 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure and kinetics of chemically cross-linked protein gels from small-angle X-ray scattering†
Shuji
Kaieda
*a,
Tomás S.
Plivelic
b and
Bertil
Halle
a
aDepartment of Biophysical Chemistry, Lund University, PO Box 124, SE-22100 Lund, Sweden. E-mail: kaieda.shuji@gmail.com
bMAX IV Laboratory, Lund University, PO Box 118, SE-22100 Lund, Sweden
First published on 23rd December 2013
Abstract
Glutaraldehyde (GA) reacts with amino groups in proteins, forming intermolecular cross-links that, at sufficiently high protein concentration, can transform a protein solution into a gel. Although GA has been used as a cross-linking reagent for decades, neither the cross-linking chemistry nor the microstructure of the resulting protein gel have been clearly established. Here we use small-angle X-ray scattering (SAXS) to characterise the microstructure and structural kinetics of gels formed by cross-linking of pancreatic trypsin inhibitor, myoglobin or intestinal fatty acid-binding protein. By comparing the scattering from gels and dilute solutions, we extract the structure factor and the pair correlation function of the gels. The protein gels are spatially heterogeneous, with dense clusters linked by sparse networks. Within the clusters, adjacent protein molecules are almost in contact, but the protein concentration in the cluster is much lower than in a crystal. At the ∼1 nm SAXS resolution, the native protein structure is unaffected by cross-linking. The cluster radius is in the range 10–50 nm, with the cluster size determined mainly by the availability of lysine amino groups on the protein surface. The development of structure in the gel, on time scales from minutes to hours, appears to obey first-order kinetics. Cross-linking is slower at acidic pH, where the population of amino groups in the reactive deprotonated form is low. These results support the use of cross-linked protein gels in NMR studies of protein dynamics and for modeling NMR relaxation in biological tissue.
1. Introduction
Glutaraldehyde (GA; 1,5-pentanedial) has been widely used during the past 50 years to immobilise and stabilise proteins through covalent intermolecular cross-links.1,2 This bifunctional reagent has been used as a fixative in studies of cell or tissue ultrastructure,3,4 to stabilise protein crystals for X-ray diffraction,5,6 and to characterise the quaternary structure of proteins in solution.7,8 Applications of GA cross-linking in immunochemistry and biotechnology include protein immobilisation on solid carriers, e.g., in affinity chromatography and biosensors, as well as carrier-free immobilisation of enzymes in solution, in amorphous precipitates, or in crystals for use as industrial biocatalysts.9–13The present study was motivated by yet another application of protein cross-linking: water NMR studies of biological systems. In a protein solution, all anisotropic nuclear spin couplings are averaged out by protein tumbling. As a result, the longitudinal relaxation of the water (1H, 2H or 17O) magnetisation only reports on molecular motions faster than the protein's tumbling time (typically, ∼10 ns). Protein cross-linking profoundly alters the NMR conditions, allowing motions on time scales up to hundreds of μs to influence the relaxation. In the NMR context, cross-linked protein gels were first used as model systems for biological tissue,14–18 wherein the proteins are largely immobilised,19 in efforts to elucidate the molecular determinants of the water 1H relaxation that governs contrast in magnetic resonance images of soft tissue. More recently, cross-linked protein gels have been used in 2H and 17O magnetic relaxation dispersion (MRD) studies of intermittent protein dynamics on the ns–μs time scale.20,21
GA reacts primarily with the protein's amino groups, in lysine side-chains and at the N-terminus, although minor involvement of other residues (arginine, histidine, tyrosine and cysteine) has been reported.22–26 Despite extensive study, the details of the reaction mechanism remain poorly understood.1,2 Besides the dialdehyde, an aqueous solution of GA contains several species in equilibrium, including hemihydrate, dihydrate, cyclic hemiacetal, polymeric forms of the hemiacetal and various aldol condensation adducts.27–32 These equilibria, which depend on pH, temperature and concentration, may account for the efficiency of GA as a cross-linking agent by allowing it to form linkers of variable length.
In quantitative MRD studies of protein dynamics, the cross-links should ideally inhibit protein tumbling without affecting the internal (conformational) dynamics of the protein. A necessary condition for this is that the protein structure is unaffected by GA cross-linking, except locally at the chemically modified residues. For protein crystals, X-ray diffraction demonstrates that the structural perturbation caused by cross-linking is indeed local.6,26 For cross-linked protein gels, the evidence is less direct, but the limited results available so far have not revealed any significant differences in internal protein dynamics between gel and solution.20
The protein gels used in MRD studies are formed by adding GA to protein solutions at concentrations where the protein molecules are separated by several water layers.20,21 But even if the protein is amply hydrated on average, the protein molecules may not be uniformly distributed. Cross-linking may well produce a gel structure with dense tightly cross-linked protein clusters connected by more dilute weakly cross-linked networks. Even if such spatial heterogeneity has little effect on the internal dynamics, water dynamics in the first hydration layer on the protein surface would be affected.20,21
To our knowledge, the structure of chemically cross-linked protein gels has not been examined directly. Such studies would have implications for the interpretation of MRD data from cross-linked protein gels and, more generally, would further our understanding of protein cross-linking by GA. A technique suitable for this task is small-angle X-ray scattering (SAXS),33,34 which can provide information about gel structure via the structure factor, essentially the Fourier transform of the protein–protein pair correlation function, as well as about the integrity of the protein's tertiary structure via the form factor.
In protein science, SAXS has proven to be useful for determining the low-resolution structure of monomeric and oligomeric proteins in dilute solution,35,36 but this technique has also been used to study protein–protein interactions in more concentrated solutions37–39 and to obtain structural information about more complex protein systems, such as casein micelles,40 gluten films41 and gels of heat denatured proteins.42
Here, we report SAXS data for GA cross-linked gels of three proteins: bovine pancreatic trypsin inhibitor (BPTI), equine skeletal muscle myoglobin (Mb) and rat intestinal fatty acid-binding protein (IFABP). MRD studies of μs protein dynamics and internal water exchange in these protein gels have already been performed (BPTI and Mb)20,21 or are currently underway (IFABP).43 For each protein, we analyse the scattering intensity profiles in terms of the inhomogeneous protein distribution in the gel. We also report time-resolved SAXS measurements of the cross-linking kinetics.
2. Experimental
2.1. Sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 3 min to remove any insoluble proteins. To prepare cross-linked samples, the protein solution was supplemented with 25% glutaraldehyde solution (Sigma). After vigorous mixing, the solution was transferred to a 1.5 mm o.d. borosilicate capillary (Hilgenberg GmbH) where the cross-linking reaction proceeded at 6 °C. Approximately 50 μl of the solution was reserved for pH measurement. SAXS measurements were also performed on protein solutions without GA. The pH of these solution samples was adjusted to match that of the corresponding cross-linked sample by adding either HCl or NaOH.
000 rpm for 3 min to remove any insoluble proteins. To prepare cross-linked samples, the protein solution was supplemented with 25% glutaraldehyde solution (Sigma). After vigorous mixing, the solution was transferred to a 1.5 mm o.d. borosilicate capillary (Hilgenberg GmbH) where the cross-linking reaction proceeded at 6 °C. Approximately 50 μl of the solution was reserved for pH measurement. SAXS measurements were also performed on protein solutions without GA. The pH of these solution samples was adjusted to match that of the corresponding cross-linked sample by adding either HCl or NaOH.
2.2. SAXS measurements
SAXS experiments were carried out at the I911-4 beamline45 of the MAX-lab synchrotron using a wavelength of 0.91 Å. The sample, contained either in a capillary (gels) or in a flow-through cell (solutions), was maintained at 20 °C or, for the kinetics experiments, at 6 °C. For each protein sample, a pure solvent sample (MilliQ water or buffer solution) was also measured. Two-dimensional SAXS images were recorded using a PILATUS 1 M detector (Dectris) with an exposure time of 10 s (kinetics series for Mb and IFABP) or 60 s (in all other cases). Control measurements were performed to ensure that the results were not compromised by radiation damage. The scattering vector q range (q = 4πsinθ/λ, where λ is the wavelength and 2θ is the scattering angle) was calibrated with a silver behenate sample. Reported scattering profiles I(q) were obtained as the difference of the azimuthally averaged SAXS 2D images from the sample and solvent.
2.3. SAXS analysis
For a sample of NP identical protein molecules of volume VP contained in a volume V, the scattering intensity in the decoupling approximation, where the orientation of a protein molecule is taken to be independent of its position and the configuration of other protein molecules, can be factorised as33,34,46| I(q) = nP(VPΔρ)2P(q)S(q), | (1) |
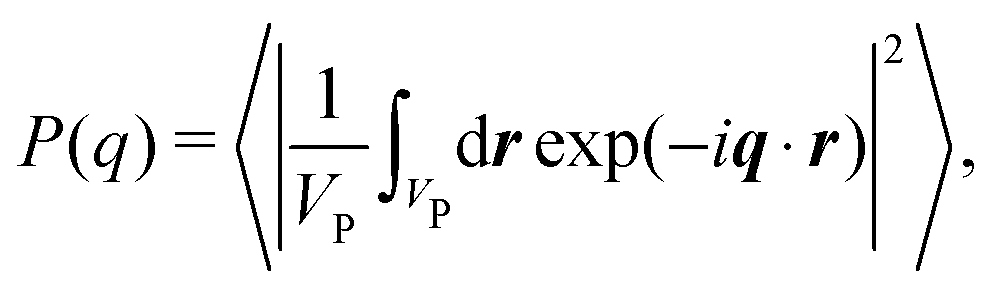 | (2) |
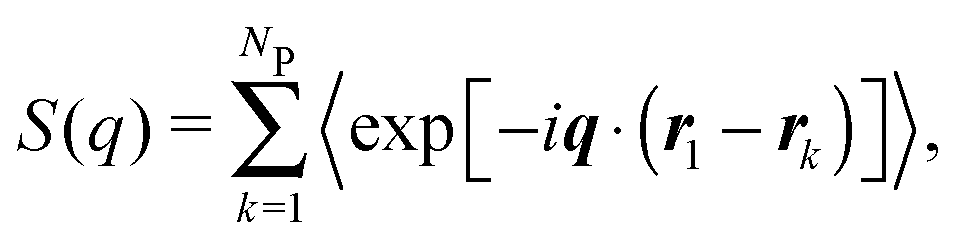 | (3) |
![[S with combining overline]](https://www.rsc.org/images/entities/i_char_0053_0305.gif) (q).46 For the samples studied here, the difference between (q) and S(q) due to non-spherical protein shape is likely to be small.
(q).46 For the samples studied here, the difference between (q) and S(q) due to non-spherical protein shape is likely to be small.
According to eqn (1), the structure factor S(q; nP) of a protein gel at concentration nP can be obtained from the corresponding SAXS intensity I(q; nP) and the intensity I(q; n0P) measured from a solution of the same protein at a concentration n0P sufficiently low that S(q; n0P) = 1:
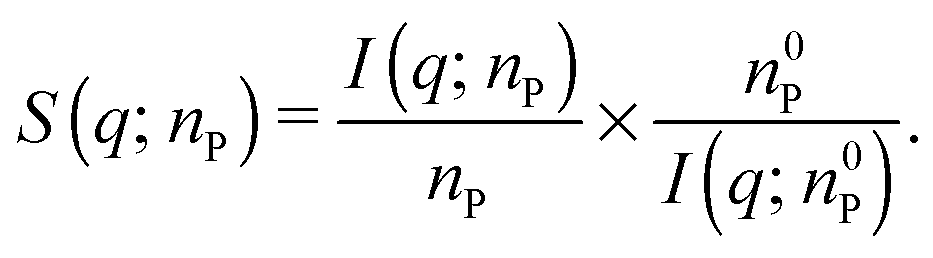 | (4) |
For each protein, the quantity IP(q; n0P) in eqn (4), hereafter referred to as the apparent form factor (AFF), was obtained by merging solution SAXS profiles recorded at two different protein concentrations, as shown in Fig. S1 in the ESI.† The two profiles were first superimposed in an intermediate q window (q = 1.9 ± 0.3, 1.5 ± 0.1 or 1.7 ± 0.1 nm−1 for BPTI, Mb or IFABP, respectively), indicated by vertical dashed lines in Fig. S1,† and a hybrid profile was constructed by merging the low-q part of the lower concentration profile with the high-q part of the higher concentration profile. At low q, where the SAXS profile is sensitive to protein–protein correlations, we thus use data from the most dilute solution, while, at high q, we benefit from the better signal-to-noise ratio of the data from the more concentrated solution. The merged profile was then smoothed with the aid of a Savitzky–Golay filter.47 For BPTI and Mb, a linear regression on a Guinier plot33 was performed in a low-q window, indicated by vertical dotted lines in Fig. S1,† and used to extrapolate the AFF to q = 0. Finally, the AFF was obtained by scaling the merged profile by a constant factor that minimises the difference between the merged solution profile and the gel profile in a high-q range (2.95–3.05, 2.9–3.1 or 3.9–4.1 nm−1 for BPTI, Mb or IFABP, respectively), where we expect that S(q) = 1. This scaling, which ensures that the gel structure factor derived from eqn (4) tends to 1 at the highest q, is justified by the different sample containers used for gels (capillary) and solutions (flow-through cell).
For an isotropic sample of identical protein molecules, the structure factor is related to the Fourier transform of the protein–protein pair correlation function (PCF), g(r). After angular integration, one obtains48
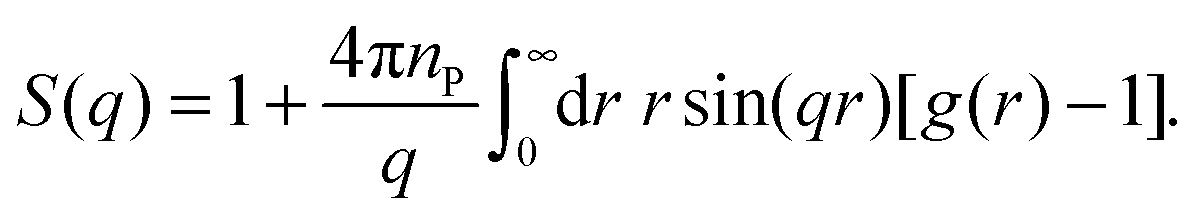 | (5) |
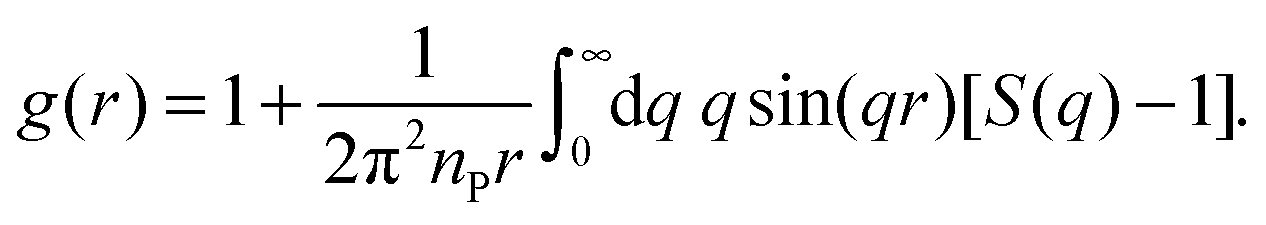 | (6) |
To obtain the PCF from eqn (6), the structure factor, which has been experimentally determined in a finite q range, must be extended to higher and lower q. First, a value qmax is selected, above which S(q) is set equal to 1. This value, qmax = 3.0, 3.0 or 4.0 nm−1 for BPTI, Mb or IFABP, respectively, is taken as the midpoint of the q range used to scale the AFF. Then a value qmin is selected (0.09, 0.065 or 0.075 nm−1 for BPTI, Mb or IFABP, respectively), below which S(q) is extrapolated by fitting a quadratic polynomial to the 20 data points (covering ∼0.07 nm−1) just above qmin. Using cubic spline interpolation, we resample S(q) with fixed spacing Δq. Finally, we zero-fill S(q) − 1 to obtain a real-space resolution Δr = 0.065 nm.
It follows from eqn (6) that even a small deviation of S(q) from 1 at high q has a large effect on g(r) at small r. Our protocol of setting S(q) = 1 above qmax thus causes g(r) to be negative at small r (Fig. S2†). This unphysical feature is removed by forcing g(r) = 0 whenever the transform produces negative values.
From the PCF, we compute the running coordination number as
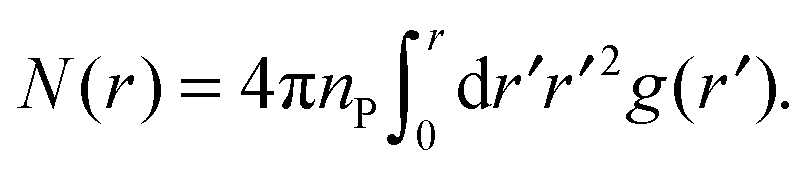 | (7) |
3. Results and discussion
3.1. Structure factor and pair correlation function
Crystal structures49–51 of the three investigated proteins, BPTI, Mb and IFABP, are shown in Fig. 1. BPTI has a pear-like shape with principal dimensions of 2.1 and 3.4 nm and volume VP = 7.792 nm3. The shape of Mb is oblate-like, with principal dimensions 2.6 and 4.0 nm and VP = 21.67 nm3. IFABP is also oblate-like with principal dimensions 2.5 and 3.6 nm and VP = 18.79 nm3. The effective diameter, σP, of a sphere of volume VP and the number, NLys, of lysine residues per protein are listed in Table 1, which also presents relevant gel sample characteristics such as the protein volume fraction ϕP and the centre-to-centre separation dPP between protein molecules assuming a cubic lattice. The protein concentration, pH and the GA/protein mole ratio, NGA, were chosen to match gel samples used in MRD studies.20,21,43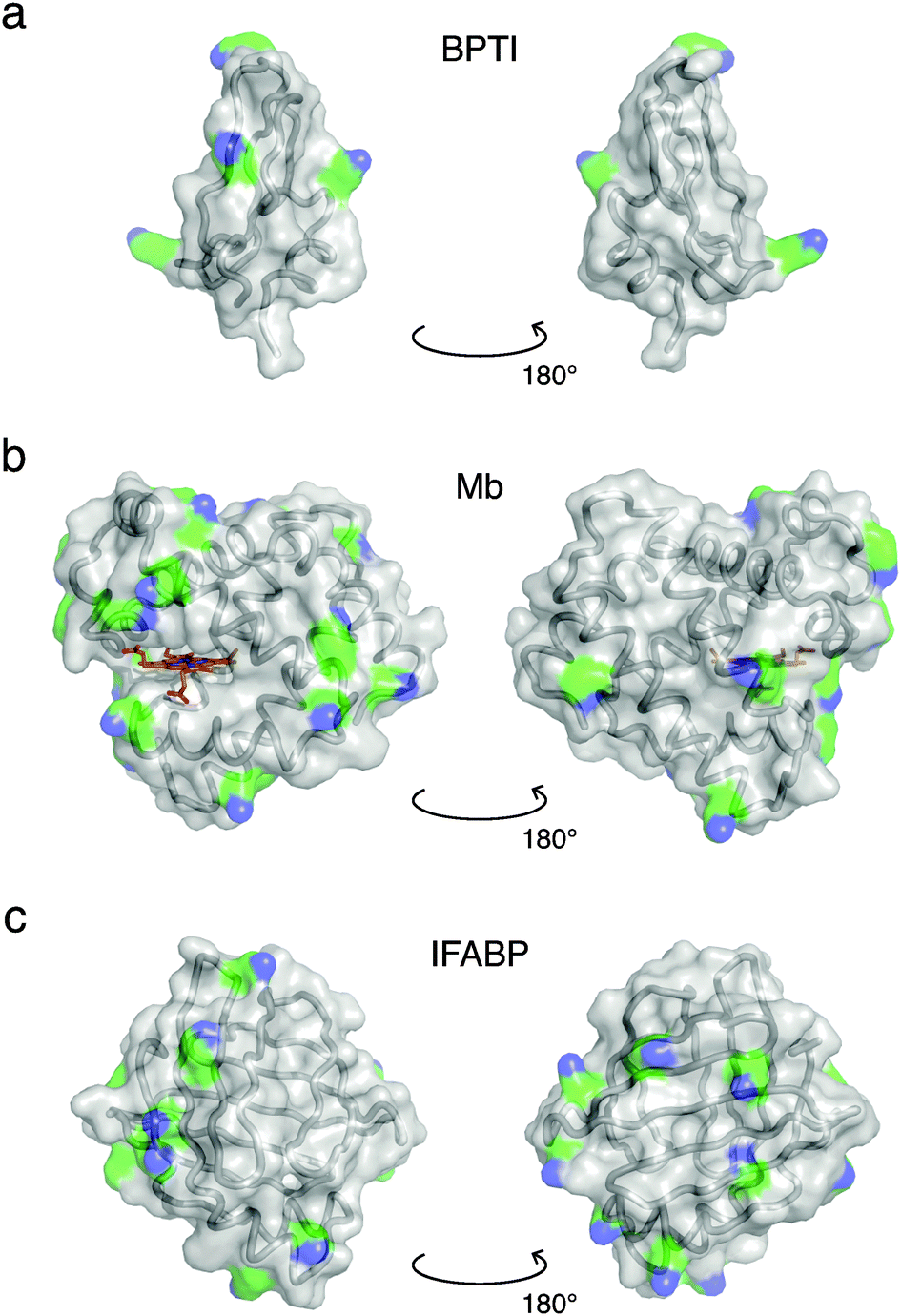 | ||
| Fig. 1 Crystal structures of BPTI (PDB ID 1bpi49), Mb (1wla50) and IFABP (1ifc51). Ribbon and surface representations are superimposed, while the haem group of Mb is shown in a stick representation (C, N, O and Fe atoms coloured orange, blue, red and brown, respectively). Lysine side-chains are coloured by element (C, green; N, blue). | ||
| Protein |
N
Lysa |
σ
Pb (nm) |
C P (mM/mg ml−1) |
ϕ
Pc (%) |
d
PPd (nm) |
pHe | Z | N GA | ξ (nm) |
k
CLh (h−1) |
|---|---|---|---|---|---|---|---|---|---|---|
a Number of lysine residues.
b Effective protein diameter.
c Protein volume fraction.
d Centre-to-centre separation in a cubic lattice, dPP = σP(ϕcp/ϕP)1/3 with 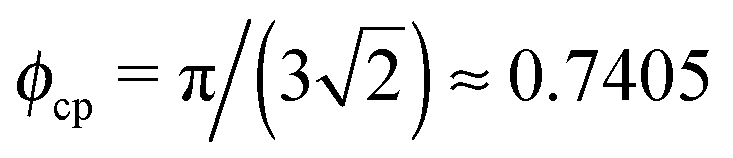 .
e pH measured in the cross-linked gel.
f Net protein charge at given pH, calculated with standard pKa values and unmodified lysines.
g Correlation length (Section 3.2).
h Cross-linking rate at 6 °C at q = 0.1 nm−1. .
e pH measured in the cross-linked gel.
f Net protein charge at given pH, calculated with standard pKa values and unmodified lysines.
g Correlation length (Section 3.2).
h Cross-linking rate at 6 °C at q = 0.1 nm−1.
|
||||||||||
| BPTI | 4 | 2.46 | 16.5/108 | 7.78 | 5.21 | 4.1 | +7.3 | 30.0 | 16 | 0.115 ± 0.002 |
| Mb | 19 | 3.46 | 1.62/28.5 | 2.11 | 11.3 | 6.8 | +3.7 | 30.0 | 157 | 18.2 ± 0.1 |
| IFABP | 15 | 3.30 | 3.83/59.4 | 4.33 | 8.50 | 7.0 | +0.2 | 30.0 | 52 | 9.65 ± 0.05 |
The concentration normalised (gel–solvent) scattering profiles of the three protein gels are shown in Fig. 2. The structure factor, S(q), was deduced from eqn (4) using an apparent form factor (AFF), I(q; n0P), constructed from SAXS profiles recorded on protein solutions at two concentrations (Section 2.3, Fig. 2 and Fig. S1†). For IFABP, the solution SAXS profile, and thus the AFF, increases sharply below q ≈ 0.2 nm−1 even at the lowest concentration (0.5 mM), indicative of protein aggregation. A crude analysis shows that this feature in the scattering profile can be rationalised by a very small fraction (∼10−5) of the IFABP molecules existing in large aggregates (effective diameter ∼ 10 σP). For each protein, the AFF constructed from solution SAXS profiles at two concentrations agrees well (at q ≥ 1 nm−1 for IFABP) with a CRYSOL52 fit based on the crystal structure of the corresponding protein. We therefore conclude that the proteins are essentially monomeric form in our solution samples.
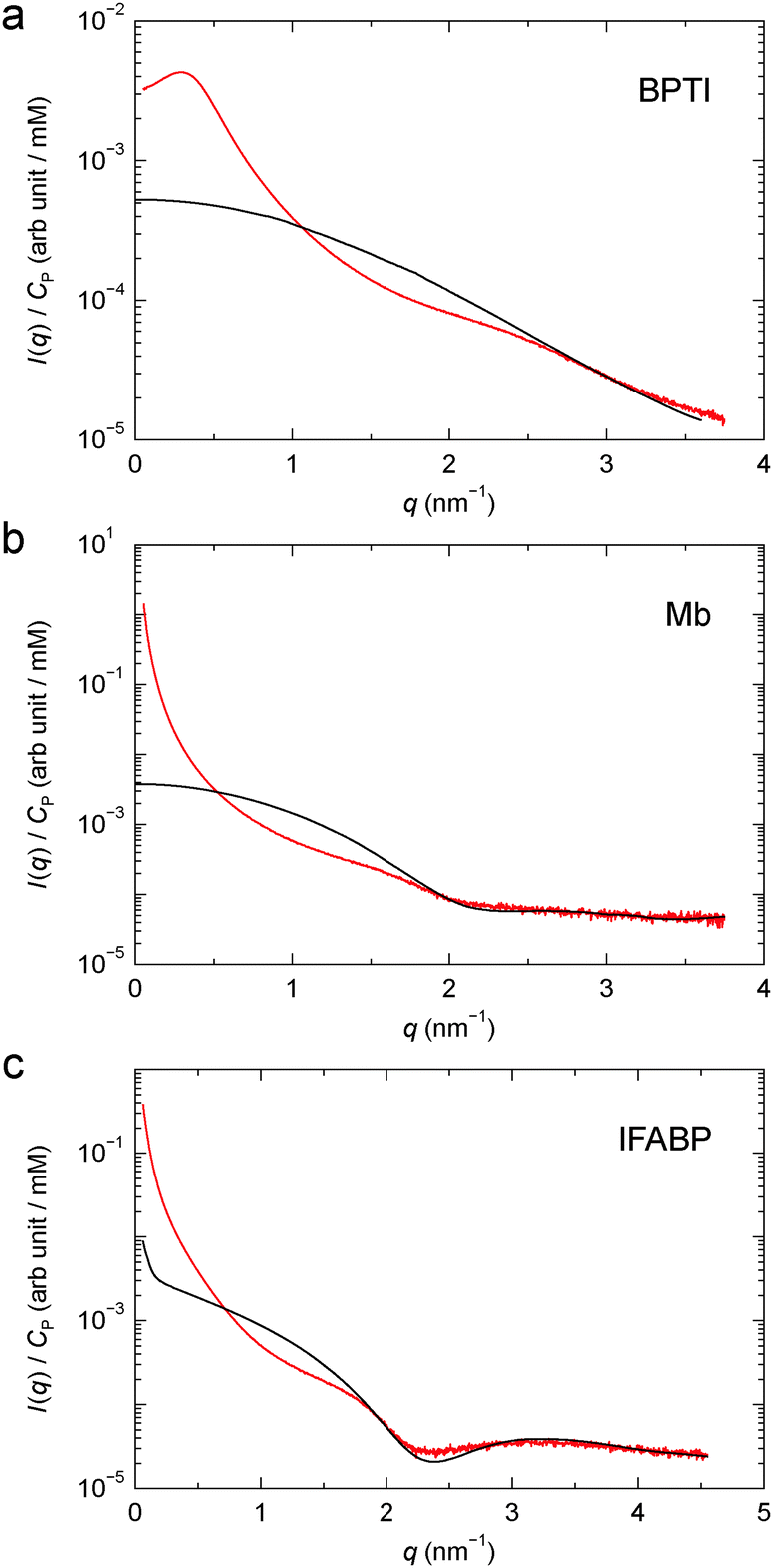 | ||
| Fig. 2 Concentration normalised scattering profile from cross-linked BPTI (a), Mb (b) and IFABP (c). The apparent form factor, derived from solution SAXS profiles as described in Section 2.3, is also shown for each protein (black curve). | ||
The close agreement between the SAXS profiles from the Mb and IFABP gels with the corresponding AFFs at high q (Fig. 2b and c), where we expect that S(q) = 1, indicates that the only effect of GA is to induce protein–protein correlations. The protein structure thus appears to be the same in solution and gel. We note that although the AFF has been scaled to agree with the gel profile at high q (Section 2.3), this scaling does not alter the shape of the AFF. For BPTI, the AFF cannot be scaled to superimpose with the gel profile over an extended high-q range (Fig. 2a), presumably because the S(q) = 1 limit is not reached in the investigated q range for this small protein. Another consequence of the limited q range is that the dip in the AFF, which reflects the size and shape of the protein molecule, is observed for Mb and IFABP (at q ≈ 2.1 and 2.3 nm−1, respectively; see Fig. 2b and c), but not for the smaller BPTI. Using the program CRYSOL52 to compute the form factor from the BPTI crystal structure 1bpi,49 we find a shallow dip at q ≈ 4.5 nm−1. Nevertheless, in the subsequent analysis of the BPTI profile, we postulate that S(q) = 1 for q ≥ 3.0 nm−1.
The structure factors of the three protein gels, deduced in this way, are shown in Fig. 3. For BPTI, S(q) shows a pronounced maximum at q ≈ 0.3 nm−1, already evident in the gel profile (Fig. 2a). In contrast, for Mb and IFABP, S(q) decreases monotonically with q from the lowest examined q value (0.06 nm−1) up to q ≈ 1 nm−1 (Fig. 3b and c).
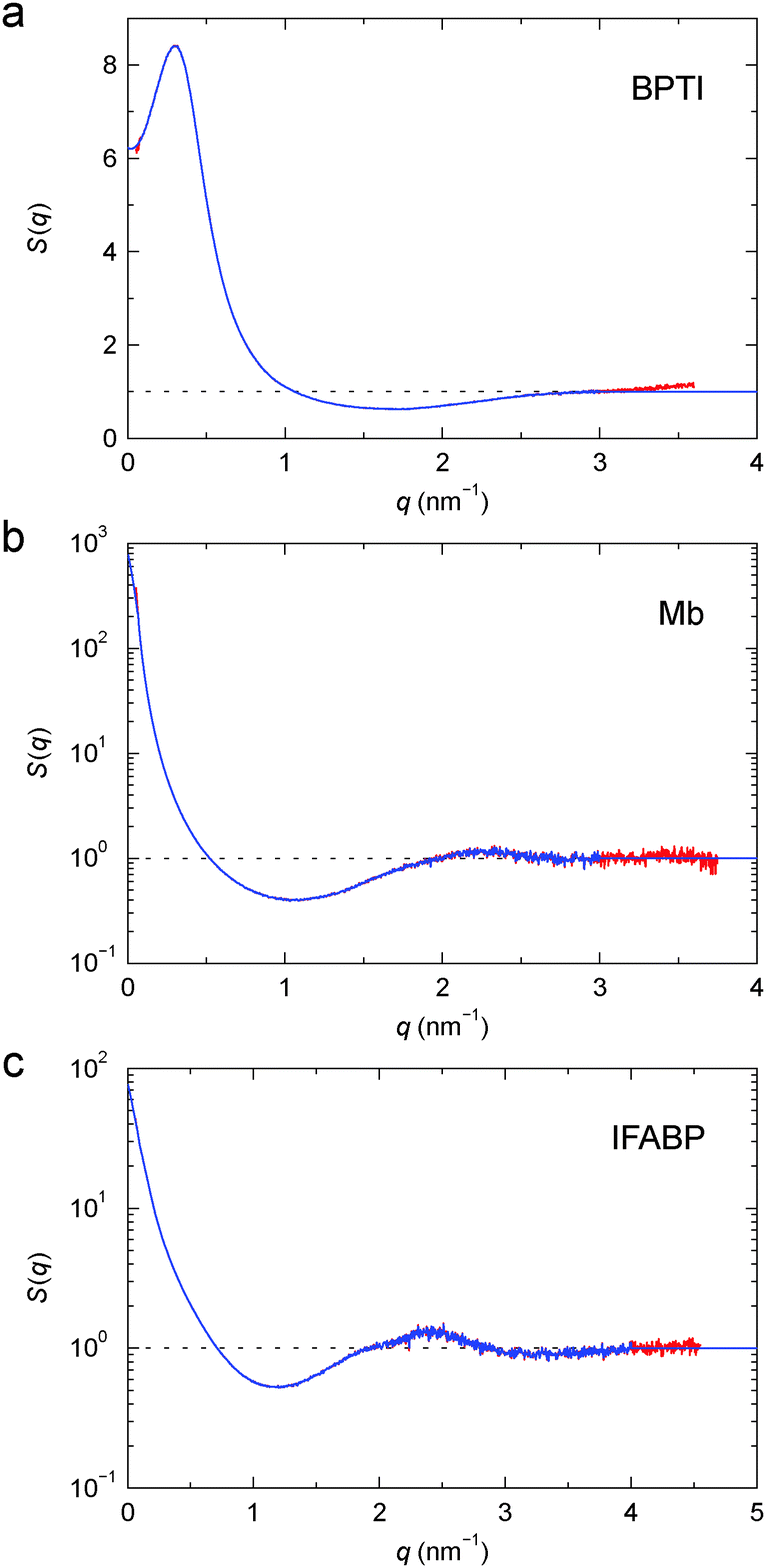 | ||
| Fig. 3 Structure factor, S(q), for cross-linked BPTI (a), Mb (b) and IFABP (c), obtained from the profiles in Fig. 2 according to eqn (4) (red) and after truncation at high q and extension to high and low q (blue). Note the linear scale in (a). | ||
To characterise the gel microstructure in real space, we transform the structure factor into a pair correlation function (PCF), g(r), with the aid of eqn (6), implemented as a fast sine transform. Before the transformation, S(q) is truncated at high q and extended to high and low q as described in Section 2.3. The modified S(q) is included in Fig. 3 as a blue curve and the resulting PCF is shown in Fig. 4. Because we set S(q) = 1 at high q, the transformation yields negative g(r) values at small r (Fig. S2†). If we force g(r) = 0 at these small r values and then inverse transform the corrected PCF according to eqn (5), we find that the back-calculated structure factor differs very little from the original one (Fig. S3†). (However, a significant deviation is seen for Mb at q ≈ 1 nm−1.) This finding is consistent with the expectation that our SAXS data (with q ≤ 4 nm−1) are insensitive to short-range (r ≲ 2 nm) structural features.
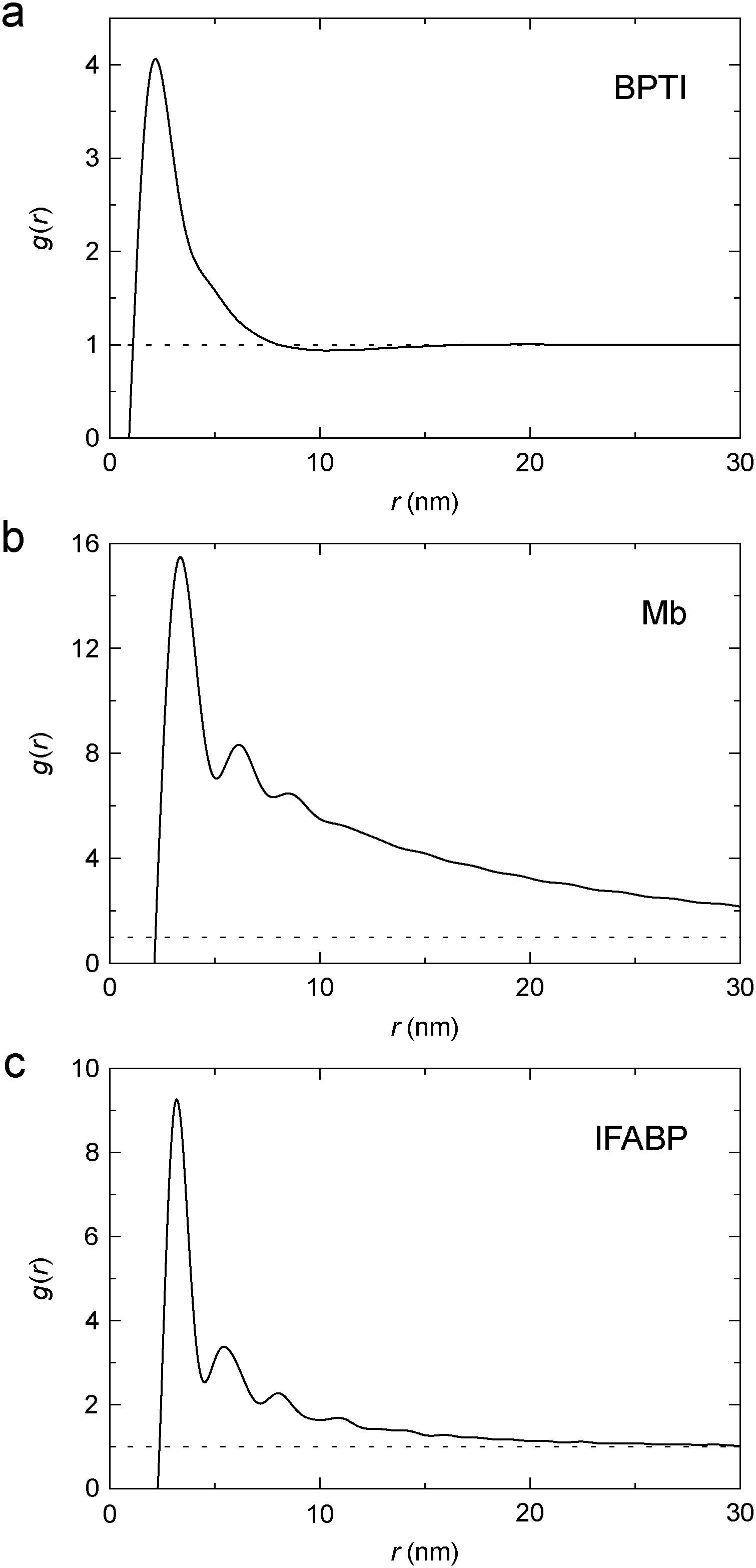 | ||
| Fig. 4 Pair correlation function, g(r), for cross-linked BPTI (a), Mb (b) and IFABP (c), obtained by Fourier transformation of S(q) (blue curves in Fig. 3). | ||
The PCF reflects the static spatial correlations in the sample, regardless of the origin of these correlations (cross-links or other interactions), and it can therefore be obtained from the structure factor without any assumptions about the structure of the gel (apart from isotropy). The analysis in Section 3.2 indicates that the protein gel is inhomogeneous on the length scale probed by our SAXS data, with dense protein clusters connected by less dense regions. This inhomogeneity complicates the interpretation of g(r). One possible approach would then be to model the protein gel as a mixture of clustered and non-clustered protein molecules. Such an approach would, however, introduce more parameters than can be justified by the data. Instead, we assume that the observed scattering is strongly dominated by clustered proteins so that the contribution from non-clustered proteins can be neglected.
The PCF is then used, along with eqn (7), to obtain the running coordination number, N(r), in the protein gel (Fig. 5). For comparison, we show N(r) for a uniform protein distribution at the same protein concentration as in the corresponding gel.
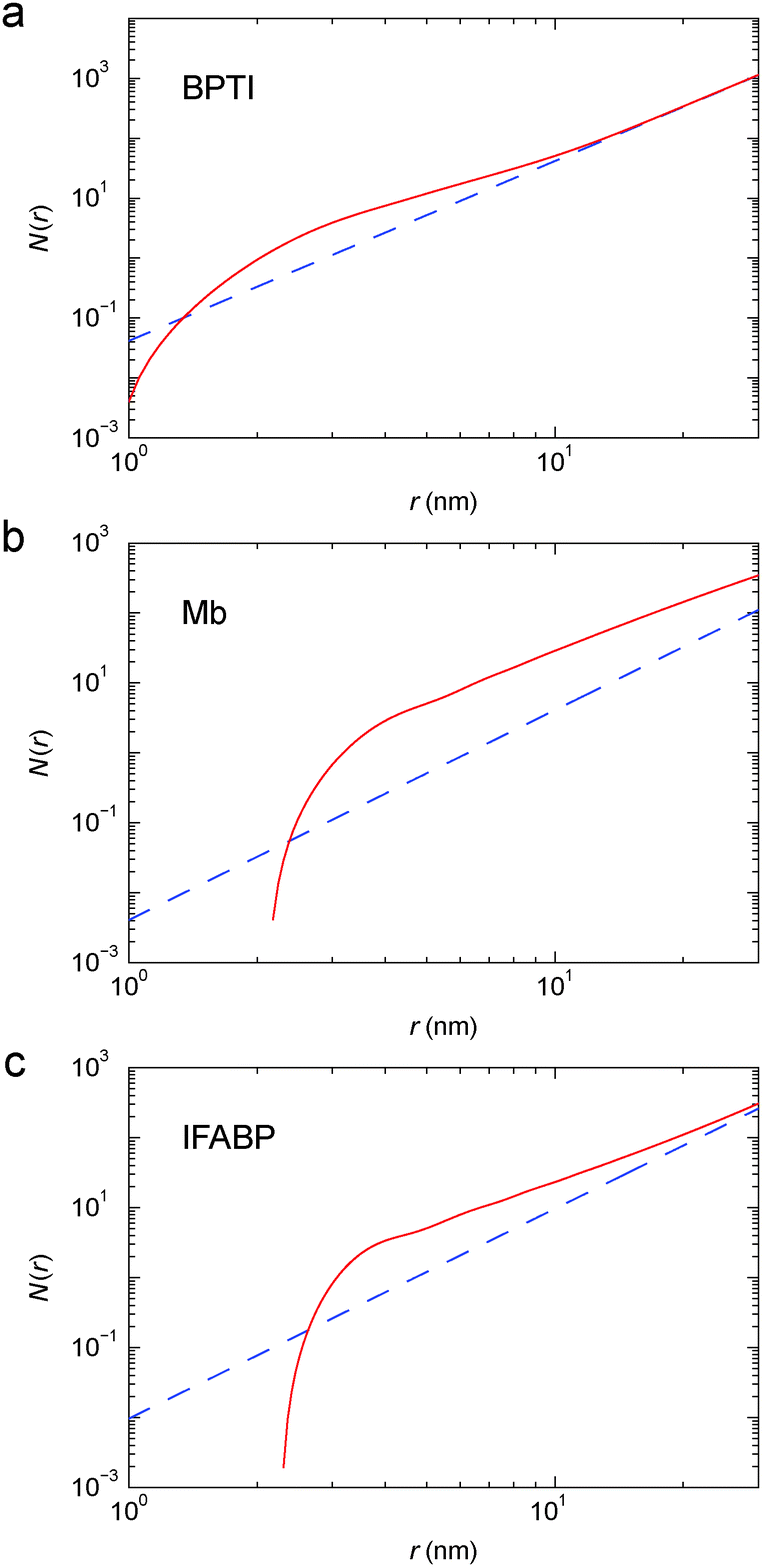 | ||
| Fig. 5 Running coordination number, N(r), for cross-linked BPTI (a), Mb (b) and IFABP (c), obtained from the corrected PCF in Fig. 4 (red solid curve) or by assuming a uniform (g(r) ≡ 1) protein distribution (blue dashed line). | ||
3.2. Microstructure of protein gels
For simple liquids, the first-shell coordination number Nc is usually obtained by integrating the PCF up to its first minimum. But, for the BPTI gel, the first minimum in g(r) is very shallow and extended (Fig. 4a), so we define Nc as the integral up to the distance rc = 8.06 nm where g(r) = 1 (on the large-r flank of the primary peak). This integration yields Nc = 31.5 (Fig. 5a). In contrast, for a uniform protein distribution and at the same BPTI concentration as in the gel, we would have Nc0 = 21.8 at rc = 8.06 nm. Within a sphere of radius 8.06 nm around a given BPTI molecule, the local protein density (concentration) defined as nP(r) = [N(r) + 1]/V(r), where V(r) is the volume of a sphere of radius r centred on the reference protein molecule, is therefore higher than the bulk density by a factor of (31.5 + 1)/(21.8 + 1) = 1.43.
From the shoulder seen at r ≈ 5 nm (Fig. 4a), it is clear that the primary peak in the PCF comprises 2, or even 3, overlapping coordination shells. If only the first shell was included, the local density would be even higher. But the local density is not uniform; presumably it is higher along the cross-linked chains than in the regions in between. Indeed, in ∼90% of the volume of the 8 nm sphere (beyond ∼3.5 nm), the running coordination number N(r) grows more slowly than for a uniform distribution (Fig. 5a).
In a log–log plot as in Fig. 5, the slope yields the fractal dimension, df, defined via the scaling relation N(r) ∝ rdf.56 Beyond ∼15 nm, N(r) exhibits bulk-like scaling with df = 3 (Fig. 5a). To make this more precise, we define a correlation length, ξ, as the distance where df has reached a value of 2.9 on its approach to the bulk value 3. Analysis of the slope in Fig. 5a then yields ξ = 16 nm for the BPTI gel.
From N(r) in Fig. 5b, we find that the first coordination shell (r < 5.0 nm) contains 5.0, the second shell (5.0 < r < 7.5 nm) 9.0 and the third shell (7.5 < r < 10.0 nm) 14.5 Mb molecules. The first three shells (r < 10 nm) thus contain 28.5 Mb molecules, whereas a uniform distribution would only have 4.0 neighbours within 10 nm. This corresponds to a local density increase by a factor of 29.5/5.0 = 5.9. The spatial heterogeneity is thus more pronounced in the Mb gel than in the more concentrated BPTI gel. Since S(0) is proportional to the mean-square fluctuation (or spatial variation) of the protein concentration, the stronger spatial heterogeneity in the Mb gel can explain the orders-of-magnitude larger S(q) at q ≈ 0 (Fig. 3).
The Mb gel analysis may not be quantitatively accurate since, for Mb, the S(q) back-calculated from the corrected (non-negative) PCF differs somewhat from the original S(q) (Fig. S3b†). The PCF becomes negative at small r (Fig. S2b†) because the negative S(q) − 1 in the range 0.5 < q < 2.0 nm−1 is not fully compensated by a slightly positive S(q) − 1 at higher q (Fig. 3b). The latter feature is not resolved in the noisy high-q part of the SAXS profile from the dilute Mb gel.
From N(r) in Fig. 5c, we find that the first coordination shell (r < 4.5 nm) contains 4.0, the second shell (4.5 < r < 7.0 nm) 6.9 and the third shell (7.0 < r < 9.5 nm) 7.8 IFABP molecules. The first three shells (r < 9.5 nm) thus contain 18.7 IFABP molecules, whereas a uniform distribution would only have 7.0 neighbours within 9.5 nm. This corresponds to a local density increase by a factor of 19.7/8.0 = 2.5, intermediate between the corresponding values of BPTI and Mb. As for Mb, the strong spatial heterogeneity in the IFABP gel should give rise to a large S(q) at q ≈ 0 (Fig. 3c).
3.3. Cross-linking kinetics
To study the kinetics of gel formation by GA cross-linking, we performed time-resolved SAXS measurements where scattering profiles were recorded at regular time intervals after mixing protein and GA solutions. Fig. 6 shows, for each protein, 16 profiles from the developing gel, along with the respective apparent form factor (Fig. S1†). The timing of each scattering profile, counted from the mixing of protein and GA solutions to the middle of the irradiation period and including a 1 min dead-time between mixing and the first irradiation period, is listed in Table S1.† Consistent with the finding that GA cross-linking alters the structure factor with little or no effect on the form factor (Section 3.1), the time-dependence of the SAXS profile is most evident in the low-q region. For Mb and IFABP, due to the fast kinetics and the limited time resolution of the experiment, we could reliably monitor the process only at q < 0.5 nm−1.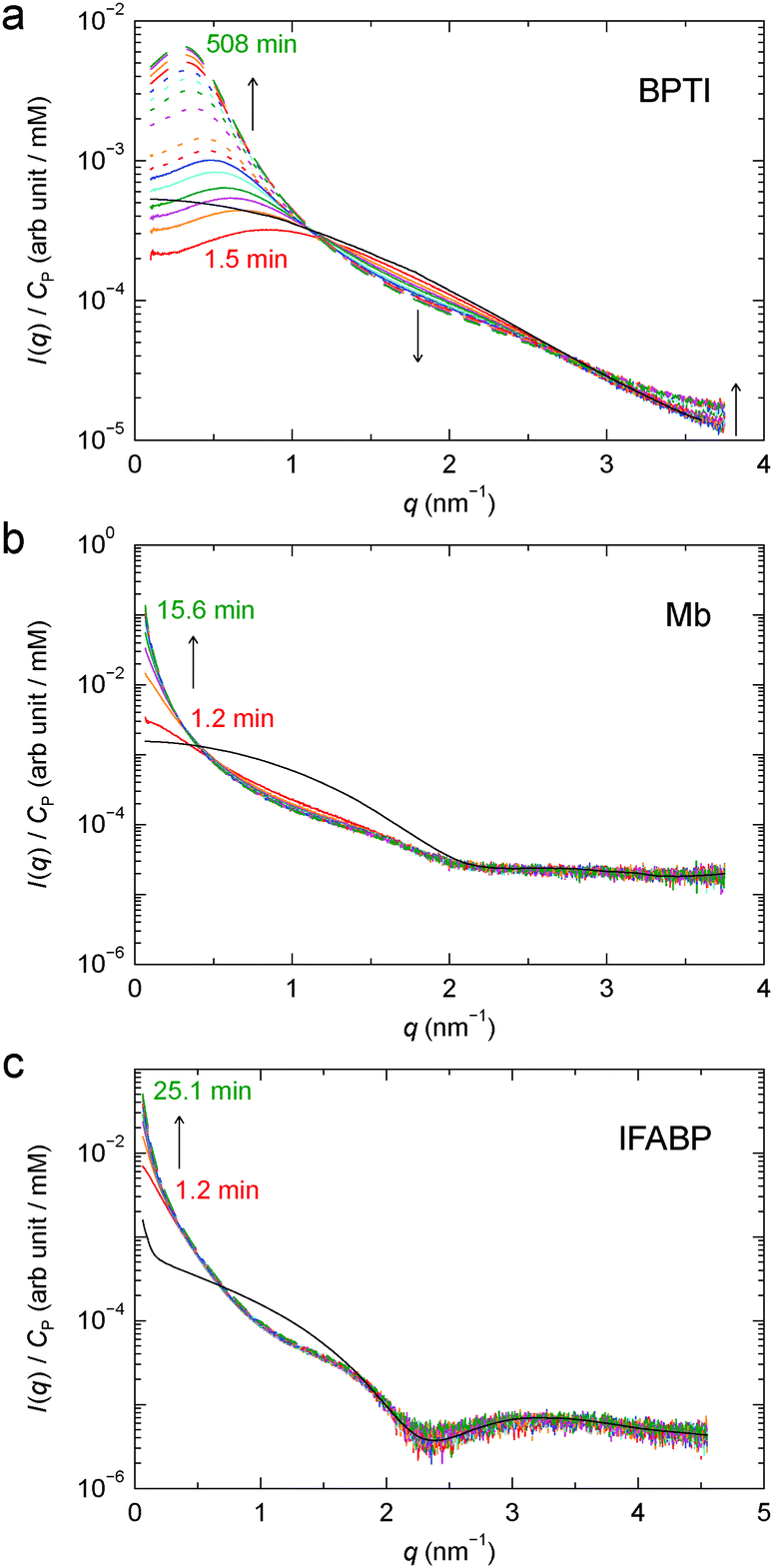 | ||
| Fig. 6 Time-resolved SAXS profiles at 6 °C from developing gels of BPTI (a), Mb (b) and IFABP (c). Time ordering is indicated by the line colour (red → orange → magenta → green → cyan → blue), the line type (solid → dotted → dashed) and arrows. The timing of the first and last profiles is given. The AFF is also shown for each protein (black solid curve). | ||
From the time-resolved SAXS data, we determine the gel formation or the cross-linking rate, kCL, by assuming first-order kinetics: I(q,t) = α(q) − β(q)exp[−kCL(q)t]. The kCL values obtained from exponential fits to the time-dependent scattering intensity at q = 0.1 nm−1 are shown in Table 1. These rates correspond to characteristic cross-linking times, τCL = 1/kCL, of 8.7 h, 3.3 min and 6.2 min for BPTI, Mb and IFABP, respectively.
Radiation damage during the multiple irradiations enhances scattering at low q, causing the last profile in the time series to overshoot the equilibrium profile in Fig. 2a. But radiation damage only makes a minor contribution to the, mainly structure-related, build-up of the low-q intensity and should therefore only give rise to a modest overestimate of kCL.
For BPTI, kCL(q) was determined as a function of q up to 2.3 nm−1, except near 1 nm−1 where the time-dependent SAXS profiles show a quasi-isosbestic point (Fig. 6a). The obtained kCL values (Fig. 7) indicate, not surprisingly, that the gel structure forms faster on shorter length scales (2π/2.0 ≈ 3 nm) than on longer length scales (2π/0.2 ≈ 30 nm). In the former case, the data suggest, in addition to the principal fast component, one or more minor slow components, but it cannot be excluded that radiation damage plays a role here.
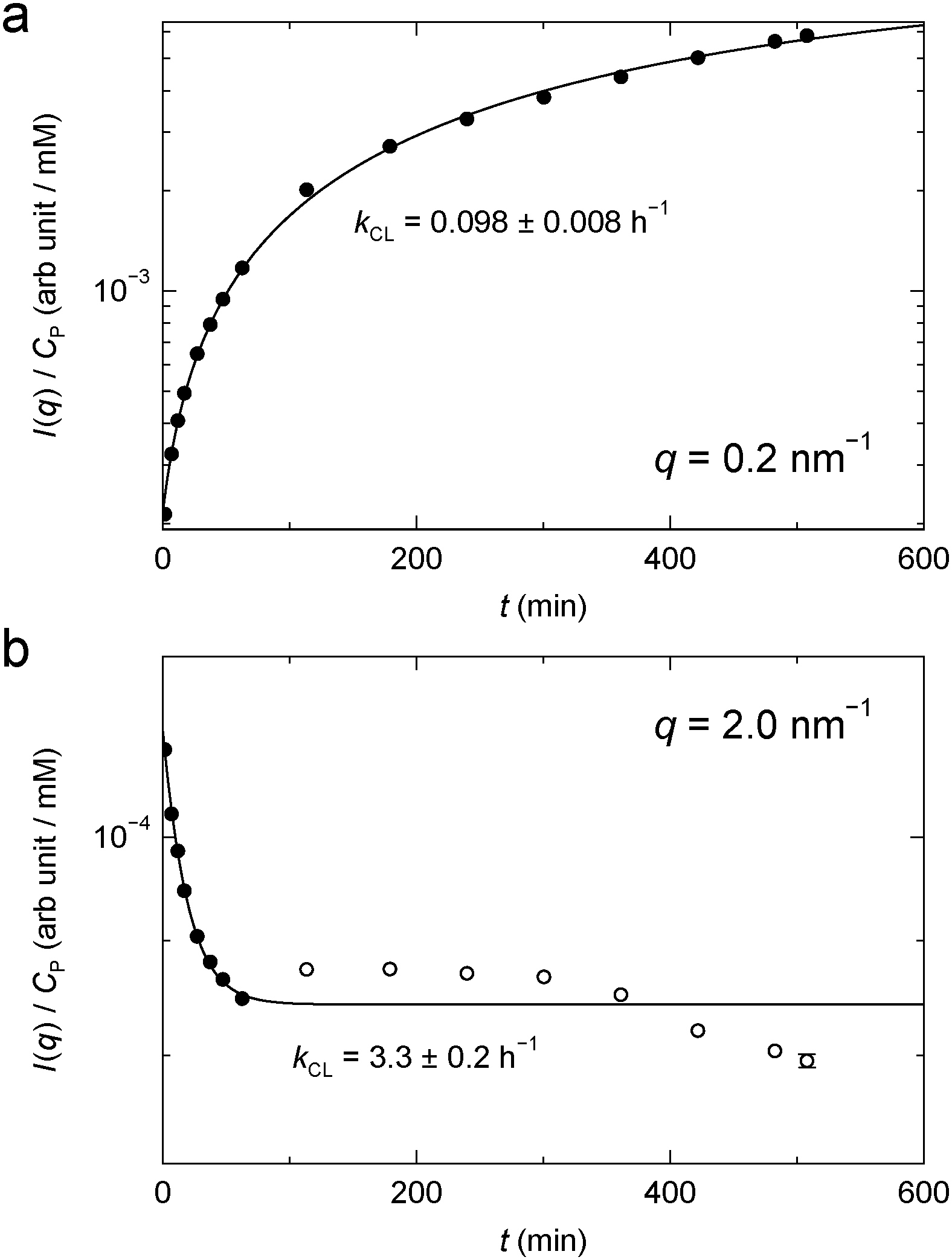 | ||
| Fig. 7 Time-dependent SAXS intensity from BPTI gel (Fig. 6a) at q = 0.2 nm−1 (a) and 2.0 nm−1 (b). The filled data points were used for the exponential fit. | ||
While we are not aware of any previous quantitative kinetic study of protein gel formation by GA cross-linking, the rate of formation of small cross-linked protein clusters has been investigated by light scattering, UV absorbance or ezymatic activity.57–60 At the much lower protein concentrations examined by these authors, the cross-linking process exhibits two distinct steps, attributed to fast cluster formation by cross-linking of the most reactive lysines followed by slower linkage of clusters.59 At the higher protein concentrations studied here, the time scales of these two processes may overlap, leading to an apparently exponential build-up of scattering intensity at low q (Fig. 7a).
The rate of gel formation depends on many factors, including protein and GA concentrations, pH, temperature and availability of primary amino groups. While it is outside the scope of this study to systematically explore these factors, we note that the BPTI gel forms 2 orders of magnitude slower than the Mb and IFABP gels. The BPTI gel differs from the two other gels in having a much higher protein concentration (Table 1). But this should accelerate gel formation, so there must be other factors at play. We suspect that the dominant factor here is the ∼3 units lower pH in the BPTI gel (Table 1), which means that the fraction of ε-amino groups in the reactive deprotonated (NH2) form1,2 is 3 orders of magnitude lower in the BPTI gel. Other, presumably less important, effects of a low pH include suppression of GA aldol condensation,1,2,27–32 which might lead to shorter cross-links and slower gel formation, and a more positive net protein charge, Z (Table 1). A larger |Z| should retard gel formation and make the clusters smaller due to intra-cluster Coulomb repulsion, but, since cross-links remove positive lysine charges, it is not clear which of the three proteins has the largest |Z|. Yet another factor that could slow down BPTI gel formation is the small number (Table 1) and uneven distribution (Fig. 1) of lysine side-chains in BPTI, which may also be responsible for the smaller size of the BPTI clusters (Section 3.2.4).
4. Conclusions
The SAXS data presented here provide quantitative information about microstructure and aggregation kinetics in GA cross-linked gels of three different proteins. While the three protein gels are qualitatively similar, the BPTI gel differs quantitatively from the Mb and IFABP gels. This difference is caused by a combination of factors, among which the most important are the higher protein concentration, the lower pH and the more limited availability of lysine amino groups in the case of BPTI. The most important conclusions derived from our analysis of the SAXS data are as follows.The native protein structure is retained in the cross-linked gel, at least at the ∼1 nm resolution afforded by the SAXS data. This conclusion follows from the close agreement at high q between the SAXS profiles from gel and dilute solution (Fig. 2). The evidence is most clear-cut for Mb and IFABP, whereas, for the smaller protein BPTI, the form factor is masked by S(q) oscillations that persist to higher q. While the lysine side-chains involved in cross-links, and perhaps some nearby side-chains as well, must be conformationally perturbed, the SAXS data rule out a significant degree of unfolding. This conclusion is consistent with the finding, from MRD measurements on BPTI,20 that the ns–μs dynamics of internal water exchange, and the rate-limiting structural fluctuations,61 are essentially the same whether the protein is free in solution or cross-linked in a gel. Furthermore, the SAXS results indicate that this is true also for Mb and IFABP.
The protein gel is spatially heterogeneous, with dense clusters linked by sparse networks. The strong spatial heterogeneity in the more dilute Mb and IFABP gels produces intense scattering at low q. The BPTI gel, with a higher concentration of smaller clusters and a shorter correlation length, is less strongly heterogeneous. The low-q scattering is therefore much weaker, allowing us to observe a peak at q ≈ 0.3 nm−1, resulting from the interplay of intra-cluster attraction and inter-cluster repulsion.
Within the clusters, adjacent protein molecules are almost in contact. The number of nearest protein neighbours, estimated by integrating over the first coordination shell (not perfectly well-defined for BPTI) in the PCF (Fig. 4), is 5 ± 1 for all three proteins. Since this exceeds the value of 2 expected for a linear chain, the protein clusters must be multiply connected. Some nearest neighbours may be cross-linked via a third protein molecule while still approaching each other almost to contact via hydrophobic attraction. Despite such close encounters, the cluster is not uniformly dense. For the BPTI gel, the protein concentration within 8 nm of a reference molecule (a spherical volume that includes 2 or 3 coordination shells) is ∼24 mM (43% above the average concentration in the gel), which is still much lower than the concentration of 136 mM in the (monomeric) BPTI crystal 1bpi.49 Yet, the close protein encounters that do occur within a cluster should lead to a stronger dynamical perturbation of water dynamics than in the hydration layer of a protein in dilute solution. At points of particularly close contact between two protein molecules, some water molecules may be trapped with survival times exceeding 1 ns, as seen for internal water molecules. Both of these phenomena, enhanced dynamical perturbation in the hydration layer and trapped water molecules with survival times in the range 1–10 ns, have been inferred from MRD studies of cross-linked proteins.20,21
Proteins with a large number of uniformly distributed lysine side-chains make larger clusters. This generalisation is based on the correlation of the range of protein–protein interactions, as reflected in the PCF (Fig. 4), with the number (Table 1) and surface distribution (Fig. 1) of lysine amino groups in the three investigated proteins. The correlation length, ξ, defined in terms of the fractal dimension, is more closely related to the typical cluster–cluster separation and therefore depends on the overall protein concentration as well as on the cluster size.
Gel formation occurs on time scales from minutes to hours under our conditions (Fig. 6). As judged by the scattering intensity at q ≤ 0.2 nm−1, gel formation appears to obey first-order kinetics (Fig. 7). The much slower gel formation for BPTI is attributed to the lower pH and the consequent lower abundance of reactive deprotonated amino groups.
Acknowledgements
We thank Hanna Nilsson for Mb purification, Annika Rogstam at Lund Protein Production Platform for IFABP preparation, Bayer Healthcare AG for a generous supply of BPTI and MAX-lab Synchrotron beamline I911-4 for beamtime under proposal ID 20120020. This work was financially supported by a project grant (to B.H.) from the Swedish Research Council and postdoctoral fellowships (to S.K.) from the Wenner-Gren Foundations and the Swedish Research Council.References
- D. R. Walt and V. I. Agayn, TrAC, Trends Anal. Chem., 1994, 13, 425–430 CrossRef CAS.
- I. Migneault, C. Dartiguenave, M. J. Bertrand and K. C. Waldron, Biotechniques, 2004, 37, 790–802 CAS.
- D. D. Sabatini, K. Bensch and R. J. Barrnett, J. Cell Biol., 1963, 17, 19–58 CrossRef CAS.
- D. Hopwood, Histochem. J., 1972, 4, 267–303 CrossRef CAS.
- F. A. Quiocho and F. M. Richards, Proc. Natl. Acad. Sci. U. S. A., 1964, 52, 833–839 CrossRef CAS.
- Y. Wine, N. Cohen-Hadar, A. Freeman and F. Frolow, Biotechnol. Bioeng., 2007, 98, 711–718 CrossRef CAS PubMed.
- C. L. Hall, J. R. Clemens, A. M. Brown and L. J. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 813–818 CrossRef PubMed.
- V. E. Fadouloglou, M. Kokkinidis and N. M. Glykos, Anal. Biochem., 2008, 373, 404–406 CrossRef CAS PubMed.
- A. F. S. A. Habeeb, Arch. Biochem. Biophys., 1967, 119, 264–268 CrossRef CAS.
- J. W. Payne, Biochem. J., 1973, 135, 867–873 CAS.
- J. J. Roy and T. E. Abraham, Chem. Rev., 2004, 104, 3705–3721 CrossRef CAS PubMed.
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307 CrossRef CAS.
- S. Talekar, A. Joshi, G. Joshi, P. Kamat, R. Haripurkar and S. Kambale, RSC Adv., 2013, 3, 12485–12511 RSC.
- J. Grad, D. Mendelson, F. Hyder and R. G. Bryant, J. Magn. Reson., 1990, 86, 416–419 CAS.
- R. G. Bryant, D. A. Mendelson and C. C. Lester, Magn. Reson. Med., 1991, 21, 117–126 CrossRef CAS.
- D. Zhou and R. G. Bryant, Magn. Reson. Med., 1994, 32, 725–732 CrossRef CAS.
- S. H. Koenig and R. D. Brown III, Magn. Reson. Med., 1993, 30, 685–695 CrossRef CAS.
- E. P. Sunde and B. Halle, J. Am. Chem. Soc., 2009, 131, 18214–18215 CrossRef CAS PubMed.
- E. Persson and B. Halle, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6266–6271 CrossRef CAS PubMed.
- E. Persson and B. Halle, J. Am. Chem. Soc., 2008, 130, 1774–1787 CrossRef CAS PubMed.
- S. Kaieda and B. Halle, J. Phys. Chem. B, 2013, 117, 14676–14687 CrossRef CAS PubMed.
- F. M. Richards and J. R. Knowles, J. Mol. Biol., 1968, 37, 231–233 CrossRef CAS.
- A. F. S. A. Habeeb and R. Hiramoto, Arch. Biochem. Biophys., 1968, 126, 16–26 CrossRef CAS.
- P. Monsan, G. Puzo and H. Mazarguil, Biochimie, 1975, 57, 1281–1292 CrossRef CAS.
- K. Okuda, I. Urabe, Y. Yamada and H. Okada, J. Ferment. Bioeng., 1991, 71, 100–105 CrossRef CAS.
- M. Salem, Y. Mauguen and T. Prangé, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2010, 66, 225–228 CrossRef CAS PubMed.
- P. M. Hardy, A. C. Nicholls and H. N. Rydon, J. Chem. Soc., Chem. Commun., 1969, 565–566 RSC.
- A. H. Korn, S. H. Feairheller and E. M. Filachione, J. Mol. Biol., 1972, 65, 525–529 CrossRef CAS.
- E. B. Whipple and M. Ruta, J. Org. Chem., 1974, 39, 1666–1668 CrossRef CAS.
- S. Margel and A. Rembaum, Macromolecules, 1980, 13, 19–24 CrossRef CAS.
- T. Tashima and M. Imai, J. Org. Chem., 1991, 56, 694–697 CrossRef CAS.
- J. Kawahara, T. Ohmori, T. Ohkubo, S. Hattori and M. Kawamura, Anal. Biochem., 1992, 201, 94–98 CrossRef CAS.
- A. Guinier and G. Fournet, Small-Angle Scattering of X-rays, Wiley, New York, 1955 Search PubMed.
- Small-Angle X-ray Scattering, ed. O. Glatter and O. Kratky, Academic Press, London, 1982 Search PubMed.
- M. H. J. Koch, P. Vachette and D. I. Svergun, Q. Rev. Biophys., 2003, 36, 147–227 CrossRef CAS.
- D. A. Jacques and J. Trewhella, Protein Sci., 2010, 19, 642–657 CrossRef CAS PubMed.
- W. Häußler, A. Wilk, J. Gapinski and A. Patkowski, J. Chem. Phys., 2002, 117, 413–426 CrossRef PubMed.
- F. Zhang, M. W. A. Skoda, R. M. J. Jacobs, R. A. Martin, C. M. Martin and F. Schreiber, J. Phys. Chem. B, 2007, 111, 251–259 CrossRef CAS PubMed.
- F. Cardinaux, E. Zaccarelli, A. Stradner, S. Bucciarelli, B. Farago, S. U. Egelhaaf, F. Sciortino and P. Schurtenberger, J. Phys. Chem. B, 2011, 115, 7227–7237 CrossRef CAS PubMed.
- C. G. de Kruif, T. Huppertz, V. S. Urban and A. V. Petukhov, Adv. Colloid Interface Sci., 2012, 171–172, 36–52 CrossRef CAS PubMed.
- R. Kuktaite, T. S. Plivelic, Y. Cerenius, M. S. Hedenqvist, M. Gällstedt, S. Marttila, R. Ignell, Y. Popineau, O. Tranquet, P. R. Shewry and E. Johansson, Biomacromolecules, 2011, 12, 1438–1448 CrossRef CAS PubMed.
- M. Pouzot, D. Durand and T. Nicolai, Macromolecules, 2004, 37, 8703–8708 CrossRef CAS.
- S. Kaieda and B. Halle, to be published.
- P. Savitsky, J. Bray, C. D. O. Cooper, B. D. Marsden, P. Mahajan, N. A. Burgess-Brown and O. Gileadi, J. Struct. Biol., 2010, 172, 3–13 CrossRef CAS PubMed.
- A. Labrador, Y. Cerenius, C. Svensson, K. Theodor and T. Plivelic, J. Phys.: Conf. Ser., 2013, 425, 072019 CrossRef.
- J. S. Pedersen, Adv. Colloid Interface Sci., 1997, 70, 171–210 CrossRef CAS.
- A. Savitzky and M. J. E. Golay, Anal. Chem., 1964, 36, 1627–1639 CrossRef CAS.
- J.-P. Hansen and I. R. McDonald, Theory of Simple Liquids, Academic Press, London, 2nd edn, 1986 Search PubMed.
- S. Parkin, B. Rupp and H. Hope, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1996, 52, 18–29 CrossRef CAS PubMed.
- R. Maurus, C. M. Overall, R. Bogumil, Y. Luo, A. G. Mauk, M. Smith and G. D. Brayer, Biochim. Biophys. Acta, 1997, 1341, 1–13 CAS.
- G. Scapin, J. I. Gordon and J. C. Sacchettini, J. Biol. Chem., 1992, 267, 4253–4269 CAS.
- D. Svergun, C. Barberato and M. H. J. Koch, J. Appl. Crystallogr., 1995, 28, 768–773 CrossRef CAS.
- C. Hamiaux, T. Prangé, M. Riès-Kautt, A. Ducruix, S. Lafont, J. P. Astier and S. Veesler, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 103–113 CrossRef CAS PubMed.
- C. Hamiaux, J. Pérez, T. Prangé, S. Veesler, M. Riès-Kautt and P. Vachette, J. Mol. Biol., 2000, 297, 697–712 CrossRef CAS PubMed.
- M. Gottschalk, K. Venu and B. Halle, Biophys. J., 2003, 84, 3941–3958 CrossRef CAS.
- P. Meakin, Adv. Colloid Interface Sci., 1988, 28, 249–331 CrossRef CAS.
- D. Hopwood, C. R. Allen and M. McCabe, Histochem. J., 1970, 2, 137–150 CrossRef CAS.
- E. F. Jansen, Y. Tomimatsu and A. C. Olson, Arch. Biochem. Biophys., 1971, 144, 394–400 CrossRef CAS.
- Y. Tomimatsu, E. F. Jansen, W. Gaffield and A. C. Olson, J. Colloid Interface Sci., 1971, 36, 51–64 CrossRef CAS.
- S. C. Mohapatra and J. T. Hsu, Biotechnol. Tech., 1994, 8, 13–16 CrossRef CAS.
- F. Persson and B. Halle, J. Am. Chem. Soc., 2013, 135, 8735–8748 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SAXS profiles from protein solutions and form factors (Fig. S1); uncorrected PCFs (Fig. S2); back-calculated structure factors (Fig. S3); calculated form factors of BPTI monomers and decamers (Fig. S4); timing for time-resolved SAXS profiles (Table S1). See DOI: 10.1039/c3cp54219j |
| This journal is © the Owner Societies 2014 |