 Open Access Article
Open Access ArticleCatalytic X–H insertion reactions based on carbenoids
Dennis
Gillingham
* and
Na
Fei
University of Basel, St. Johanns-Ring 19, Basel, CH-4056, Switzerland. E-mail: dennis.gillingham@unibas.ch; Fax: +41 (0)61 267 09 76; Tel: +41 (0)61 267 11 48
First published on 13th February 2013
Abstract
Catalysed X–H insertion reactions into diazo compounds (where X is any heteroatom) are a powerful yet underutilized class of transformations. The following review will explore the historical development of X–H insertion and give an up-to-date account of the metal catalysts most often employed, including an assessment of their strengths and weaknesses. Despite decades of development, recent work on enantioselective variants, as well as applying catalytic X–H insertion towards problems in chemical biology indicate that this field has ample room for innovation.
 Dennis Gillingham and Na Fai | Dennis Gillingham (left) was born in Newfoundland, Canada. He studied at the Memorial University of Newfoundland with Graham Bodwell, receiving a BSc in 2001. After completing a PhD at Boston College with Amir Hoveyda, he moved to the ETH Zürich for a post-doctoral stint in Don Hilvert's lab. In November 2010 he took up his current position as an assistant professor at the University of Basel. His research interests broadly encompass new synthetic methods, but with a particular focus on nucleic acids. |
Na Fei (Right) was born in Rizhao, China. She received her Bachelor's degrees in Chemistry from Shandong Agricultural University in 2008. Later she joined Shaozhong Wang's group at Nanjing University for her MSc studies. She started her PhD work in Prof. Dennis Gillingham's group at the University of Basel in 2011, where she is developing new methods for the alkylation of nucleic acids. |
Key learning points1. X–H insertion is an underexploited process with great potential for further development.2. Metal–carbenoids are less reactive than is typically believed and are compatible with a variety of common functional groups. 3. The availability of diverse ligand classes has reinvigorated the study of X–H insertion. Many metals behave poorly or are completely inactive without the right ligand. 4. Recent breakthroughs in copper- and iron-catalysed X–H insertion should stimulate further development with these metals. 5. X–H insertion can be carried out in water, opening the door to applications in chemical biology. |
1. Introduction
The carbon–heteroatom bond is a ubiquitous motif in both natural and man-made molecules. The field of chemical synthesis is therefore heavily reliant on methods to construct this bond type. The wide application of the palladium-catalyzed C–N and C–O couplings, developed primarily by Buchwald and Hartwig, is a case-in-point.1 Another class of carbon–heteroatom transformations that has undergone significant development but is still underutilized by the broader community are carbenoid based X–H insertions (XHIs), where X can be nitrogen, oxygen, sulfur, selenium, phosphorus, or a halogen. In these reactions a metal–carbenoid is typically generated in situ from a diazo precursor and then reacts with an X–H bond to deliver, either in concerted or stepwise fashion, the insertion product (see Scheme 1). The rather scant use of XHI reactions in the synthetic community belies the favourable characteristics of the process. For example the rhodium, copper, ruthenium, and iron catalysed versions of this reaction are tolerant to a range of reaction conditions and functional groups, and can accommodate a variety of heteroatom donors. Recently a number of enantioselective processes for carbenoid-based N–H and O–H insertions have been developed, again expanding the potential applicability of these methods.2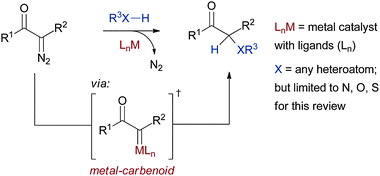 | ||
| Scheme 1 General scheme for XHI. | ||
Given its long history, it is surprising that XHI remains underdeveloped. After the seminal observations3 and the subsequent demonstration by Merck that XHI could be used industrially (see panel D in Fig. 1),4 it would seem logical that all aspects of this reaction would have been quickly refined, yet its development throughout the 80's and 90's was sporadic. The predominant focus in the carbenoid field was instead cyclopropanation and C–H insertion. A major reason for the sluggish development of XHI is likely that there are good ways to achieve the substitution of polar X–H bonds through classical uncatalysed nucleophilic displacement reactions. Over the past two decades, however, the catalysis field has proven in countless cases that the properties of templating and turn-over offered by a catalyst can deliver benefits that no stoichiometric process can. Tuning catalyst structure can offer the prospect of controlling chemo-, diastereo-, enantio-, regio-, and site-selectivity. A good catalyst serves as a template and chaperone for the substrates, guiding them over a single slice of the reaction hypersurface. Therefore, good stoichiometric methods to achieve a certain reaction do not diminish the importance of catalyst development. Chemists today are working with larger, more diverse, and structurally more complex molecules than ever before. Achieving selective reactions on these large molecules presents a task in chemoselectivity not likely to be overcome through uncatalysed processes. Progress in the XHI field on this front, i.e. tackling catalysis in complex systems, is discussed in the last section on applications to chemical biology.
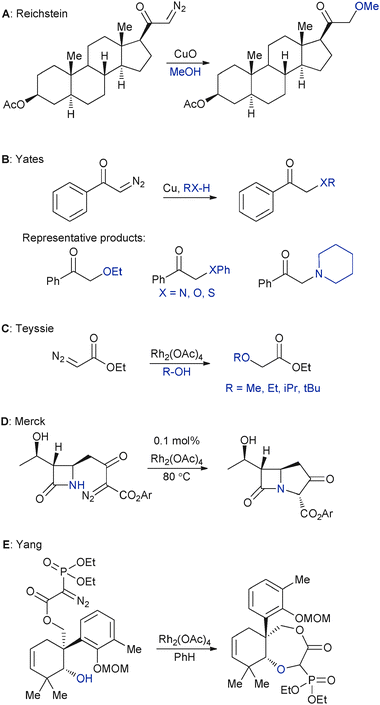 | ||
| Fig. 1 Seminal observations (panels A–D) and a recent application (panel E) in XHI chemistry. | ||
A number of excellent reviews have appeared on the topic of XHI but the comprehensive ones are more than a decade old,5 and the recent ones tend to focus on isolated aspects of the reaction.6 Furthermore, applications of XHI geared towards chemical biology have never been covered. This review will introduce XHI by referring to seminal studies and thereafter give a whistle-stop tour through the field, discussing only the highlights. Examples will be limited to XHIs where X = N, O, and S. This exclusion is not meant to dismiss the importance of other XHIs, but the focus of the present review is on education, the chosen examples are meant to be representative of the field. The reader is referred to the older reviews for a comprehensive look at the historical development of XHI and the full variety of potential insertion processes.5
2. Early milestones in XHI
Although isolated examples of XHI had been reported earlier,3 it was an unexpected observation by Casanova and Reichstein reported in 1950 that likely spurred the development of transition metal-catalysed XHI.7 They found that a pregnenolone derivative containing an α-diazo ketone motif was converted to an α-methoxy ketone instead of the intended Wolf rearrangement product when treated with copper(I) oxide in methanol (Panel A, Fig. 1). Shortly thereafter Yates described the first systematic study of XHI employing finely divided copper as a catalyst.8 He showed that a variety of X–H bonds such as those of thiophenol, aniline, piperidine, and ethanol could react with α-diazoketones to deliver the XHI products (Panel B, Fig. 1). This paper was also the first to intimate a reaction mechanism involving a carbene-type intermediate. Nozaki, Noyori, and coworkers later put forth an explicit model for the copper-stabilized carbene possibility in their ground-breaking papers on asymmetric catalysis.9 In 1973 two seminal papers were published in the metal–carbenoid catalysis field: First Salomon and Kochi established that, even in cases of nominal catalysis by Cu(II) or Cu(0), Cu(I) is the catalytically active oxidation state in Cu–carbenoid chemistry.10 And second Teyssie reported that rhodium(II) acetate (Rh2(OAc)4) is an excellent catalyst for the insertion of diazo compounds into hydroxylic bonds.11 The Teyssie study in particular ushered in a new phase in metal–carbenoid chemistry, with the exceptional reactivity and high-turnover (approximately 600 turnovers in 4 h) of Rh2(OAc)4 making it a subject of intense study for XHI in the years that followed.5 An important practical application of the rhodium(II) chemistry developed by Teyssie is the Merck synthesis of thienamycin from 1980 (Panel D, Fig. 1).4 A key step in their synthesis of this antibiotic employs rhodium(II) catalysed N–H insertion. The synthesis is a landmark for many reasons, but in the context of this review germane points include: the creative use of catalytic N–H insertion in total synthesis, and the demonstration that N–H insertion can be highly efficient even with strained molecules such as β-lactams. Recently Yang's group exploited Rh2(OAc)4-catalysed intramolecular O–H insertion in their breathtaking total synthesis of Maoecrystal V (Panel E, Fig. 1),12 again proving that XHI is a simple and elegant way to achieve carbon–heteroatom transformations.3. Mechanistic aspects of XHI with carbenoids
A concerted mechanism is generally accepted for reactions of carbenoids with non-polar bonds (i.e. as in cyclopropanation, C–H insertion, or Si–H insertion).13 With polar XHIs however the weight of evidence seems to prefer the stepwise ylide mechanism shown in Scheme 2. A more granulated picture than this generic scheme is complicated by the fact that each transition metal can lead to a slightly different pathway, or at least a change in the rate-determining step (RDS).14 Further confounding the matter is that the kinetically relevant step in many cases, certainly with rhodium, is the loss of nitrogen to deliver the metal–carbenoid, rendering the actual XHI difficult to investigate. Nevertheless the broad strokes of the pathway in Scheme 2 seem correct; it is the relative importance of the elementary steps that remain unclear.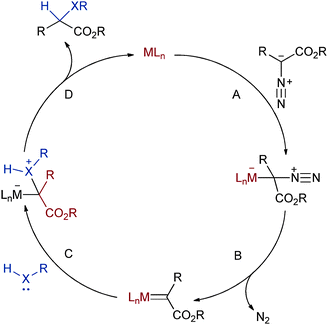 | ||
| Scheme 2 General mechanism for XHI. | ||
Difficulties notwithstanding, a number of experimental and computational studies lend insight into the rhodium(II)-, copper(I)-, and iron(III)-catalysed reactions. These will be discussed in turn in the following paragraphs. For the remainder of the present review, unless explicitly stated otherwise, any reference to step A, B, C, or D refer to the steps shown in Scheme 2.
3.1 Mechanistic studies of copper-catalysed XHIs
Whereas with rhodium(II) catalysis the RDS is typically step B in the general mechanism, according to calculations copper(I) has similar transition state energies for steps A, B, and D.14,15 Therefore one might expect the RDS with copper(I) systems to vary depending on the specific copper salt and ligand set employed. We will see shortly that this is indeed borne out in the experimental results. First, however, Scheme 3 provides a closer look at the reaction pathway with copper catalysts. Steps A, B, C, and D again refer to the same general steps as Scheme 2; when available, experimental results that lend credence to each elementary step are highlighted in the boxes. The top line of Scheme 3 shows some potential coordination modes of substrates or other Lewis bases that can potentially slow or even halt copper catalysis by blocking the crucial substrate binding; a crystal structure has been obtained of one of these coordination modes with α-diazophenanthrone (top right structure in Scheme 3).16 Coordination of the negatively polarized carbon of the diazo substrate to the Lewis acidic free copper (step A) is the first productive step in the catalytic cycle. Loss of nitrogen from the resulting intermediate, as shown in step B, then delivers the copper–carbenoid. A limited number of such copper–carbenoids (structure 2 in Scheme 3 is a representative example) have been characterized spectroscopically (X-ray, NMR).16 When treated with olefins many of these stable carbenoids undergo rapid cyclopropanation, confirming their relevance in the catalytic cycle.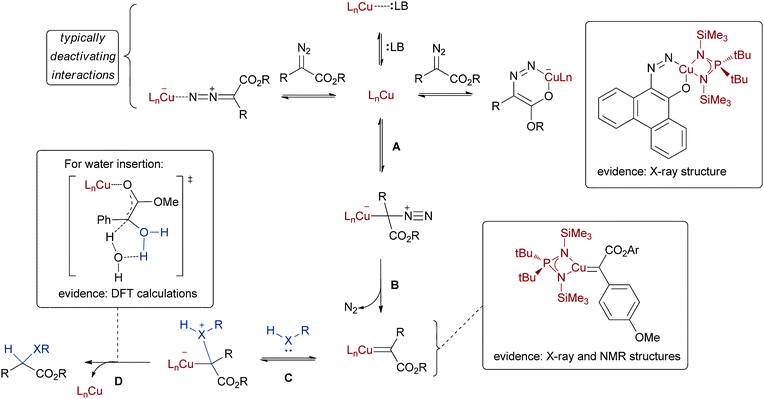 | ||
| Scheme 3 Mechanistic analysis of copper(I) catalysed XHI reaction. | ||
As mentioned earlier, it seems a number of the transition states in the elementary steps for XHI with copper complexes are close in energy. Fructos and coworkers have discovered an example of how this can be exploited to control selectivity in copper–carbenoid transfer.17 A common problem in metal–carbenoid reactions is a rapid dimerization of the diazo starting material. However, they have shown that if the catalyst is the copper(I)-N-heterocyclic carbene complex shown in Scheme 4 no reactivity towards ethyldiazoacetate (EDA) is observed (top line in Scheme 4). Only when both substrates are present does a rapid consumption of EDA ensue, leading to the expected XHI or cyclopropanation products (bottom line in Scheme 4). An explanation for these observations might be that coordination of the substrate lowers the energy of the transition state leading to copper–carbenoid. This is a stark contrast to most other catalysts, where coordination typically inhibits carbenoid formation. Although no further mechanistic studies have been reported on this system, these observations highlight the ability of ligands to dictate the course of catalytic reactions.
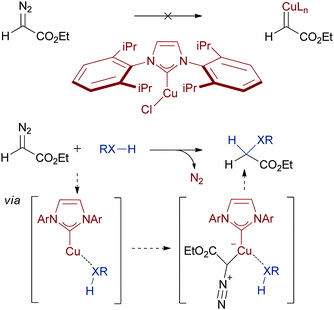 | ||
| Scheme 4 Controlling copper catalysis with ligands. | ||
3.2 Mechanistic studies of rhodium(II)-catalysed XHI
Detecting intermediates in the rhodium(II)-catalysed reaction has proven more difficult than with copper. Therefore the bulk of the current mechanistic understanding with rhodium(II) comes from computational studies and kinetic analysis. A detailed kinetic treatment of various rhodium(II) complexes by Pirrung and Morehead has shown that only one of the two available axial coordination sites is catalytically active at any given time (see Scheme 5).13 They also found that Lewis basic inhibitors may occupy the second coordination site, and that this interaction impedes catalysts. Although they did not study XHI processes, their results are particularly pertinent to the current topic because substrates for XHI necessarily contain a Lewis basic and therefore potentially inhibitory functional group. An outline of the kinetic scenario that addresses inhibition is shown in Scheme 5. Although the main results of Morehead and Pirrung's kinetic analysis are simple to understand, the arrival at these results is reasonably complex and may require the reader to review mixed inhibition enzyme kinetics.18 In brief, the starting rhodium(II) complex has two vacant axial coordination sites that can either bind to the substrate (Km), or be inhibited by Lewis bases (I in Scheme 5, paths Ki1 and Ki2). The substrate-bound rhodium complex (from path Km) can then lose nitrogen to deliver the rhodium–carbenoid (path kcat). Alternatively, a rhodium complex where one axial site is bound by an inhibitor and the other a substrate molecule could also ultimately lead to the requisite rhodium–carbenoid (βkcat). A kinetic analysis of initial reaction rates at various inhibitor and substrate concentrations allows one to tease out the important parameters in Scheme 5. They showed that the βkcat pathway does not contribute to catalysis, and that the αKi1 pathway is a minor contributor to the total reaction flux, at least at inhibitor concentration of 50 mM or less. Hence, most catalytic turnovers occur according to the path at the top of Scheme 5. These results are important for XHI because it may be that in the presence of the strongly Lewis basic substrates required for XHI the Ki1 → αKm → αKi1 → kcat pathway is a significant contributor to catalysis. Finally it is important to note that in all reactions they examined the loss of nitrogen was the RDS.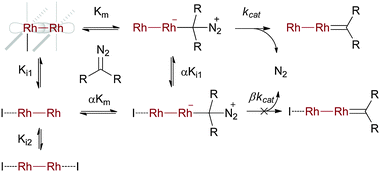 | ||
| Scheme 5 Kinetic analysis of the effect of inhibitors on rhodium-catalysed reactions. | ||
Although they have never been observed, the relevance of ylides in rhodium(II)-based XHI processes (step C in Scheme 2) can be inferred from examples where the putative ylide intermediate has been trapped with imine electrophiles19 or even coaxed into rearrangement pathways rather than the [1,2]-proton shift shown in step D of the mechanistic scheme.20 These experiments not only provide evidence for the existence of ylide intermediates, but also suggest that, in line with computational results,14 the [1,2]-proton shift has a surprisingly high activation barrier.
3.3 Mechanistic studies on iron(III)-catalysed XHIs
Although iron has been used less extensively than copper and rhodium for catalytic XHI, the iron(III)–corrole and porphyrin systems developed independently by Gross21 and Woo22 for N–H insertion indicate that iron(III) catalysis proceeds by a unique mechanism. A carbenoid-free pathway is favoured by Gross, while a ligand acceleration mechanism is advanced by Woo.As shown in Scheme 6 the iron(III) complexes can deliver N–H insertion products with up to 1000 turnovers in a matter of minutes. Intriguingly, EDA in the presence of the iron catalyst is completely unreactive. Only when the amine is added does a rapid evolution of nitrogen occur. In addition other classes of substrates that typically participate in reactions with metal–carbenoids, such as olefins or alcohols, are either completely unreactive or many orders of magnitude slower than the N–H insertion process. The possibility that the nitrogen-containing substrate is simply an ancillary ligand that promotes rapid carbene formation seems unlikely given that competition experiments where olefins and amines are present lead almost exclusively to N–H insertion.21 Gross contends that this suggests a carbenoid-free pathway involving direct nucleophilic displacement by the amine of the dinitrogen leaving group on an iron-bound EDA molecule. On the other hand, Woo has observed that homo-coupling of EDA is accelerated in the presence of Lewis basic amines, and this would seem to support an amine-accelerated carbenoid pathway.22 Further experiments are needed to clarify this mechanistic dichotomy.
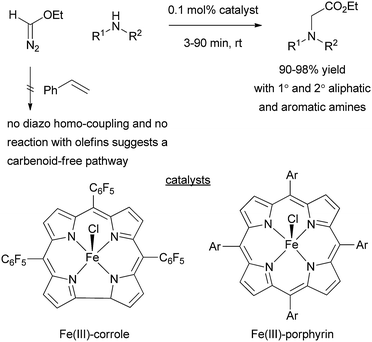 | ||
| Scheme 6 Iron(III) corrole and porphyrin complexs display unique reactivity consistent with a non-carbenoid pathway, or a carbene pathway that is activated by the presence of amine ligands. | ||
4. General points on catalysts for XHI
Shown in Fig. 2 is the periodic table of carbenoid insertion catalysts.23 Each metal in Groups 8–11 has been used to generate and transfer carbenoids. The font scaling in the table is meant to convey a qualitative assessment of how broadly applied each metal has been in catalytic reactions; elements in the smallest font (Co, Ni, Pd, Pt, Os, Ir) have been applied in carbenoid chemistry but have seen little or no use in XHI. Clearly many metals are active in the reaction, but it is copper(I) and rhodium(II) that have proven, as yet, most versatile.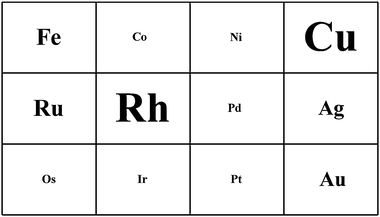 | ||
| Fig. 2 Metals for carbenoid-transfer reactions. The font scaling is meant to qualitatively convey the effectiveness of each metal. | ||
Rh2(OAc)4 was first used by Teyssie in 1973 to decompose EDA to produce a rhodium–carbenoid intermediate, subsequent O–H insertion then delivers ethers.11 Later they reported that rhodium carboxylates were also efficient catalysts for N–H and S–H insertion.24 These seminal observations set the stage for forty years of intensive study with rhodium(II) catalysts. As a result the substrate scope has been widely expanded to include aliphatic amine, aniline, amide, alcohols, phenols, thiols and silanes (see Table 1).3
| Metal |
|
|
|
Strengths | Weaknesses |
|---|---|---|---|---|---|
| a The narrow substrate range with these metals may simply be a reflection of underdevelopment. | |||||
| Rh |

|

|

|
• High turn-over numbers and frequencies | • Expensive |
| • Broad substrate scope | • Competing C–H insertion and β-elimination | ||||
| • Biomolecule modification | • Moderate stereocontrol | ||||
| Cu |

|

|

|
• Cheap | • Susceptible to inhibition by Lewis bases |
| • Chemoselective | |||||
| • Enatioselective variant available | |||||
| • Reactivity highly sensitive to ligands | |||||
| Rua |

|

|

|
• Chemoselective | • Expensive |
| • Rich coordination chemistry | • Narrow substrate scopea | ||||
| Fea |

|

|

|
• Cheap | • Narrow substrate scopea |
| • Low toxity | |||||
| • Reactivity highly sensitive to ligands | |||||
| • Enantioselective variant available | |||||



Attesting to the practicality of rhodium(II) systems is the Merck synthesis of thienamycin shown in panel D of Fig. 1. Nevertheless, despite such early promise, rhodium(II) chemistry has not been a panacea for XHI applications. The seeming preference for a metal-free ylide pathway (see Fig. 3) has thwarted attempts to create enantioselective processes. Additionally the facility with which rhodium carbenoids undergo C–H insertion29 and β-elimination30 is a problem with certain substrates.
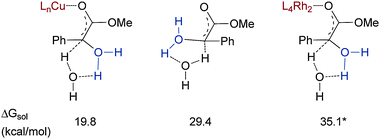 | ||
| Fig. 3 Comparison of the metal-associated and metal-free transition states for proton transfer in copper- and rhodium-catalysed O–H insertion. *Note that the energy of the rhodium-associated species has been adjusted to place it on the same energy scale as the copper structure. | ||
The above mentioned issues with rhodium have led to a renaissance in the application of copper catalysts to tackle current unsolved problems in XHI. Although first reported to be useful for XHI reactions in the early fifties,7,8 XHI chemistry with copper remained underutilized until recently – likely as a result of the harsh reaction conditions, low insertion yields, and sparing solubility of the copper complexes. Research with copper was therefore largely abandoned for rhodium(II) catalysts. In 2002, a report from Pérez and co-workers demonstrated that copper(I) complexes with homoscorpionate ligands could catalyse the insertion of EDA into N–H bonds of amines and amides in high yields under mild conditions.31 The electronic interaction between the copper and the heterocyclic ligand not only enhanced its stability, but also improved its reactivity and selectivity in the XHI reaction as a result of its unique structure. The great strides made in catalysis over the past fifty years are leveraged largely on the development of new ligands that can tune the properties of its bound metal. The report that the homoscorpionate ligands can improve copper(I)–carbenoid chemistry served to remind the community that these catalysts had a great deal of unrealized potential. Since then developments with copper(I) have been rapid and substantial.6,25,32
Ruthenium, one of rhodium's direct neighbours in the periodic table, was first introduced to catalyze ethyl diazoacetate insertion into S–H and N–H bonds in 1997.33 Recent results on ruthenium catalysed N–H34 and O–H insertion35 reactions, reported by the Che and Lacour groups respectively, demonstrate that ruthenium complexes can sometimes offer unique reactivity in comparison with other catalysts. Although a relative newcomer in XHI, the favourable properties of ruthenium (its similar reactivity to rhodium, lower cost, more available oxidation states, and rich coordination chemistry) suggest a bright future.
Given the costs associated with the rarer transition metals, processes based on iron would be well-received by potential users. The Woo and Gross groups have independently shown that iron(III)–corrole and iron(III)–porphyrin complexes are excellent catalysts for N–H insertion into a variety of amines and diazo substrates.21,36 Furthermore, the recent application of iron–spirobisoxaline complexes in highly efficient enantioselective O–H insertion should stimulate further developments with this practical alternative to the precious metal catalysts.37
The substrate range and specificity of rhodium(II)5 and copper(I)38 systems have already been reviewed or are covered in other sections of this review. Therefore the remainder of this section will simply give an overview in tabular format of the breadth of substrates and reaction types each catalyst has so far been applied to (see Table 1).
5. Enantioselective XHI processes
Enantioselective XHI processes have been advanced a great deal in the last five years. Since both the early work5 and recent developments in this area have been reviewed,38 the following section will attempt more to shed light on crucial aspects of the process rather than a blow-by-blow of developments. The earliest example of an asymmetric XHI process is the chiral auxiliary approach of Kagan.39 They determined that copper cyanide promoted N–H insertion into substrates containing chiral ester side-chains such as menthyl or phenylethylamine leading to amino acid products in up to 26% optical purity. Strangely, after this report research into copper-based processes for asymmetric XHI ended for almost two decades. It was in 2004 when Bachmann and coworkers demonstrated up to 27% ee in N–H insertions catalysed by copper(I)–bisoxazoline complexes that the community re-evaluated the potential of copper(I).32 Again it was likely the discovery of the incredible catalytic abilities of rhodium(II) acetate that focused the field's attention on creating asymmetric catalysts based on rhodium(II).11 Additionally, over the ensuing years the successes with chiral rhodium catalysts for enantioselective C–H insertion probably gave the impression that an efficient enantioselective XHI was tantalizingly close.40 Ultimately a renaissance in copper-based approaches has been the key to achieving highly enantioselective XHI reactions. A tenable explanation for the poor ability of rhodium(II) complexes in promoting enantioselectivity in O–H insertion has been put forth by the Yu group based on DFT calculations (B3LYP functional) of both the copper(I) and rhodium(II)-catalysed reaction paths.14 They found that a crucial difference in the stereogenesis is that whereas copper complexes prefer to remain bound to the substrate during protonation, the rhodium catalysts favour dissociation and hence cannot transfer chiral information. Shown in Fig. 3 are the calculated free energies of the proton transfer transition states that led to this mechanistic hypothesis. The copper-associated structure is nearly 10 kcal mol−1 lower in energy than the metal-free transition state, which in turn is approximately 6 kcal mol−1 lower in energy than the rhodium associated transition state.Whatever the mechanistic rationale, chiral copper complexes are currently the best choice for achieving a variety of enantioselective XHI processes. Shown in Table 2 are the ligand systems that have proven most effective thus far.6,25–28 From their complex structures it is clear that one of the drawbacks with current methods is that the ligand syntheses are demanding. Before a method can become practical, however, it is first essential that the concept is established, and the systems shown have indeed opened the door to new developments in the area of XHI with copper catalysts.
| a See ref. 25. b See ref. 26. c See ref. 6. d See ref. 27. e See ref. 28. | |||||
|---|---|---|---|---|---|
| Ligand |

|

|

|

|

|
| Preferred metal salt | Cu(OTf)2 | CuBr | CuCl or Cu(MeCN)4PF6 | (CuOTf)2·PhH | CuCl |
| Reactions studied | O–H insertiona | N–H insertionb | O–H,c N–H,c and S–H insertionc | O–H insertiond | N–H insertion (1° and 2° amines)e |
| ee range (%) | 20–98 | 80–94 | Typically 60–90 | 50–90 | 70–98 |
The spiro-bisoxazoline ligands developed by Zhou have proven especially versatile in XHI processes.6 A look at the minimized 3D structures and the conformation that would be required for bidentate chelation with copper(I) presents a perplexing picture. In the minimal energy conformer (left-most structure in Fig. 4) the nitrogens prefer to be more than 6 Å apart, precluding bidentate chelation. In the rotomer that would be required for copper chelation there is only 3.0 Å between the two nitrogens (middle structure in Fig. 4). Based on typical Cu–N bond lengths in similar complexes, this space would be a tight squeeze for copper coordination. With most ligands a simple conformational adjustment would readily occur to accommodate metal binding, but the rigid spiro motif in these ligands prohibits this possibility. Recently the Zhou group has put forth an explanation for this peculiarity that involves a bimetallic copper complex which creates a C2-symmetric environment with the help of two ligand molecules (right-most structure in Fig. 4).6 The Cu–Cu distance in the X-ray structure is 2.78 Å, suggesting the possibility of a cooperative role for both metal centres in catalysis. Certainly two ligand molecules are involved since a small but measurable positive non-linear effect is observed. A kinetic analysis of the Zhou system such as the one conducted by Pirrung and Morehead13 for rhodium(II) would be of interest since it could identify whether both coppers were independently active, cooperative, or susceptible to mixed inhibition.
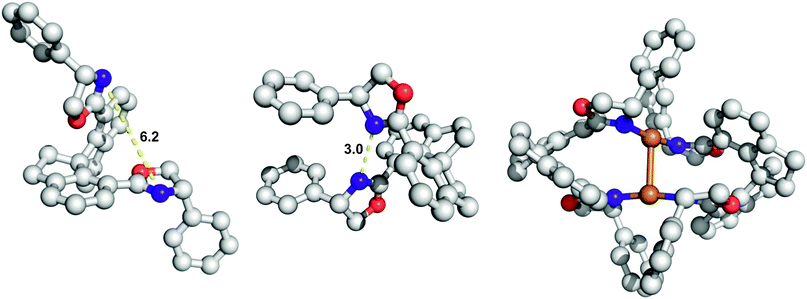 | ||
| Fig. 4 Right: lowest energy conformer for the Zhou ligand (MM2 minimization). Middle: oxazole rotomers that would be required for bidentate chelation to copper. Right: crystal structure of the bimetallic complex determined by Zhou. | ||
Despite the preference for the metal-free ylide pathway in XHI, enantioselective processes based on rhodium(II) may yet be realized. A recent report demonstrating that an achiral rhodium(II) carbene can be intercepted by an imine activated with a chiral Bronsted acid indicates that cooperative rhodium(II)–Bronsted acid catalysis may offer an alternative approach towards enantioselective XHI.42 Indeed Saito and coworkers have found that cinchona alkaloid additives can deliver enantiomeric excesses of up to 50% in the O–H insertion of α-diazoesters with water catalysed by achiral rhodium(II) catalysts.41 The Zhou group have also recently discovered that BINOL-based chiral phosphoric acids deliver high levels of enantioinduction in N–H insertion reactions.42 These promising initial findings suggest that XHI coupled with enantioselective protonation is an area poised for further development.
6. Application of metal–carbenoid XHI in chemical biology
6.1 Challenges in chemical biology
Chemical Biology uses the concepts and tools of chemistry to study, perturb, or retool biological systems. Although this field has been around for decades in a variety of guises, it has only recently come-of-age and settled into its own field of study. This maturing is, as is always the case in scientific progress, coupled with advances in other fields. Today it is possible to carry out precise chemical reactions on large complex molecules (e.g. in proteins or carbohydrates) in increasingly complex settings (e.g. in the cytosol, or on the cell surface). The technical hurdles that have historically hampered the development of such chemistry have been removed by advancements in synthetic chemistry, molecular biology, and analytical techniques such as NMR, X-ray crystallography, HPLC analysis, and fluorescence spectroscopy. For example, today a chemist can take a native protein, modify it with some chemical reaction and then characterize at atomic resolution the site of modification often in a matter of days.Despite these advances, an area where significant development is still required is in the controlled chemical modification of native biomolecules such as proteins, carbohydrates, and nucleic acids. Bioorthogonal chemistry, first articulated by Bertozzi, is an approach that involves introducing unnatural functional groups into target substrates.43 The selective reaction between these functional groups must not be seen in natural systems, and must not interfere with endogenous biological chemistry. Once the bioorthogonal functional group is introduced into the biomolecular target, the selective chemical reaction can then be used to label, modify, probe, or pull-down the molecule of interest. While this strategy has proven useful, the inherent need to create bioorthogonal functional groups in both reaction partners can be cumbersome. In natural systems a great variety of enzymes act simultaneously on their cognate substrates without interfering with one another; such complete catalyst control is the ideal form of reaction orthogonality. Given the right catalysts and ligands it should be possible for chemists to imitate the kind of selectivity achieved by enzymes, but progress in creating artificial catalysts to selectively target native biomolecular structures has thus far been modest. Nevertheless, much of the progress achieved to date has been with catalysts for XHI.
6.2 Application of XHI in selective protein labeling
Examination of protein active sites by diazo modification is nearly as old as the discovery of copper–carbenoid chemistry. Rajagopalan and coworkers44 as well as Delpierre and Fruton45 discovered almost simultaneously that the active-site carboxyl residue of pepsin is modified with diazo compounds at least 103–104 fold faster in the presence of large excesses of copper(II) salts (see Fig. 6). They used this selective reaction to aid in mechanistic studies on the role of the carboxylate residues in pepsin catalysis. Intriguingly the reaction does not proceed efficiently with just any carboxylate, only the active site aspartate that has the unusually high pKa characteristic of aspartate proteases is reactive. Although the mechanistic underpinnings of the reaction were unknown at the time, such reactivity with carboxylates is reasonable considering that XHI would prefer the protonated form of the acid. These authors also observed a short induction period before protein modification began, consistent with the necessity to generate copper(I) from a sacrificial diazo-coupling before effective catalysis can ensue.10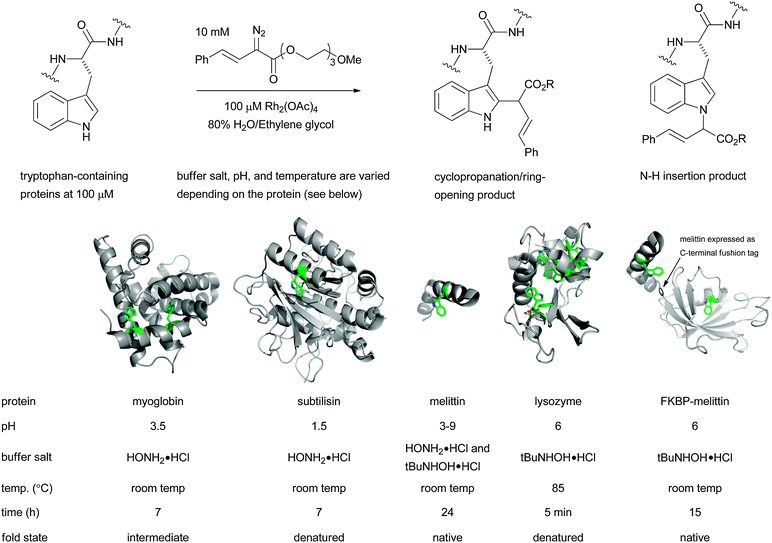 | ||
| Fig. 5 Rhodium catalysed tryptophan labelling can be used to modify a variety of proteins. | ||
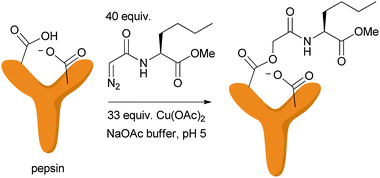 | ||
| Fig. 6 Earliest example of carbenoid chemistry to modify proteins. | ||
It was many years later before the potential of rhodium(II) complexes to promote XHI reactions for applications in chemical biology was recognized. Antos and Francis reported in 2004 that tryptophans in myoglobin and subtilisin could be modified at low pH using α-diazo esters and dirhodium tetraacetate (see Fig. 5).46 The reaction seems to proceed by a mixture of XHI and cyclopropanation of the tryptophan indole moiety. During this study they also discovered that hydroxylamine hydrochloride was a uniquely effective buffer salt for the reaction. They speculated that its role was to bind the metal centre and stabilize reactive intermediates. In their follow-up study they identified t-butylhydroxyl amine as an even better buffer salt and with it they could carry out reactions over a broad pH range (3–9) with little loss in efficiency or selectivity.46 Again the role of t-butylhydroxyl amine remains mysterious. Their new reaction conditions lead to smooth alkylation of tryptophan residues on a variety of proteins, including a case with FKBP where a tryptophan-containing C-terminal melittin tag was engineered into a protein to allow selective labelling with rhodium–carbenoid chemistry (see Fig. 5). This strategy is conceptually similar to the classical approach of introducing a cysteine into a target protein for subsequent reaction. For the metal-mediated tryptophan labelling to proceed efficiently its side-chain must be solvent accessible. This can be achieved by selecting proteins with solvent accessible tryptophans, or by denaturing with pH or temperature.
While the selective alkylation of tryptophans is an important advance, the Francis system requires large amounts of the rhodium complex and is limited to tryptophans. It would be ideal to be able to modify nearly any desired amino acid side-chain with the site-selectivity controlled by the catalyst. The Ball lab has developed a technique where the reactive rhodium–carbenoid is generated with a rhodium complex bound between two carboxylate residues in a α-helix.47 The interaction of the catalyst containing helix with its partner helix in a coiled coil motif brings the catalyst into a microenvironment completely controlled by the coiled coil registry (see Fig. 7). The local concentration of the rhodium vis-à-vis its binding partner is in the molar range, although its concentration in bulk solution is high micromolar. The rate accelerations achieved through high effective molarity allowed these researchers to target not only aromatic residues, but also nearly every amino acid side-chain that contains a nucleophilic hetereoatom. It is important to reiterate that although many amino acid side-chains can be targeted, selectivity can be precisely controlled through applying the well understood binding rules of the coiled coil motif. When they tested variants where the helical-wheel model predicted a large distance between the reactive side-chain and the catalyst no reaction was observed (see top of Fig. 7). The Ball system leverages proximity induced catalysis as the control element rather than bioorthogonal reactivity.47 Therefore the potential applications of this technique are numerous. In principle modifying any new protein of interest would only require identifying a ligand that binds specifically to the target region in the protein. While this may sound difficult it is important to bear in mind that computational methods are at the level of being able to predict with high degrees of confidence protein tertiary structure and binding interactions.48
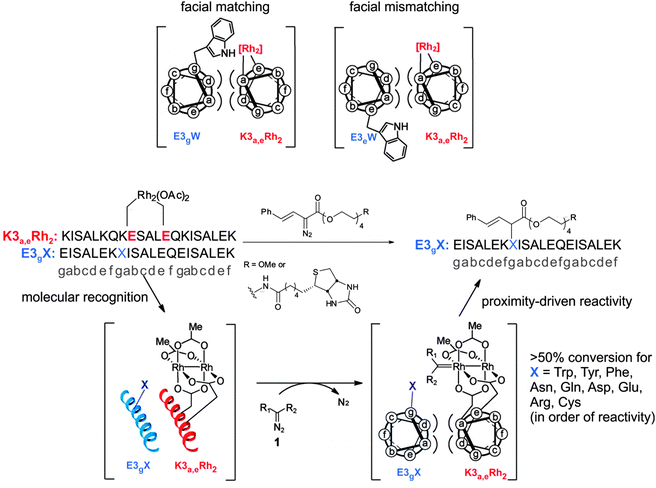 | ||
| Fig. 7 Top: the coiled–coil interaction induces proximity between catalyst and substrate in the matched case, and disposes them away from each other in the mismatched case. Bottom: this proximity-induced selectivity enables the specific targeting of a variety of protein side-chains (adapted with permission from ref. 47). | ||
Ho and coworkers have developed a ruthenium(II) porphyrin complex for XHI reactions in aqueous media and applied it to the modification of the N-terminus or reactive cysteines in peptides and proteins.49 The large hydrophobic porphyrin core of this complex is rendered water soluble by virtue of four pendant β-D-glucose moieties. The system is highly active, delivering complete conversion in 1 h at biological pH and temperature with micromolar concentrations of both substrates and only 10 mol% of the catalyst. The ruthenium(II) porphyrin is not only effective for XHI but also a variety of other metal–carbenoid reactions. Unlike the iron–porphyrin system it seems that reactions catalysed by this ruthenium–porphyrin complex proceed by a metal–carbenoid intermediate.
6.3 Application of XHI in nucleic acid labeling
Our group recently reported that rhodium–carbenoids can target the exocyclic N–H group of various nucleic acids (see Table 3).50 Introducing unnatural chemical motifs into native nucleic acid structures is important for a myriad of reasons. For example recent work has shown that chemical changes in the nucleobases of short interfering RNAs can alter their susceptibility to nucleases as well as their detection by the immune system.51 In addition, the discovery that DNA and RNA methylation patterns are an important component of gene regulation (i.e. epigenetics) means that chemical methods to selectively target nucleic acid structures will be of great value in studying and perturbing this regulation pathway.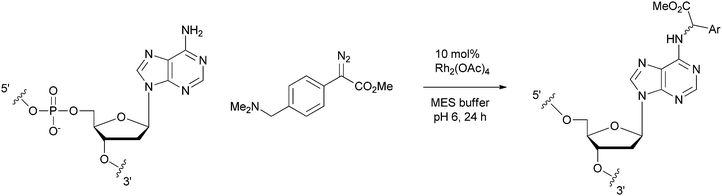
| Entry | Sequence | Structure | Conva (%) | Product | Significance |
|---|---|---|---|---|---|
| a Conversion based on the oligonucleotide; the diazo substrate is typically completely consumed when nucleic acids are present. b Diazo substrate also not consumed. | |||||
| 1b | d(T)4 | Single-stranded | 0 | No reaction | Ts are not alkylated |
| 2b | r(U)4 | Single-stranded | 0 | No reaction | Us are not alkylated |
| 3 | d(TAT) | Single-stranded | 20 | N6-A alkylation | As are targeted |
| 4 | d(TCT) | Single-stranded | 16 | N4-C alkylation | Cs are targeted |
| 5 | d(TTTATTTGTTTCTTT) | Single-stranded | 37 | Multiple alkylations | Longer sequences can be targeted |
| 6 | d(CGAACGTTTTTCGTTCG) |

|
0 | No reaction | Double-stranded sequences are unreactive |
| 7 | d(CGAACGTTTTTCGTTCGA) |

|
21 |
|
3′ Overhang regions can be targeted |
| 8 | r(CUAGCAUUUUUUGCUAGA) |

|
33 |
|
Short hairpin RNAs can be targeted |
The dirhodium(II) tetraacetate catalyst we employed was selective for exocyclic N–Hs in the nucleobases, but otherwise delivers unspecific alkylation in single-stranded nucleic acids (compare entries 1–5 in Table 3). In contrast studies on hairpin sequences revealed that double-stranded stretches were unreactive (entry 6). It seems that if the N–H bonds are engaged in Watson–Crick base-pairing they are unavailable for reaction with the rhodium. This selectivity profile was exploited to target certain unpaired regions in DNAs and RNAs (see entries 7 and 8).
The turn-over frequency of the rhodium(II) catalyst was dramatically affected by the nucleic acid sequence. For example while reaction with d(TAT) required 24 h to deliver complete consumption of the diazo substrate, reaction with d(TCT) under otherwise identical conditions led to complete diazo consumption in 3 h. This is not simply an effect of the inherent nucleophilicity of the substrate since a mix experiment with both substrates also leads to sluggish conversion of the diazo compound. Based on the kinetic analysis of rhodium(II) catalysis shown in Scheme 4, these results suggest that either the pre-equilibriums leading up to catalysis, or the αKil or βkcat turnover pathways are affected by nucleic acid ligands. We are currently investigating if and to what extent these possibilities are operative.
6.4 XHI for synthesis of small-molecule probe compounds
The study of enzyme and protein chemistry has been greatly facilitated by the availability of small-molecules probes that can bind reliably to a protein of interest. The standard design principle is to select a molecule that binds the target protein and then append a reporter group. Often these reporter tags contain things like fluorophores, a biotin tag, radiolabels, isotopic labels, photoactivatable groups, or digoxygenin – all molecular moieties that open the door to a variety of powerful analytical techniques. Chemical methods to create these binder-reporter conjugates must be simple and reliable. The advent of the copper-catalysed variant of the Huisgen cycloaddition has had a substantial impact on this area of research. As mentioned in Section 6.1 this method does have the drawback that both substrates need to be chemically manipulated to incorporate azide and alkyne motifs in preparation for the conjugation reaction. Reactions that target common functional groups in a selective way would represent a valuable contribution to this area. Moreover, the availability of a battery of reliable yet chemically orthogonal conjugation reactions also enables the creation of multifunctional reporter probes.The Romo group has recently developed a selection of diazo substrates designed for use in the synthesis of probe molecules in conjunction with XHI.52 They demonstrated that rhodium–carbenoid based O–H and N–H insertion can be used to create functional conjugates with a variety of biologically active natural products (see Fig. 8). Furthermore, the ester side-chains of the diazo substrates included alkynyl groups to facilitate further modification by the copper-catalysed azide alkyne cycloaddition. Shown in Fig. 8 is the collection of natural products that have been demonstrated to be compatible with this technique. Most of these molecules contain multiple potential reactive sites and yet practical yields of the mono-etherification products can be obtained.
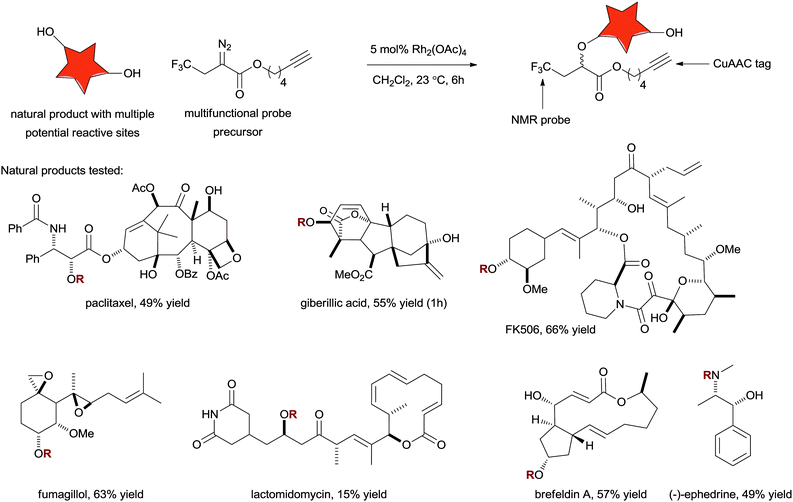 | ||
| Fig. 8 An assortment of complex natural products with diverse functionality can be converted to probe compounds in a single step using Rh-catalysed O–H and N–H insertion. | ||
7. Conclusions
Despite its long history the full potential of the XHI reaction is unrealized. Although the earliest observations of XHI were made with copper, up until the end of the nineties rhodium(II) catalysts were almost exclusively employed. A consequence of this extensive development is that the limitations of rhodium(II) catalysts are well delineated. The renaissance of copper(I) catalysts and the discovery of the unique reactivity of ruthenium(II) and iron(III) are therefore timely developments which may enable researchers to overcome the unmet challenges of XHI with rhodium(II). The cases described in this review where a certain ligand dramatically alters a catalyst's reactivity or selectivity or both reveal a second parameter that can be adjusted to control XHI, reiterating the significance of ligand development for tackling challenges in any catalytic reaction.Chemical intuition might suggest that metal–carbenoids are too reactive to be exploited in complex environments and/or with protic solvents. Counter to this intuition are the recent developments applying XHI to the modification of intact proteins, nucleic acids, and complex natural products. These results hint at a prolific future for the XHI reaction in fields such as chemical biology and nanotechnology, where selective catalysis in complex settings is essential.
Notes and references
- Catalyzed Carbon–Heteroatom Bond Formation, ed. A. Yudin, Wiley-VCH, Weinheim, 2010 Search PubMed.
- C. J. Moody, Angew. Chem., Int. Ed., 2007, 46, 9148–9150 CrossRef CAS.
- D. J. Miller and C. J. Moody, Tetrahedron, 1995, 51, 10811–10843 CrossRef CAS and references therein.
- T. N. Salzmann, R. W. Ratcliffe, B. G. Christensen and F. A. Bouffard, J. Am. Chem. Soc., 1980, 102, 6161–6163 CrossRef.
- M. P. Doyle and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley, New York, 1998 Search PubMed.
- S.-F. Zhu and Q.-L. Zhou, Acc. Chem. Res., 2012, 45, 1365–1377 Search PubMed and references therein.
- R. Casanova and T. Reichstein, Helv. Chim. Acta, 1950, 33, 417–422 Search PubMed.
- P. Yates, J. Am. Chem. Soc., 1952, 74, 5376–5381 CrossRef CAS.
- H. Nozaki, H. Takaya, S. Moriuti and R. Noyori, Tetrahedron, 1968, 24, 3655–3669 CrossRef CAS and references therein.
- R. G. Salomon and J. K. Kochi, J. Am. Chem. Soc., 1973, 95, 3300–3310 CrossRef CAS.
- R. Paulissen, H. Reimlinger, E. Hayez, A. J. Hubert and P. Teyssié, Tetrahedron Lett., 1973, 14, 2233–2236 CrossRef.
- J. X. Gong, G. A. Lin, W. B. Sun, C. C. Li and Z. Yang, J. Am. Chem. Soc., 2010, 132, 16745–16746 CrossRef CAS.
- M. C. Pirrung, H. Liu and A. T. Morehead, J. Am. Chem. Soc., 2002, 124, 1014–1023 CrossRef CAS.
- Y. Liang, H. Zhou and Z.-X. Yu, J. Am. Chem. Soc., 2009, 131, 17783–17785 CrossRef CAS.
- J. M. Fraile, J. I. García, V. Martínez-Merino, J. A. Mayoral and L. Salvatella, J. Am. Chem. Soc., 2001, 123, 7616–7625 CrossRef CAS.
- I. V. Shishkov, F. Rominger and P. Hofmann, Organometallics, 2009, 28, 1049–1059 CrossRef CAS and references therein.
- M. R. Fructos, T. R. Belderrain, M. C. Nicasio, S. P. Nolan, H. Kaur, M. M. Díaz-Requejo and P. J. Pérez, J. Am. Chem. Soc., 2004, 126, 10846–10847 CrossRef CAS.
- A. Cornish-Bowden, Fundamentals of Enzyme Kinetics, Wiley-VCH, Weinheim, 4th edn, 2012 Search PubMed.
- H. Huang, X. Guo and W. Hu, Angew. Chem., Int. Ed., 2007, 46, 1337–1339 CrossRef CAS.
- Z. Li, B. T. Parr and H. M. L. Davies, J. Am. Chem. Soc., 2012, 134, 10942–10946 Search PubMed.
- I. Aviv and Z. Gross, Chem.–Eur. J., 2008, 14, 3995–4005 CrossRef CAS.
- H. M. Mbuvi, E. R. Klobukowski, G. M. Roberts and L. K. Woo, J. Porphyrins Phthalocyanines, 2010, 14, 284–292 Search PubMed and references therein.
- M. R. Fructos, T. R. Belderrain, P. de Frémont, N. M. Scott, S. P. Nolan, M. M. Díaz-Requejo and P. J. Pérez, Angew. Chem., Int. Ed., 2005, 44, 5284–5288 CrossRef CAS.
- R. Paulissen, E. Hayez, A. J. Hubert and P. Teyssie, Tetrahedron Lett., 1974, 15, 607–608 CrossRef.
- T. C. Maier and G. C. Fu, J. Am. Chem. Soc., 2006, 128, 4594–4595 CrossRef.
- E. C. Lee and G. C. Fu, J. Am. Chem. Soc., 2007, 129, 12066–12067 CrossRef CAS.
- T. Osako, D. Panichakul and Y. Uozumi, Org. Lett., 2011, 14, 194–197 Search PubMed.
- Z. Hou, J. Wang, P. He, J. Wang, B. Qin, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2010, 49, 4763–4766 CrossRef CAS.
- C. J. Moody and R. J. Taylor, Tetrahedron Lett., 1987, 28, 5351–5352 CrossRef CAS.
- T. Goto, K. Takeda, N. Shimada, H. Nambu, M. Anada, M. Shiro, K. Ando and S. Hashimoto, Angew. Chem., Int. Ed., 2011, 50, 6803–6808 CrossRef CAS.
- M. E. Morilla, M. M. Diaz-Requejo, T. R. Belderrain, M. C. Nicasio, S. Trofimenko and P. J. Perez, Chem. Commun., 2002, 2998–2999 RSC.
- S. Bachmann, D. Fielenbach and K. A. Jorgensen, Org. Biomol. Chem., 2004, 2, 3044–3049 RSC.
- E. Galardon, P. Le Maux and G. Simonneaux, J. Chem. Soc., Perkin Trans. 1, 1997, 2455–2456 RSC.
- Q.-H. Deng, H.-W. Xu, A. W.-H. Yuen, Z.-J. Xu and C.-M. Che, Org. Lett., 2008, 10, 1529–1532 Search PubMed.
- M. Austeri, D. Rix, W. Zeghida and J. Lacour, Org. Lett., 2011, 13, 1394–1397 CrossRef CAS.
- L. K. Baumann, H. M. Mbuvi, G. Du and L. K. Woo, Organometallics, 2007, 26, 3995–4002 CrossRef CAS.
- S.-F. Zhu, Y. Cai, H.-X. Mao, J.-H. Xie and Q.-L. Zhou, Nat. Chem., 2010, 2, 546–551 CrossRef CAS.
- X. Zhao, Y. Zhang and J. Wang, Chem. Commun., 2012, 48, 10162–10173 RSC.
- J.-F. Nicoud and H. B. Kagan, Tetrahedron Lett., 1971, 12, 2065–2068 CrossRef.
- H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861–2904 CrossRef CAS.
- H. Saito, R. Iwai, T. Uchiyama, M. Miyake and S. Miyairi, Chem. Pharm. Bull., 2010, 58, 872–874 CrossRef CAS.
- B. Xu, S.-F. Zhu, X.-L. Xie, J.-J. Shen and Q.-L. Zhou, Angew. Chem., Int. Ed., 2011, 50, 11483–11486 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS.
- T. G. Rajagopalan, W. H. Stein and S. Moore, J. Biol. Chem., 1966, 241, 4295–4297 Search PubMed.
- G. R. Delpierre and J. S. Fruton, Proc. Natl. Acad. Sci. U. S. A., 1966, 56, 1817–1822 Search PubMed.
- J. M. Antos, J. M. McFarland, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2009, 131, 6301–6308 CrossRef CAS and references therein.
- Z. T. Ball, Acc. Chem. Res., 2012 DOI:10.1021/ar300261h , and references therein.
- S. J. Fleishman, T. A. Whitehead, D. C. Ekiert, C. Dreyfus, J. E. Corn, E.-M. Strauch, I. A. Wilson and D. Baker, Science, 2011, 332, 816–821 CrossRef CAS.
- C.-M. Ho, J.-L. Zhang, C.-Y. Zhou, O.-Y. Chan, J. J. Yan, F.-Y. Zhang, J.-S. Huang and C.-M. Che, J. Am. Chem. Soc., 2010, 132, 1886–1894 CrossRef CAS.
- K. Tishinov, K. Schmidt, D. Häussinger and D. G. Gillingham, Angew. Chem., Int. Ed., 2012, 51, 12000–12004 Search PubMed.
- H. Peacock, R. V. Fucini, P. Jayalath, J. M. Ibarra-Soza, H. J. Haringsma, W. M. Flanagan, A. Willingham and P. A. Beal, J. Am. Chem. Soc., 2011, 133, 9200–9203 CrossRef CAS.
- S. Chamni, Q.-L. He, Y. Dang, S. Bhat, J. O. Liu and D. Romo, ACS Chem. Biol., 2011, 6, 1175–1181 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |