 47, 9311 CrossRef CAS .
- P.-F. Fu and T. Marks, J. Am. Chem. Soc., 1995, 117, 10474 CrossRef .
- K. Koo, P.-F. Fu and T. J. Marks, Macromolecules, 1999, 32, 981 CrossRef CAS .
- K. Koo and T. J. Marks, J. Am. Chem. Soc., 1999, 121, 8791 CrossRef CAS .
- K. Koo and T. J. Marks, J. Am. Chem. Soc., 1998, 120, 4019 CrossRef CAS .
-
S. E. Thomas, Organic Synthesis: The Roles of Boron and Silicon, Oxford Chemistry Primers, New York, 1991, p. 47 Search PubMed .
- G. Xu and T. C. Chung, J. Am. Chem. Soc., 1999, 121, 6763 CrossRef CAS .
- T. C. Chung and G. Xu, Macromolecules, 2001, 34, 8040 CrossRef CAS .
- T. C. Chung and J.-Y. Dong, J. Am. Chem. Soc., 2001, 123, 4871 CrossRef CAS .
- J.-Y. Dong, Z. M. Wang, H. Hon and T. C. Chung, Macromolecules, 2002, 35, 9352 CrossRef CAS .
- J.-Y. Dong and T. C. Chung, Macromolecules, 2002, 35, 1622 CrossRef CAS .
- T. C. Chung, H. L. Lu and C. L. Li, Macromolecules, 1994, 27, 7533 CrossRef CAS .
- H. Kaneko, S. Kojoh, N. Kawahara, S. Matsuo, T. Matsugi and N. Kashiwa, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 5103 CrossRef CAS .
- T. Shiono and K. Soga, Macromol. Rapid. Commun., 1992, 13, 371 CrossRef CAS .
- K. Matyaszewski, J. Saget, J. Pyun, M. Schlögl and B. Rieger, J. Macromol. Sci., Part A: Pure Appl. Chem., 2002, A39, 901 CrossRef .
- T. C. Chung and D. Rhubright, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 2759 CrossRef CAS .
- B. Lu and T. C. Chung, Macromolecules, 1998, 31, 5943 CrossRef CAS .
- B. Lu and T. C. Chung, Macromolecules, 1999, 32, 2525 CrossRef CAS .
- T. Matsugi, S. Kojoh, N. Kawahara, S. Matsuo and H. Kaneko, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3965 CrossRef CAS .
- H. Kaneko, J. Saito, N. Kawahara, S. Matsuo and T. Matsugi, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 812 CrossRef CAS .
- S. Förster and T. Plantenberg, Angew. Chem., Int. Ed., 2002, 41, 688 CrossRef .
- I. W. Hamley, Prog. Polym. Sci., 2009, 34, 1161 CrossRef CAS .
- M. Xenidou and N. Hadjichristidis, Macromolecules, 1998, 31, 5690 CrossRef CAS .
- H.-C. Kim, S. Park and W. D. Hinsberg, Chem. Rev., 2010, 110, 146 CrossRef CAS .
- J. Rodríguez-Hernández, F. Chécot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691 CrossRef .
- Y. Yagci and M. A. Tasdelen, Prog. Polym. Sci., 2006, 31, 1133 CrossRef CAS .
- T. C. Chung, Prog. Polym. Sci., 2002, 27, 39 CrossRef CAS .
- K. V. Bernaerts and F. E. Du Prez, Prog. Polym. Sci., 2006, 31, 671 CrossRef CAS .
- Y. Doi and T. Koyama, Makromol. Chem., 1985, 186, 11 CrossRef CAS .
- K. Inoue and K. Matyaszewski, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 496 CrossRef .
- A. Dix and S. Ptacek, Macromol. Symp., 2006, 236, 186 CrossRef CAS .
- Q. Yi, G. Fan, X. Wen, J.-Y. Dong and C. C. Han, Macromol. React. Eng., 2009, 3, 91 CrossRef CAS .
- T. C. Chung, H. L. Lu and W. Janvikul, Polymer, 1997, 138, 1495 CrossRef .
- T. C. Chung and H. L. Lu, J. Mol. Catal. A: Chem., 1997, 115, 115 CrossRef CAS .
- K. Zhang, Z. Ye and R. Subramanian, Macromolecules, 2008, 41, 640 CrossRef CAS .
- W.-J. Wang, P. Liu, B.-G. Li and S. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3024 CrossRef CAS .
- Y. Zhao, X. Shi, H. Gao, L. Zhang, F. Zhu and Q. Wu, J. Mater. Chem., 2012, 22, 5737 RSC .
- Selective end-functionalisation by the hydroxy-functionalised monomer is only observed for allyl alcohol. In case of longer methylene spacers the polar monomer is incorporated in the polymer chain as well.
- A. Leblanc, J.-P. Broyer, C. Boisson, R. Spitz and V. Monteil, Pure Appl. Chem., 2012, 84(10), 2113 CrossRef CAS .
- A. Leblanc, E. Grau, J.-P. Broyer, C. Boisson, R. Spitz and V. Monteil, Macomolecules, 2011, 44, 3293 CrossRef CAS .
- H. Yasuda, F. Furo, H. Yamamoto and A. Nakamura, Macromolecules, 1992, 25, 5115 CrossRef CAS .
- J. Gromada, T. Chenal, A. Mortreux and F. Leising, J. Mol. Catal. A: Chem., 2002, 182/183, 523 CrossRef .
- K. Tanaka, M. Furo and E. Ihara, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 1382 CrossRef CAS .
- G. Desurmont, Y. Li and H. Yasuda, Organometallics, 2000, 19, 1811 CrossRef CAS .
- G. Desurmont, T. Tokimitsu and H. Yasuda, Macromolecules, 2000, 33, 7679 CrossRef CAS .
- M. D. K. Ingal, S. J. Joray, S. J. Duffy, D. P. Long and P. A. Biaconi, J. Am. Chem. Soc., 2000, 122, 7845 CrossRef .
- G. Desurmont, M. Tanaka, Y. Li, H. Yasuda, T. Tokimitsu, S. Stone and A. Yanagase, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4095 CrossRef CAS .
- H. Frauenrath, S. Balk, H. Keul and H. Höcker, Macromol. Rapid. Commun., 2001, 22, 1147 CrossRef CAS .
- S. Collins and D. G. Ward, J. Am. Chem. Soc., 1992, 114, 5460 CrossRef CAS .
- V. Kotzabasakis, S. Mourmouris, M. Pitsikalis, N. Hadjichristidis and D. J. Lohse, Macromolecules, 2011, 44, 1952 CrossRef CAS .
- K. Kempe, A. Krieg, C. R. Becer and U. S. Schubert, Chem. Soc. Rev., 2012, 41, 171 RSC .
- E. Espinosa, M. Glassner, C. Boisson, C. Barner-Kowollik and F. D'Agosto, Macromol. Rapid. Commun., 2011, 32, 1447 CrossRef CAS .
- N. Hadjichristidis, H. Iatrou, M. Pitsikalis and J. Mays, Prog. Polym. Sci., 2006, 31, 1068 CrossRef CAS .
- C. Feng, Y. Li, D. Yang, J. Hu, X. Zhang and X. Huang, Chem. Soc. Rev., 2011, 40, 1282 RSC .
- M. Hong, J.-Y. Liu, B.-X. Li and Y.-S. Li, Macromolecules, 2011, 44, 5659 CrossRef CAS .
- H. L. Lu, S. Hong and T. C. Chung, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2795 CrossRef CAS .
- N. B. Bowden, M. Dankova, W. Wiyatno, C. J. Hawker and R. M. Waymouth, Macromolecules, 2002, 35, 9246 CrossRef CAS .
- Y. Inoue, T. Matsugi, N. Kashiwa and K. Matyaszewski, Macromolecules, 2004, 37, 3651 CrossRef CAS .
- J. Imuta, Y. Toda and N. Kashiwa, Chem. Lett., 2001, 710 CrossRef CAS .
- C. Cao, J. Zou, J.-Y. Dong, Y. Hu and T. C. Chung, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 429 CrossRef CAS .
- T. C. Chung and W. Janvikul, J. Organomet. Chem., 1999, 581, 176 CrossRef CAS .
-
D. Baskaran and A. H. E. Müller, in Controlled and Living Polymerisations, ed. A. H. E. Müller and K. Matyaszewski, Wiley-VCH Verlag GmbH & Co., Weinheim, 2009, p. 1 Search PubMed .
- E. Drent and P. H. M. Budzelaar, Chem. Rev., 1996, 96, 663 CrossRef CAS .
- C. Bianchini and A. Meli, Coord. Chem. Rev., 2002, 225, 35 CrossRef CAS .
-
A. Sen, Synthesis of alkene-carbon monoxide copolymers and cooligomers, Kluwer Academic Publishing, Dordrecht, 2003 Search PubMed .
-
M. P. Doyle, M. A. McKervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, John Wiley & Sons, New York, 2000 Search PubMed .
- X. Zhou and K. Shea, J. Am. Chem. Soc., 2000, 122, 11515 CrossRef CAS .
-
P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed .
- Of course, if we include the energy load and the byproducts generated in their synthesis, diazocompounds are less ‘green’ in terms of atom and energy efficiency.
-
M. Regitz and G. Maas, Diazocompounds – Properties and Synthesis, Academic Press, Orlando, 1986 Search PubMed .
- Z. Zhang and J. Wang, Tetrahedron, 2008, 64, 6577 CrossRef CAS .
- J. R. Fulton, V. K. Aggarwal and J. de Vicente, Eur. J. Org. Chem., 2005, 1479 CrossRef CAS .
- P. Muller, Acc. Chem. Res., 2004, 37, 243 CrossRef .
-
P. W. N. M. van Leeuwen, Homogeneous Catalysis: Understanding the Art, Kluwer Academic Publishers, Dordrecht, 2004 Search PubMed .
-
P. W. N. M. van Leeuwen, K. Morokuma and J. H. van Lenthe, Theoretical aspects of homogeneous catalysis, Kluwer Academic Publishers, Dordrecht, 1995 Search PubMed .
- N. M. G. Franssen, A. J. C. Walters, J. N. H. Reek and B. de Bruin, Catal. Sci. Technol., 2011, 1, 153 CAS .
- E. Jellema, A. L. Jongerius, J. N. H. Reek and B. de Bruin, Chem. Soc. Rev., 2010, 39, 1706 RSC .
- E. Ihara, Adv. Polym. Sci., 2010, 231, 191 CrossRef CAS .
- A. Loose, J. Prakt. Chem., 1909, 124, 185 Search PubMed .
- N. Imoto and T. Nakaya, J. Macromol. Sci., Rev. Macromol. Chem., 1972, C7(1), 1 CrossRef .
- A. G. Nasini, Makromol. Chem., 1961, 44, 550 CrossRef .
- A. G. Nasini and G. T. L. Saini, Chimia, 1962, 127 CAS .
- A. G. Nasini, G. Saini and L. Trossarelli, Pure Appl. Chem., 1962, 4, 255 CrossRef CAS .
- G. W. Cowell and A. Ledwith, Q. Rev., Chem. Soc., 1970, 24, 119 RSC .
- H. Werner and J. H. Richards, J. Am. Chem. Soc., 1968, 90, 4976 CrossRef CAS .
- F. D. Mango and I. Dvouretzky, J. Am. Chem. Soc., 1966, 88, 1654 CrossRef CAS .
- E. Ihara, M. Kida, M. Fujioka, N. Haida, T. Itoh and K. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1536 CrossRef CAS .
- K. Sheshadri, S. V. Atre, Y.-T. Tao, M.-T. Lee and D. L. Allara, J. Am. Chem. Soc., 1997, 119, 4698 CrossRef .
- W. Guo and K. Jennings, Langmuir, 2002, 18, 3123 CrossRef CAS .
- D. Bai and K. Jennings, J. Am. Chem. Soc., 2006, 127, 3048 CrossRef .
- J. Lin, C. Chang, C. J. Jenks, M. X. Yank, T. H. Wentzlaff and B. E. Bent, J. Catal., 1994, 147, 250 CrossRef CAS .
- J. P. Hindermann, G. J. Hutchings and A. Kiennemann, Catal. Rev. Sci. Eng., 1993, 35, 1 CAS .
- G. D. Buckley, L. H. Cross and N. H. Ray, J. Chem. Soc., 1950, 2714 RSC .
- K. Lorey, J. Prakt. Chem., 1929, 124, 185 CrossRef .
- E. Ihara, N. Haida, M. Iio and K. Inoue, Macromolecules, 2003, 36, 36 CrossRef CAS .
- E. Ihara, Y. Ishiguro, N. Yoshida, T. Hiraren, T. Itoh and K. Inoue, Macromolecules, 2009, 42, 8608 CrossRef CAS .
- E. Ihara, H. Takahashi, M. Akazawa, T. Itoh and K. Inoue, Macromolecules, 2011, 44, 3287 CrossRef CAS .
- E. Ihara, M. Fujioka, N. Haida, T. Itoh and K. Inoue, Macromolecules, 2005, 38, 2101 CrossRef CAS .
- E. Ihara, Y. Goto, T. Itoh and K. Inoue, Polym. J., 2009, 41, 1117 CrossRef CAS .
- E. Ihara, T. Hiraren, T. Itoh and K. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1638 CrossRef CAS .
- E. Ihara, T. Hiraren, T. Itoh and K. Inoue, Polym. J. (Tokyo), 2008, 40, 1094 CrossRef CAS .
- E. Ihara, K. Saiki, Y. Goto, T. Itoh and K. Inoue, Macromolecules, 2010, 43, 4589 CrossRef CAS .
- E. Ihara, Y. Hara, T. Itoh and K. Inoue, Macromolecules, 2011, 44, 5955 CrossRef CAS .
- E. Jellema, P. H. M. Budzelaar, J. N. H. Reek and B. de Bruin, J. Am. Chem. Soc., 2007, 129, 11631 CrossRef CAS .
- E. Jellema, A. L. Jongerius, A. J. C. Walters, J. M. M. Smits, J. N. H. Reek and B. de Bruin, Organometallics, 2010, 29, 2823 CrossRef CAS .
- E. Jellema, A. L. Jongerius, G. Alberda van Ekenstein, S. D. Mookhoek, T. J. Dingemans, E. M. Reingruber, A. Chojnacka, P. J. Schoenmakers, R. Sprenkels, E. R. H. van Eck, J. N. H. Reek and B. de Bruin, Macromolecules, 2010, 43, 8892 CrossRef CAS .
- M. Rubio, E. Jellema, M. A. Siegler, A. L. Spek, J. N. H. Reek and B. de Bruin, Dalton Trans., 2009, 8970 RSC .
- D. G. H. Hetterscheid, C. Hendriksen, W. I. Dzik, J. M. M. Smits, E. R. H. van Eck, A. E. Rowan, V. Busico, M. Vacatello, V. Van Axel Castelli, A. Segre, E. Jellema, T. G. Bloemberg and B. de Bruin, J. Am. Chem. Soc., 2006, 128, 9746 CrossRef CAS .
- N. M. G. Franssen, K. Remerie, T. Macko, J. N. H. Reek and B. de Bruin, Macromolecules, 2012, 45, 3711 CrossRef CAS .
- A. I. Olivos-Suarez, M. P. del Río, K. Remerie, J. N. H. Reek and B. de Bruin, ACS Catal., 2012, 2, 2046 CrossRef .
- N. M. G. Franssen, J. N. H. Reek and B. de Bruin, Dalton Trans., 2013 10.1039/c3dt32941k , accepted.
- B. Bantu, K. Wurst and M. R. Buchmeiser, J. Organomet. Chem., 2007, 692, 5272 CrossRef CAS .
- A. J. C. Walters, E. Jellema, M. Finger, P. Aarnoutse, J. M. M. Smits, J. N. H. Reek and B. de Bruin, ACS Catal., 2012, 246 CrossRef CAS .
- M. Finger, J. N. H. Reek and B. de Bruin, Organometallics, 2011, 30, 1094 CrossRef CAS .
- M. Finger, M. Lutz, J. N. H. Reek and B. de Bruin, Eur. J. Inorg. Chem., 2012, 1437 CrossRef CAS .
- A. J. C. Walters, O. Troeppner, I. Ivanović-Burmazović, C. Tejel, M. P. del Río, J. N. H. Reek and B. de Bruin, Angew. Chem., Int. Ed., 2012, 51, 5157 CrossRef CAS .
- N. M. G. Franssen, M. Finger, J. N. H. Reek and B. de Bruin, Dalton Trans., 2013, 42, 8970 RSC .
|
| This journal is © The Royal Society of Chemistry 2013 |
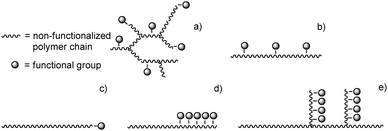
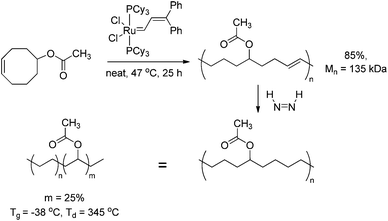
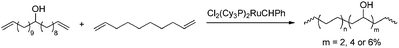

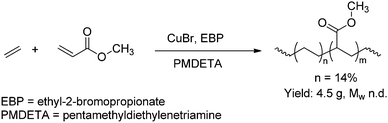
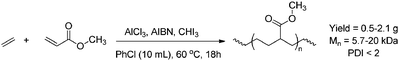
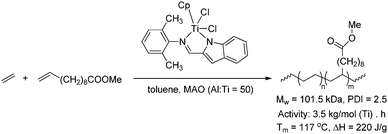
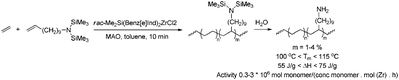
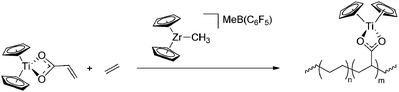
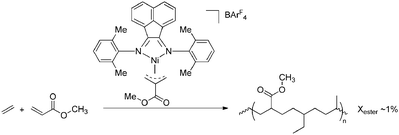
![Synthesis of low-Mw copolymers of ethene and methylacrylate obtained with a Ni catalyst bearing a [N–O] ligand.](/image/article/2013/CS/c3cs60032g/c3cs60032g-s21.gif)
![Copolymerisation of ethene and methyl methacrylate catalysed by a Ni[N–O] complex.](/image/article/2013/CS/c3cs60032g/c3cs60032g-s22.gif)
![Synthesis of ethene–methyl acrylate copolymers showing consecutive acrylate insertions, obtained via coordination/insertion polymerisation using a Pd[P–O] catalyst.](/image/article/2013/CS/c3cs60032g/c3cs60032g-s23.gif)
![Pd[P–O]-catalysed copolymerisation of ethene and N-isopropylacrylamide (NIPAM).](/image/article/2013/CS/c3cs60032g/c3cs60032g-s24.gif)
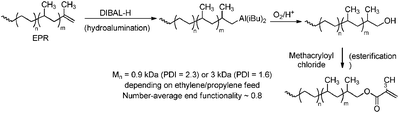
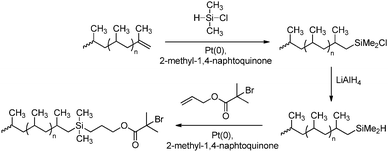
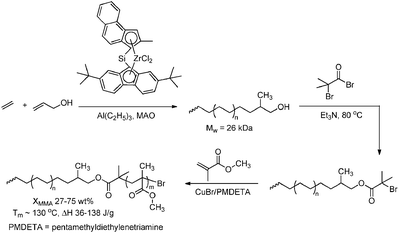
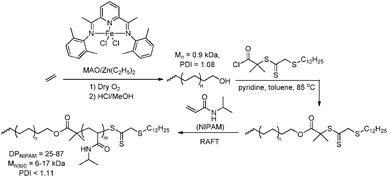
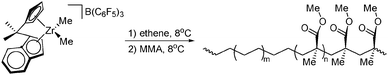
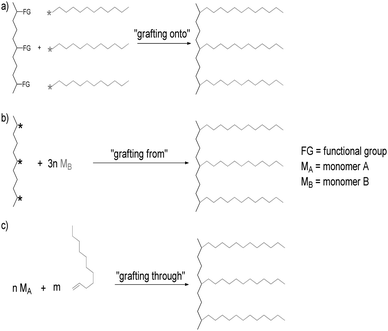
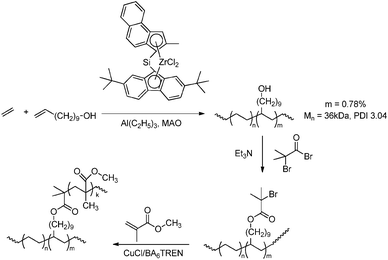
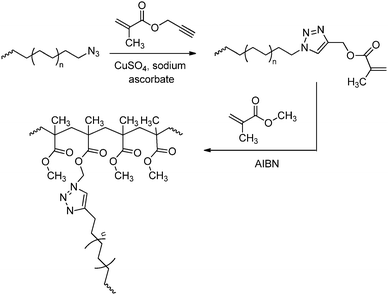
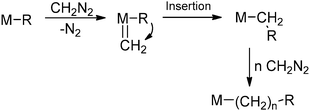
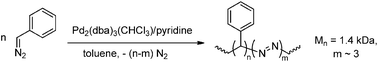
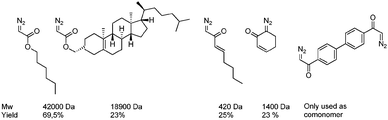
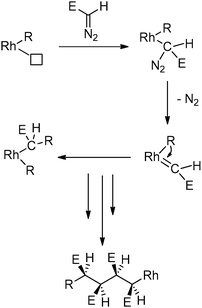
![Random and [homo-A]-[random-B > A] block copolymers synthesised via Rh-mediated carbene polymerisation.](/image/article/2013/CS/c3cs60032g/c3cs60032g-s53.gif)