 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA multinuclear solid state NMR, density functional theory and X-Ray diffraction study of hydrogen bonding in Group I hydrogen dibenzoates†
Gregory J. Reesa, Stephen P. Daya, Alberth Larib, Andrew P. Howesa, Dinu Iugaa, Mateusz B. Pitakc, Simon J. Colesc, Terry L. Threlfallc, Mark E. Lightc, Mark E. Smithad, David Quigleya, John D. Wallis*b and John V. Hanna*a
aDepartment of Physics, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: j.v.hanna@warwick.ac.uk; Fax: +44 (0)247 6150954; Tel: +44 (0)247 6150806
bSchool of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham, NG11 8NS, UK. E-mail: john.wallis@ntu.ac.uk; Fax: +44 (0)115 8488077; Tel: +44 (0)115 8488053
cNational Crystallography Service, School of Chemistry, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: S.J.Coles@soton.ac.uk; Fax: +44 (0) 238 0596723; Tel: +44 (0) 238 0596721
dLancaster University, Bailrigg, Lancaster LA1 4YW, UK. E-mail: m.e.smith@lancaster.ac.uk; Fax: +44 (0)152 4592001; Tel: +44 (0)152 592001
First published on 6th September 2013
Abstract
An NMR crystallographic approach incorporating multinuclear solid state NMR (SSNMR), X-ray structure determinations and density functional theory (DFT) are used to characterise the H bonding arrangements in benzoic acid (BZA) and the corresponding Group I alkali metal hydrogen dibenzoates (HD) systems. Since the XRD data often cannot precisely confirm the proton position within the hydrogen bond, the relationship between the experimental SSNMR parameters and the ability of gauge included plane augmented wave (GIPAW) DFT to predict them becomes a powerful constraint that can assist with further structure refinement. Both the 1H and 13C MAS NMR methods provide primary descriptions of the H bonding via accurate measurements of the 1H and 13C isotropic chemical shifts, and the individual 13C chemical shift tensor elements; these are unequivocally corroborated by DFT calculations, which together accurately describe the trend of the H bonding strength as the size of the monovalent cation changes. In addition, 17O MAS and DOR NMR form a powerful combination to characterise the O environments, with the DOR technique providing highly resolved 17O NMR data which helps verify unequivocally the number of inequivalent O positions for the conventional 17O MAS NMR to process. Further multinuclear MAS and static NMR studies involving the quadrupolar 7Li, 39K, 87Rb and 133Cs nuclei, and the associated DFT calculations, provide trends and a corroboration of the H bond geometry which assist in the understanding of these arrangements. Even though the crystallographic H positions in each H bonding arrangement reported from the single crystal X-ray studies are prone to uncertainty, the good corroboration between the measured and DFT calculated chemical shift and quadrupole tensor parameters for the Group I alkali species suggest that these reported H positions are reliable.
Introduction
The characterisation of hydrogen bonds between organic or biological molecules studied by 1H, 13C and 15N solid state NMR has become very common in the past few decades.1–6 Hydrogen bonding has a critical role in the higher order structure formation in proteins, DNA, RNA, polymers and in more general crystal packing and self-assembly phenomena;7–9 it is interesting to note that a new definition of hydrogen bonding has been published recently by an IUPAC task force.10 Hydrogen bonding is commonly studied by infra-red (IR) spectroscopy, solution state NMR and neutron diffraction.11–13 Neutron diffraction is a facile method of measuring the hydrogen bonding interaction as it gives precise atomic positions and bond lengths.14 The IR stretching frequencies have been shown to be proportional to these distances determined by neutron diffraction, however it suffers from the limitation of resolution when multiple hydrogen bonded sites are present in the sample.15–17 Solid state NMR (SSNMR) is a powerful technique for the elucidation of the local structure by providing a description of the short-range environment around each nucleus, which naturally complements the longer-range structural information obtained by diffraction techniques.Most SSNMR studies of H bonding arrangements have traditionally focussed on the more straightforward spin-½ nuclei (i.e.1H, 13C, 15N) that are typically engaged in this structural motif.3,4,18,19 However, with the availability of much higher magnetic field strengths and a greater understanding of the experimental methodologies required to treat quadrupolar (I > ½) nuclei, 17O static and magic-angle-spinning (MAS) NMR have now become a routine analytical tool for many chemical and materials systems, including those which are characterised by H bonding schemes.20–27
Earlier work on small organic molecules and biological systems has attempted to describe the ‘strength’ of the H bond using the precise measurement of one or more NMR interaction parameter(s).18,19,28–35 From both the 1H solution and solid state MAS NMR studies the 1H isotropic chemical shift (δiso) of the carboxylic acid protons was shown to vary substantially over a 10–25 ppm range as the deshielding (i.e. tendency to higher ppm values) increases with the increased H bond character.28,30–32 For example, previous 1H measurements have reported a value of δiso 12.7 ppm for the carboxyl proton in benzoic acid (BZA).31
Other studies have focussed on the 13C carboxylic acid chemical shift anisotropy (CSA) and the sensitivity of specific tensor components towards the H bonding phenomena. As depicted in Fig. 1, the δ11 element of the CSA tensor represents the least shielded (most downfield) component of the 13C static lineshape, and it has been shown to shift to a lower ppm value (upfield) upon deprotonation and shift to a higher ppm value when protonated. The δ33 element is the most shielded (i.e. most upfield) component and it has been demonstrated to be located within the plane of the CO2H functional group, while the δ22 element is aligned along the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and varies substantially with the H bond length.4,18 In a similar fashion, the 17O CS and EFG tensorial elements of the 17O SSNMR resonance exhibit similar trends and have been reported to exhibit considerable variation with the decreasing H bond length. Preceding work on carboxylic acid-bearing systems has determined that the δ11 element of the O–H moiety, and the δ22 element of the CO moiety comprising the CO2H group are aligned nearly parallel with the strong H bond vector, and they shift away and become misaligned as the H bond strength decreases (i.e. H bond length increases).20,35 Wu et al. have previously undertaken 17O MAS NMR studies on a range of small molecules which included benzoic acid (BZA) and potassium hydrogen dibenzoate (KHD) and measured the electric field gradient and chemical shift tensors. These studies confirmed previous nuclear quadrupole resonance (NQR) measurements, and from the deconvolution of these tensorial interactions it was deduced that the hydrogen bond distance was reflected in the anisotropic parameters.24,25
O bond and varies substantially with the H bond length.4,18 In a similar fashion, the 17O CS and EFG tensorial elements of the 17O SSNMR resonance exhibit similar trends and have been reported to exhibit considerable variation with the decreasing H bond length. Preceding work on carboxylic acid-bearing systems has determined that the δ11 element of the O–H moiety, and the δ22 element of the CO moiety comprising the CO2H group are aligned nearly parallel with the strong H bond vector, and they shift away and become misaligned as the H bond strength decreases (i.e. H bond length increases).20,35 Wu et al. have previously undertaken 17O MAS NMR studies on a range of small molecules which included benzoic acid (BZA) and potassium hydrogen dibenzoate (KHD) and measured the electric field gradient and chemical shift tensors. These studies confirmed previous nuclear quadrupole resonance (NQR) measurements, and from the deconvolution of these tensorial interactions it was deduced that the hydrogen bond distance was reflected in the anisotropic parameters.24,25
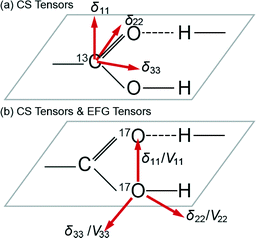 | ||
| Fig. 1 A schematic representation of the (a) 13C chemical shift (CS) tensor, and (b) 17O chemical shift and electric field gradient (EFG) tensors involved in carboxylic hydrogen bonded systems. | ||
In this work, a combined multinuclear solid state nuclear magnetic resonance (SSNMR), X-ray diffraction (XRD) and density functional theory (DFT) approach to the study of the H bonding in benzoic acid (BZA) and the full series of its corresponding Group I alkali metal (Li, Na, K, Rb and Cs) hydrogen dibenzoate/dihydrogen tribenzoate salts is presented. The Li, Rb and Cs hydrogen dibenzoate (HD) and the Na dihydrogen tribenzoate (DT) variants are a novel and unreported family of structures which represent how the increasing ionic radius of a monovalent cation can perturb the hydrogen bonding arrangement. From this structural series, the corresponding influences of both the H bonding arrangement and the Group I metal cation size upon the 1H, 13C and 17O NMR tensorial interactions can be measured, with the long-range periodicity and short-range hydrogen bonding arrangement around the metal site being rationalised by density functional theory (DFT) calculations.
Tightly H bonded systems can be classified into two predominant groups differentiated by the specific types of intermolecular bonding between carboxylic groups; these have been confirmed by work on the relevant crystal structures.36 Type A occurs when two carboxyl groups are related by a symmetry element in the crystallographic frame, hence there is effectively one O environment present due to the proton oscillation between these adjacent O positions with the averaged H position sitting symmetrically disposed between them. Type B is relevant when the two carboxyl groups are chemically inequivalent and thus not symmetry related; in this case the proton is disposed closer to one of the carboxylate groups effectively creating two different oxygen positions (as in the case of BZA). The hydrogen dibenzoate (HD) system has the potential to form H bonds between the carboxylate and carboxylic acid groups, and have been notionally described as ‘negative charge assisted hydrogen bonds’.37 The aim of this study is to invoke an NMR crystallography approach to analyse the true type of H bonding arrangement in the Group I alkali metal (Li, K, Rb and Cs) hydrogen dibenzoate salts, although corresponding studies on the NaTD are not reported since only a few viable crystals suitable for an X-ray structural analysis were isolated. Through the use of accurate energy minimization and geometry optimisation algorithms in fully periodic CASTEP 6.01 DFT code, the refinement of the proton position in each H bonding scheme and the subsequent NMR parameter calculation should provide an accurate description when constrained against experimental 1H MAS NMR measurements and XRD data. This primary level of description will facilitate a more involved analysis of the 13C CSA tensor, and the 17O CSA and EFG tensors, as the increasing ionic radius of the monovalent Group 1A cation directly perturbs both the H bonding arrangement and the carboxylate group.
Results and discussion
Crystallography
The crystallographic structure determination of BZA has been previously completed by numerous workers, with the relatively recent neutron diffraction study performed at 20 K by Wilson et al.38 being chosen for the DFT calculations. The hydrogen dibenzoates of Li, K, Rb and Cs were prepared by the reaction of BZA with half an equivalent of the corresponding metal hydroxide under conditions where the product slowly crystallised from the reaction mixture. The corresponding reaction with sodium hydroxide gave crystals of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 benzoate:benzoic acid complex better referred to as a dihydrogen tribenzoate (DT) complex.39 The Li, Rb and Cs hydrogen dibenzoates (HD) and the NaDT complex, have not been previously reported. The crystal structures of these materials were determined at a low temperature (typically 120 K) and their coordination motifs and crystal packing schemes are given in Fig. 2 and 3, respectively, with their structural parameters summarised in Table 1. A reproducible method of crystallising the NaDT complex was not identified, with only a few viable crystals being isolated for an X-ray structural analysis; hence, no SSNMR studies have not been reported for this system. All the structures contain M2O2 rings where M is the alkali metal ion and the O atoms come from two benzoate anions. These are roughly square in shape, and formed into one dimensional polymers in the case of the Li and Na salts, and into two dimensional polymers for K, Rb and Cs so that the coordination number is 6 or 8, the former being the one commonly isolated for RbHD.
:2 benzoate:benzoic acid complex better referred to as a dihydrogen tribenzoate (DT) complex.39 The Li, Rb and Cs hydrogen dibenzoates (HD) and the NaDT complex, have not been previously reported. The crystal structures of these materials were determined at a low temperature (typically 120 K) and their coordination motifs and crystal packing schemes are given in Fig. 2 and 3, respectively, with their structural parameters summarised in Table 1. A reproducible method of crystallising the NaDT complex was not identified, with only a few viable crystals being isolated for an X-ray structural analysis; hence, no SSNMR studies have not been reported for this system. All the structures contain M2O2 rings where M is the alkali metal ion and the O atoms come from two benzoate anions. These are roughly square in shape, and formed into one dimensional polymers in the case of the Li and Na salts, and into two dimensional polymers for K, Rb and Cs so that the coordination number is 6 or 8, the former being the one commonly isolated for RbHD.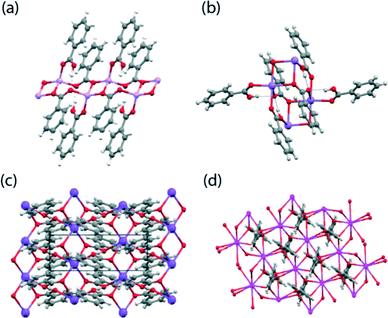 | ||
| Fig. 2 The local coordination motifs for the (a) LiHD, (b) NaDT, (c) KHD and (d) monoclinic CsHD systems. | ||
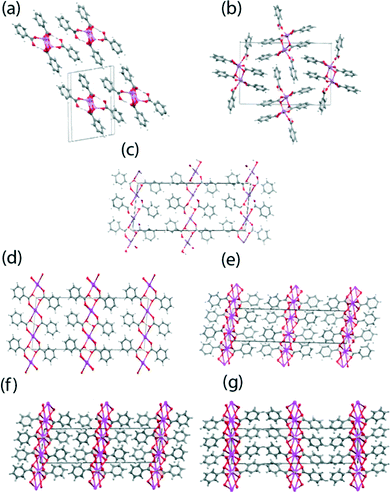 | ||
| Fig. 3 The crystal packing schemes for the (a) LiHD, (b) NaDT, (c) KHD, (d) RbHD (6 coordinate monoclinic), (e) RbHD (8 coordinate monoclinic), (f) CsHD (monoclinic) and (g) CsHD (orthorhombic) systems. | ||
| Sample | LiH(PhCO2)2 LiHD | Na(PhCO2)·2(PhCO2H) NaDT | KH(PhCO2)2 KHD | RbH(PhCO2)26-coordinate RbHD(6) | RbH(PhCO2)28-coordinate RbHD(8) | CsH(PhCO2)2 monoclinic CsHD(m) | CsH(PhCO2)2 orthorhombic CsHD(o) |
|---|---|---|---|---|---|---|---|
| Crystal system | triclinic | monoclinic | monoclinic | monoclinic | monoclinic | monoclinic | orthorhombic |
| a/Å | 5.3600(3) | 5.8077(1) | 29.586(2) | 29.347(4) | 29.0315(9) | 28.9095(8) | 28.3393(19) |
| b/Å | 8.5693(7) | 14.4331(4) | 3.7964(3) | 3.8697(5) | 6.9630(2) | 7.0509(2) | 7.0215(3) |
| c/Å | 13.4999(11) | 22.0139(6) | 11.1214(8) | 11.3973(16) | 6.6156(2) | 6.7779(2) | 6.9066(4) |
| α/° | 98.830(4) | 90 | 90 | 90 | 90 | 90 | 90 |
| β/° | 93.800(5) | 97.047(2) | 97.096(9) | 95.478(7) | 99.297(2) | 99.970(2) | 90 |
| γ/° | 95.524(5) | 90 | 90 | 90 | 90 | 90 | 90 |
| V/Å3 | 607.80(8) | 1831.33(8) | 1239.59(16) | 1288.4(3) | 1319.75(7) | 1360.73(7) | 1374.31(14) |
| Space group | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) | P21/c | C2/c | C2/c | C2/c | C2/c | Pbcn |
| Z | 2 | 4 | 4 | 4 | 4 | 4 | 4 |
| T/K | 120 | 120 | 120 | 100 | 120 | 120 | 150 |
| R(F, F2 > 2Θ) | 0.054 | 0.041 | 0.043 | 0.025 | 0.028 | 0.046 | 0.031 |
| Coordination number at M | 4 | 6 | 6 | 6 | 8 | 8 | 8 |
| O–H–O/Å, | 1.03(3) | 0.87(2) | 0.84(3) | 1.2374(13) | 1.249(13) | 1.236(8) | 1.233(5) |
| 1.59(3) | 1.80(2) | 1.62(3) | 1.2374(13) | 1.249(13) | 1.236(8) | 1.233(5) | |
| O–O/Å; | 2.6128(17) | 2.6641(14) | 2.456(3) | 2.475(3) | 2.456(3) | 2.457(3) | 2.457(3) |
| O–H–O/° | 171(3) | 173(3) | 174(4) | 180* | 159(6) | 167(6) | 170(7) |
| O–H–O/Å, | 0.92(2) | ||||||
| 1.64(2) | |||||||
| O–O/Å; | 2.5625(14) | ||||||
| O–H–O/° | 177(2)o | ||||||
| O–C–O/Å | 1.257(2) | 1.2482(15) | 1.244(2) | 1.243(2) | 1.233(3) | 1.229(2) | 1.225(3) |
| 1.273(2) | 1.2847(15) | 1.293(2) | 1.298(2) | 1.298(3) | 1.302(3) | 1.299(4) | |
| 1.231(2) | 1.2273(16) | ||||||
| 1.316(2) | 1.3194(16) | ||||||
| 1.2192(17) | |||||||
| 1.3262(17) | |||||||
| M2O2 rings: | 1.919(3) | 2.3326(10) | 2.6895(14) | 2.8206(13) | 2.9641(18) | 3.0631(13) | 3.063(2) |
| M–O x2/Å; | 1.981(3) | 2.3859(10) | 2.8110(14) | 2.9688(13) | 2.9956(19) | 3.0660(14) | 3.090(2) |
| angle at M and O/° | 92.22(13) | 84.00(4) | 90.09(6) | 92.89(5) | 81.11(5) | 80.91(4) | 80.86(6) |
| 87.78(13) | 96.00(4) | 95.39(6) | 99.42(5) | 98.89(5) | 99.09(4) | 99.14(6) | |
| 87.26(4) | 83.85(4) | 3.1561(19) | 3.2230(14) | 3.231(2) | |||
| 3.210(2) | 3.2459(15) | 3.286(2) | |||||
| 73.77(5) | 75.15(4) | 74.80(6) | |||||
| 106.23(5) | 104.85(4) | 105.20(6) | |||||
| M–O | 1.918(3) | 2.3322(11) | 2.7641(14) | 2.9448(15) | |||
| outside M2O2 ring/Å | 1.975(3) | 2.3436(11) | |||||
| 2.6461(11) | |||||||
| 2.6693(10) |
For LiHD two Li atoms and an O atom from each of two benzoate anions form a centrosymmetric Li2O2 grouping, and the second oxygen atoms from the two benzoates act as coordinating ligands to the next Li2O2 ‘squares’ in the +a and −a directions to form the coordination polymer, in which the ‘squares’ are linked by an eight-membered ring involving four O, two C and two Li atoms (see Fig. 2(a) and 3(a)). A benzoic acid molecule completes the structure, with the carbonyl oxygen atom providing the fourth coordination site at the Li position, and the −OH group forming a H bond with the benzoate O atom. The CO and C–O bonds of the benzoic acid are clearly distinguished (1.231(2) and 1.316(2) Å). There are thus four distinct O atom environments: an OH, a CO group which coordinates one Li, and two benzoate O atoms, one of which coordinates one Li ion and forms a H bond, while the second one coordinates two Li ions as part of the Li2O2 square. The NaDT crystal structure is related to the LiHD structure but an additional benzoic acid molecule coordinates Na ions of adjacent “squares” by its carbonyl O and by its hydroxyl O, so expanding the metal coordination number to 6. The OH group of this benzoic acid also forms a hydrogen bond with the linking benzoate O atom which is thus involved in two roughly perpendicular H bonds as well as coordinating one Na ion, while the second benzoate O atom is just involved in a Na2O2 square (see Fig. 2(b) and 3(b)).
The crystal structures of the K, Rb and Cs hydrogen dibenzoates comprise two-dimensional coordination polymers with tight hydrogen bonds in which the O⋯O separations lie in the narrow range of 2.456–2.475 Å; these distances are significantly shorter than in the Li and Na salts (2.562–2.664 Å). In the monoclinic KHD salt the six-coordinate K ions are organised in layers perpendicular to the a axis (see Fig. 2(c) and 3(c)). There are chains of coplanar K2O2 motifs which share K ions running along the b direction, with the line of K ions lying on a two-fold axis. In contrast the M2O2 motifs in the Li and Na salts are centrosymmetric and do not share metal ions. The benzoate O atoms belonging to the K2O2 motifs are not involved in H bonding, while the second benzoate O atoms coordinate K ions in adjacent lines and so eight-membered rings are formed. The acidic H atom is located between two O atoms of this ring. These two O atoms are 2.456(3) Å apart, and related by a centre of symmetry. Difference electron density maps showed two positions for this H atom, which were refined as each 50% occupied (O–H: 0.84(3)); H⋯O: 1.62(3) Å, angle at H: 174(4)°). RbHD is isostructural to KHD and contains six-coordinate Rb ions (Fig. 3(d)). The difference electron density map did not show clear evidence for two positions for the (O)H atom and so the H atom is positioned on a centre of symmetry between the two O atoms which are 2.475(3) Å apart. The H atom refined with a large isotropic displacement parameter (0.083(17) Å2), indicative of some disorder in its position. On one occasion a crystal of a polymorph with eight-coordinate Rb ions was obtained (Fig. 3(e)) and this is isostructural with the monoclinic Cs analogue described below. Powder diffraction data shows that the six-coordinate polymorph is predominant in our samples of RbHD. The CsHD analogue has two polymorphs, one crystallising in the monoclinic C2/c space group and the other crystallising in the orthorhombic space group Pbcn, although their overall crystal packing arrangements are very similar (see Fig. 3(f) and 3(g)). The eight-coordinate Cs ions are organised in layers and coordinated by benzoate anions such that each metal ion is involved in four centrosymmetric Cs2O2 rhombus-like motifs, two formed by bonds to non-H bonded benzoate O atoms and two by longer bonds to the H bonded benzoate O atoms. In the monoclinic phase the H bonds between these latter rings involve two O atoms 2.457(3) Å apart, and these two O atoms subtend an angle of 44.49(4)° at the Cs position. A similar observation was made for the isostructural Rb polymorph. For the orthorhombic phase of CsHD the unit cell lengths are very similar to those of the monoclinic phase, but the β angle has decreased by ~10° (90 v 99.97°). There is a significant amount of crystallographic evidence for such symmetrical O–H–O hydrogen bonds (vide supra), and it is notable that we have both symmetrical and unsymmetrical tight O–H–O hydrogen bonds among the K, Rb and Cs hydrogen dibenzoates, though we emphasize that for the Rb and Cs salts, there is less certainty in the interpretation of the electron density in the O–H–O region, and the high isotropic displacement parameter for the H atom in these cases may indicate variation in the position of the H atom. The nearest comparison is to the neutron diffraction studies on K and Cs hydrogen trifluoroacetates which both show short symmetrical O–H–O hydrogen bonds with an O⋯O separation of 2.436 Å and isotropic displacement parameters of 0.016 Å2 for both salts at 20 K.39 Future neutron diffraction studies on the hydrogen dibenzoate salts will provide useful further insights. More complete descriptions of these crystal structures are available in the ESI.†
Solid State NMR
The 1H MAS NMR data from BZA and the Group I (Li, K, Rb, Cs) HD systems is shown in Fig. 4, and the measured and GIPAW DFT calculated 1H chemical shift tensor information is summarised in Table 2. Each 1H MAS NMR spectrum exhibits multiple resonances which represent the individual proton environments in each sample, with the majority of the resonances in each spectrum belonging to the aromatic region. However, there is a distinct downfield resonance at δiso > 10 ppm which is indicative of a deshielded proton participating in a H bond arrangement. The BZA and LiHD systems reveal resonances at δiso 13.2 and δiso 13.0 ppm, respectively, which represent comparatively weak H bonding scenarios. The shift of δiso 13.2 ppm for BZA obtained from a fast 1H MAS experiment at 11.7 T differs from a previously reported value of δiso 12.7 ppm which was obtained using the 1H CRAMPS method, and probably suffers from poorer resolution due to much slower MAS frequencies and a lower magnetic field strength.31 An increase in the monovalent cation ionic radius causes a dramatic downfield shift in the resonance position to δiso 20.8, 20.2 and 20.9 ppm for the KHD, RbHD and CsHD salts, respectively. These large deshielded shifts are indicative of tight H bonds which has been facilitated by the changing coordination around the alkali metal centre (from the initial BZA dimeric structure) as the increasing size of the alkali cation influences the type of H bonding observed. From Table 1 the coordination number around the Li metal centre is 4, around the K and Rb centres it is 6, and around the Cs position it is 8. The increasing ionic radius of each metal centre impacts upon the resultant strength of the each H bond.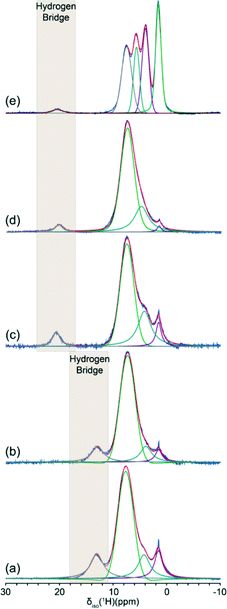 | ||
| Fig. 4 Fast 1H MAS NMR data (νr = 27.5 kHz, ν0 =500.1 MHz) acquired from (a) BZA, (b) LiHD, (c) KHD, (d) RbHD and (e) CsHD. The hydrogen bonded proton is marked in the shaded grey region. | ||
| Sample | δiso(MAS)1Ha | δiso1H | δiso(MAS)13Cb | δiso(CSA)13Cc | δ1113C | δ2213C | δ3313C | Ωd | Κd | Δδe | ηδe | δiso(CSA)13Cf | δ1113C | δ2213C | δ3313C | Ω | κ | Δδ | η |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Directly measured from the experimental 1H MAS NMR data.b Directly measured from the experimental 13C CPMAS NMR data.c Calculated as the average of the experimentally derived δ11, δ22, δ33 values; δiso(CSA) = (δ11 + δ22 + δ33)/3.d Herzfeld–Berger shift convention: δ11 ≥ δ22 ≥ δ33, δiso = (δ11 + δ22 + δ33)/3, Ω = (δ11 − δ33), κ = 3(δ22 − δiso)/ (δ11 − δ33) (1 ≥ κ ≥ −1).e Haeberlen shift convention: |δ33 − δiso| ≥ |δ11 − δiso| ≥ |δ22 − δiso|, δiso = (δ11 + δ22 + δ33)/3, Δδ = δ33 − 1/2(δ11 + δ22) = 3/2(δ33 − δiso), ηδ = (δ22 − δ11)/ (δ33 − δiso) (1 ≥ ηδ ≥ 0).f Calculated as the average of the NMR-CASTEP calculated δ11, δ22, δ33 values; δiso(CSA) = (δ11 + δ22 + δ33)/3.g Determined by root-mean-square (RMS) statistical analysis. | |||||||||||||||||||
| CASTEP | CASTEP | CASTEP | CASTEP | ||||||||||||||||
| Site | O⋯1HO | −13COOH | −13COOH | −13COOH | −13COOH | −13COOH | −13COOH | ||||||||||||
| Error (ppm/±) | ±0.25 | ±0.5 | ±2g | ±2 | ±2 | ±2 | ±2 | ±0.05 | ±2 | ±0.05 | |||||||||
| BZA | 13.2 | 14.5 | 172 | 172 | 223 | 187 | 107 | 116 | 0.38 | −98 | 0.54 | 172 | 246 | 174 | 97 | 149 | 0.03 | −113 | 0.96 |
| LiHD | 13.0 | 13.2 | 174 | 174 | 227 | 189 | 105 | 122 | 0.38 | −103 | 0.54 | 172 | 241 | 180 | 95 | 146 | 0.16 | −115 | 0.79 |
| KHD | 20.8 | 21.2 | 176 | 175 | 237 | 179 | 108 | 130 | 0.12 | −101 | 0.86 | 176 | 247 | 178 | 103 | 176 | 0.04 | −109 | 0.95 |
| RbHD | 20.2 | 20.7 | 176 | 176 | 243 | 178 | 107 | 135 | 0.05 | −104 | 0.93 | 175 | 243 | 184 | 98 | 145 | 0.18 | −115 | 0.77 |
| CsHD | 20.9 | 21.1 | 173 | 173 | 242 | 169 | 109 | 133 | −0.10 | 103 | 0.86 | 173 | 233 | 192 | 93 | 140 | 0.42 | −120 | 0.51 |
The ab initio GIPAW DFT calculations of the 1H chemical shift tensor parameters using the CASTEP code confirms these observations (see Table 2). They accurately predict the experimentally observed downfield shift in δiso of ~8 ppm upon moving from BZA and LiHD to the K, Rb and Cs HD structures bearing the larger cations. Furthermore, they also corroborate the observation that the BZA dimer has a marginally more deshielded proton when directly compared to the LiHD. For the purposes of this study, the H bonding in the BZA and LiHD systems can be described as weak while the bonds in the heavier Group I alkali metal HD analogues (KHD, RbHD and CsHD) can be described as strong.
The corresponding 13C MAS NMR data from these systems were acquired using the cross-polarisation (CP) technique and isotopic 13C labelling of the carboxylic acid functionality to maximise the accuracy of the 13C CSA measurements. Each 13C CPMAS NMR measurement captured the CSA manifold under slow MAS conditions, with the data being acquired at spinning frequencies of νr = 0.8, 1.5 and 12 kHz to allow a detailed sideband analysis to be undertaken. These 13C CPMAS NMR spectra are shown in Fig. 5 and the measured chemical shift tensor data are collated in Table 2. The 13C CSA data yields tensorial information for the CO2H site which has previously been shown to be proportional to the H bond strength.3,4,18 This is clearly demonstrated by the δ22 tensor element in this series of data which is aligned along the CO bond and prone to variation with the H bond length. From Table 2 it can be observed that δ22 = 187.1 and 189.2 ppm for the more weakly H bonded BZA and LiHD systems, respectively, and this element systematically shifts upfield to δ22 = 179.3, 178.4 and 168.6 ppm for the more strongly H bonded KHD, RbHD and CsHD systems, respectively. The 13C isotropic chemical shift values measured directly by simulation of each fast MAS spectrum (δiso(MAS)) and by averaging the individual tensorial components (δiso(CSA) = (δ11 + δ22 + δ33)/3) are in excellent agreement, thus verifying the accuracy of the sideband analysis. However, neither of these isotropic values, or the other CSA tensor elements δ11 and δ33, exhibit a systematic correlation with the H bonding strength or increasing cation size.
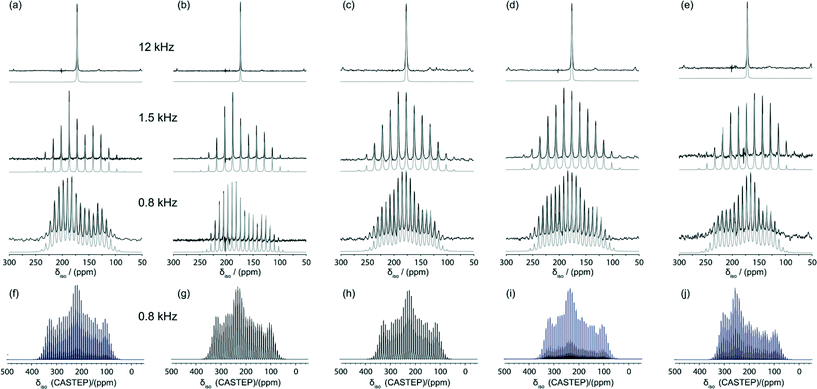 | ||
| Fig. 5 13C MAS NMR data using variable MAS frequencies of νr = 12, 1.5 and 0.8 kHz (ν0 = 100.3 MHz) obtained from (a) BZA, (b) LiHD, (c) KHD, (d) RbHD and (e) CsHD. The respective Simpson41 simulations of the CASTEP parameters at νr = 0.8 kHz (ν0 = 100.3 MHz) for (f) BZA, (g) LiHD, (h) KHD, (i) RbHD and (j) CsHD are shown below. | ||
The GIPAW DFT calculations of the 13C chemical shift tensor parameters using the CASTEP code are also summarised in Table 2. Although the calculated values of δiso(CSA) agree well with δiso(MAS) and δiso(CSA) values derived from measurement, there is some disparity between the trends displayed by the individual tensor elements. In particular, the calculated δ22 values display a trend that is opposite to that of its δ22 experimental counterpart. This suggests that the calculated δ11, δ22 and δ33 values possess systematic errors that cancel when considering the total average value (i.e. δiso(CSA)), and these values may be influenced by the large amplitude motion and delocalised nature of the H position in the H bond. Furthermore, the measured and calculated composite parameters such as Ω, κ, Δδ and ηδ also show no discernible correlation, as evidenced in Table 2 and the direct comparisons shown in Fig. 5. As motion is deemed to be the primary source of this error, SEDC (dispersion correction) calculations were invoked and they show no improvement on the original data trends (see ESI†).
The O sites present in the hydrogen dibenzoate systems which participate directly in the H bond are an excellent SSNMR indicator of local site symmetry, with the anisotropic terms elucidated from 17O MAS NMR data providing a useful insight into the strength of the hydrogen bond.2017O enriched BZA was prepared from α,α,α-trichlorotoluene and H217O in a microwave reactor, and this was a precursor for the preparation of the 17O labelled HD and DT systems. The 17O MAS NMR data acquired from these labelled complexes, obtained using a spinning frequency νr = 27.5 kHz, are shown in Fig. 6 together with their total simulation and deconvolution. In general, simple carboxylic acids contain two chemically inequivalent O environments, namely the carbonyl (CO) and hydroxyl (O–H) positions. It would be expected that both these positions would yield very different 17O isotropic chemical shifts (δiso), quadrupole coupling constants (CQ) and asymmetry parameters (ηQ). Previous work by Wong et al. has suggested that the CO groups have 17O isotropic shifts in the range δiso 310–340 ppm, while those of the O–H group are more shielded and shifted upfield to a δiso 170–190 ppm range. Due to the low point symmetry of the O position in the CO environment it was suggested that the 17O CQ values for this moiety will be >7 MHz, while CQ values for the O–H moiety will be situated in a 6.5–7 MHz range.20
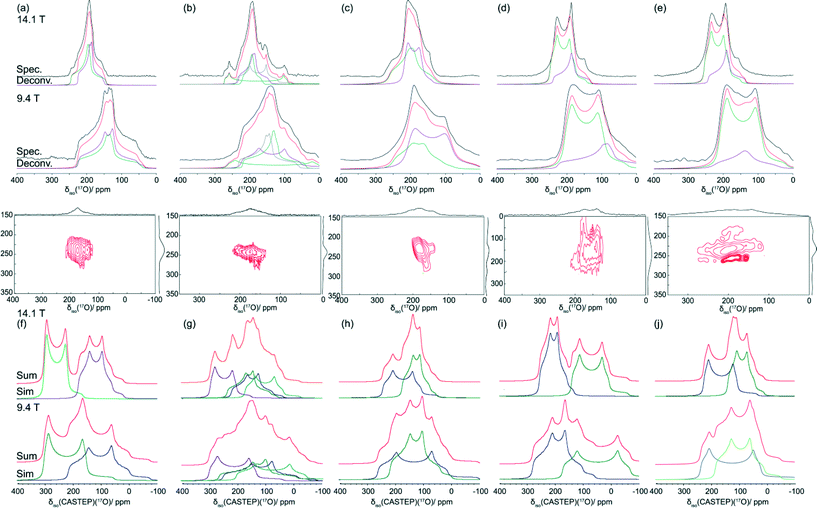 | ||
| Fig. 6 17O MAS NMR data acquired at B0 = 14.1 and 9.4 T (νr = 27.5 kHz, ν0 = 81.33 and 54.22 MHz) for (a) BZA, (b) LiHD, (c) KHD, (d) RbHD and (e) CsHD are compared with 17O central transition lineshapes simulated from CASTEP derived δiso, CQ and ηQ values using Simpson for (f) BZA, (g) LiHD, (h) KHD, (i) RbHD and (j) CsHD. The deconvolution of these experimental and calculated 17O MAS NMR data are shown in colour with each spectrum presented in green and purple, with the sum of the fits in red. The 17O 2D 3QMAS data (B0 = 14.1 T) is shown for all systems to demonstrate the lack of resolution exhibited in the F1 dimension and the limited analytical value it offers in the simulation and understanding of the 1D MAS NMR data. | ||
The 17O MAS NMR data from enriched BZA reported by Wu suffered from signal-to-noise limitations and was evaluated as a single site spectrum. The subsequent lineshape analysis reported that the O positions were characterised by one set of parameters (δiso 230 ppm, CQ = 5.7 MHz, ηQ = 1.0), thus implying that BZA participates in a type A H bonding arrangement.25 More recently published results by Hagaman et al. have shown that there are indeed two sites present characterised with the parameters δiso 236 ppm, CQ = 5.4 MHz, ηQ = 0.99 and δiso = 233 ppm, CQ = 5.6 MHz, ηQ = 0.98.40 This suggests that these O sites are inequivalent and the proton in not equally distributed along the H bond thus invoking a type B H bond classification, as observed in the crystal structure. The 17O MAS NMR results for enriched BZA presented in Fig. 6(a) and Table 3 corroborate Hagaman's analysis with a deconvolution into two sites (δiso 236 ppm, CQ = 6.0 MHz, ηQ = 0.92 and δiso 225 ppm, CQ = 5.6 MHz, ηQ = 0.81), with 17O enrichment necessary for the high quality simulations of the 9.4 and 14.1 T data. Small discrepancies may be caused by a CSA contribution to the lineshape given that the BZA sample in his study possesses a higher 17O content (Hagaman ~35%, this work ~86%) and differing MAS frequencies (Hagaman 15–20 kHz, this work 27.5 kHz). The 3QMAS spectra of 17O enriched BZA in Fig. 6(a) sample shows two observable sites, however due to the significant overlap and broadening in the F1 dimension it is not possible to deconvolute these resonances and elucidate EFG tensor information. This broadening is probably induced by large amplitude motion in the H bond arrangement; as observed in Fig. 3 this is appears to be a phenomenon that effects the quality of all 3QMAS data in this series. This observed broadening infers that the timescale of the H motion must be similar to the timescale of the NMR experiment (i.e. microseconds), thus interfering with the 1H decoupling from both the 13C and 17O nuclei. The GIPAW DFT results also confirm the presence of two O positions (see Table 3), although the NMR CASTEP calculations predict slightly larger CQ values of 7.63 and 7.45 MHz and smaller associated ηQ values of 0.43 and 0.10 for the CO and O–H groups, respectively. In addition, the effect of H motion is readily observed in the correlation between the measured and NMR CASTEP calculated 17O δiso values; while a good agreement is exhibited for the OH group there appears to be very large discrepancy of ~100 ppm observed for the CO moiety.
| Sample | O Site | δiso(MAS)17Oa ppm/±3.0 | CQ17Oa MHz/±0.5 | ηQ17Oa /±0.1 | δiso(PQ)17Ob ppm/±3.0 | PQ17Ob MHz/±0.5 | δiso(MAS)17O CASTEP | CQ17O CASTEP | ηQ17O CASTEP | PQ17Oc CASTEP |
|---|---|---|---|---|---|---|---|---|---|---|
| a Measured from simulation of the experimental 17O MAS NMR data.b Measured from the treatment of the variable B0 field DOR data.c Calculated using NMR-CASTEP derived CQ and ηQ values. | ||||||||||
| BZA | 1 | 236.5 | 5.96 | 0.92 | 262.8 | 8.54 | 336.0 | 7.45 | 0.10 | 7.46 |
| 2 | 224.8 | 5.61 | 0.81 | 242.5 | 7.66 | 229.9 | 7.63 | 0.43 | 7.86 | |
| LiHD | 1 | 315.8 | 12.36 | 0.08 | 303.4 | 10.04 | 328.7 | 7.50 | 0.18 | 7.54 |
| 2 | 236.4 | 7.94 | 0.40 | 257.4 | 9.12 | 270.5 | 7.65 | 0.70 | 8.25 | |
| 3 | 236.3 | 6.21 | 0.88 | 257.2 | 9.67 | 260.8 | 7.96 | 0.55 | 8.35 | |
| 4 | 220.9 | 4.70 | 0.88 | 251.2 | 9.64 | 229.2 | 8.54 | 0.31 | 8.68 | |
| KHD | 1 | 257.6 | 7.40 | 0.69 | 285.9 | 8.28 | 287.0 | 8.63 | 0.30 | 8.76 |
| 2 | 227.4 | 6.41 | 0.16 | 187.1 | 3.71 | 222.2 | 6.45 | 0.58 | 6.80 | |
| RbHD | 1 | 255.7 | 6.96 | 0.32 | 286.8 | 6.54 | 0.56 | 6.87 | ||
| 2 | 239.7 | 6.37 | 0.99 | 206.8 | 8.85 | 0.23 | 8.92 | |||
| CsHD | 1 | 233.6 | 6.44 | 0.18 | 352.8 | 12.27 | 277.7 | 8.72 | 0.14 | 8.75 |
| 2 | 226.7 | 5.51 | 0.99 | 254.5 | 11.07 | 195.2 | 6.86 | 0.42 | 7.06 | |
The single crystal XRD structure determination of the LiHD complex discussed above has been shown to support four crystallographic independent O sites. From the 17O MAS NMR data acquired at 9.4 and 14.1 T (see Fig. 6(b)) four distinct sites can be observed and simulated with confidence. The O sites represented by the two smaller CQ values (i.e. 6.21 and 4.70 MHz) are tentatively assigned to the carbonyl and hydroxyl environments of the benzoic acid, respectively, while the two sites with the larger CQ values (i.e. 12.36 and 7.94 MHz) are assigned to the central Li2O2 moiety.
The Group I alkali metal HD systems supporting the larger cations (i.e. K, Rb, Cs) revert back to a two O environment model comprised of K–O–K and K–O–H species. A previous study of the KHD complex by Wu et al. reveals two O sites, with the K–O–K position characterised by the 17O NMR parameters δiso 287 ppm, CQ = 8.30 MHz, ηQ = 0.23 and the K–O–H position characterised by δiso 213 ppm, CQ = 5.90 MHz, ηQ = 0.55.24 The simulations of the experimental 17O MAS NMR data acquired for the KHD sample in this study (see Fig. 6(c)) reveals the K–O–K species to be described by δiso 257 ppm, CQ = 7.40 MHz, ηQ = 0.69 and for the K–O–H species δiso 227 ppm, CQ = 6.41 MHz, ηQ = 0.16. Hence, for KHD it can be surmised that some discrepancies exist in the 17O isotropic chemical shift and EFG parameters between these studies, particularly for the downfield K–O–K bonded O position. GIPAW DFT calculations for the KHD 17O NMR parameters (see Table 3) are in reasonable agreement with the experimentally determined parameters from this study for the downfield K–O–K bonded O site, and exhibit an excellent agreement with the upfield K–O–H bonded O arrangement. This suggests that the XRD and SSNMR descriptions of the KHD system performed in this work are accurate and are well-constrained by the other methodologies in this combined SSNMR, XRD and DFT approach. From Fig. 6(d) and 6(e) it can be observed that the larger RbHD and CsHD systems reveal very similar 17O MAS NMR spectra. This is expected as the local O environments described by the XRD structure determination are all comparable. The 17O isotropic chemical shifts are confined to a range of δiso 227–256 ppm, and similarly the CQ values fall within a narrow range of 5.51–6.97 MHz. As shown in Table 3, the GIPAW DFT calculated 17O NMR parameters appear to suffer from reduced accuracy when compared to the experimental values. These inaccuracies could be attributed to limitations with the 17O, 87Rb and 133Cs pseudopotentials, and relativistic effects may be contributing to the electronic description of the large Rb+ and Cs+ cations. Nevertheless, the actual H bonding arrangements in RbHD and CsHD are accurately described by the precise relationship between the 1H MAS NMR data and the corresponding GIPAW DFT predictions of the 1H δiso values.
The double rotation (DOR) experiment involves spinning the sample about two angles to enable averaging of both the P2 and P4 terms of the 4th order Legendre polynomial describing the angular dependences of the 2nd order quadrupolar interaction.42,43 This reduces the first order quadrupole broadening by rotation around the more familiar magic angle of 54.7° with respect to B0, and also reduces the second order broadening by spinning at a second angle of 30.6° with respect to the magic angle.35,44–46 An advantage of the DOR technique is that the external B0 dependence of the observed (featureless) centre-of-gravity shift (δcg) can be used to deduce information about the 17O isotropic chemical shift and EFG parameters. For a spin I = 5/2 nucleus it can be derived:
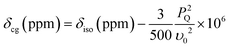 | (1) |
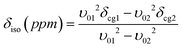 | (2) |
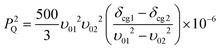 | (3) |
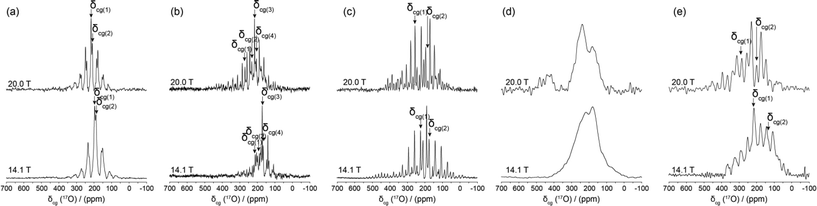 | ||
| Fig. 7 17O DOR NMR data (νr(inner) = 1.2–1.8 kHz, νr(outer) = 5.8–8.8 kHz, ν0 = 81.33 and 115.25 MHz) measured from the (a) BZA, (b) LiHD, (c) KHD, (d) RbHD and (e) CsHD systems. The centre-of-gravity shift (δcg) for each resonance is indicated in all spectra. | ||
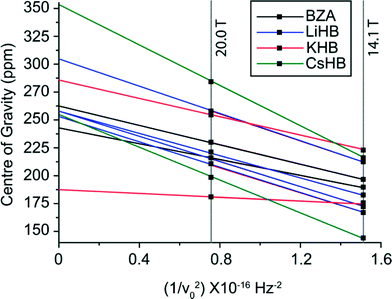 | ||
| Fig. 8 The B0 variation of the centre-of-gravity shifts (δcg) measured from the 17O DOR data for BZA and the Group I alkali HD complexes. | ||
The resolution achieved by the 17O DOR NMR experiment is excellent (FWHM ~230 Hz, ~2 ppm) as highlighted by the BZA, LiHD and KHD data in Fig. 7(a)–7(c); these data serves to confirm the number of chemically inequivalent O positions in each system. 1H decoupling during acquisition was not employed as the motion of the H bonded proton is on a similar timescale to the NMR experiment which effectively recoupled a component of the 1H–17O dipolar interaction, thus broadening each resonance. The same phenomenon was observed during the acquisition of the 17O MAS NMR data presented in Fig. 6. As depicted in Fig. 8, the δiso value is effectively the centre of gravity at an infinite magnetic field strength. For BZA values of δiso 262.8 ppm and δiso 242.5 ppm are determined for the carbonyl and hydroxyl species, respectively, which are marginally higher than δiso values determined by conventional MAS methods (see Table 3). The quadrupole parameters CQ and ηQ are degenerate within the expression for PQ and they cannot be independently determined; since ηQ can vary as 0 ≤ ηQ ≤ 1 the maximum error in CQ generated by this uncertainty is ~15%. The PQ values for BZA determined from the 17O DOR NMR method are 8.54 and 7.66 MHz for the carbonyl and hydroxyl moieties, respectively. Since there is a tendency for higher ηQ values approaching 1 (see corresponding 17O MAS NMR determined values in Table 3) then the scaling of these PQ values by ~15% yields CQ values of 7.18 and 6.51 MHz. Given the complexities of both the 17O MAS and DOR data which possess both quadrupole and CSA contributions which are further affected by inherent H motion, then the overall agreement is acceptable.
The 17O DOR NMR results for LiHD and KHD (see Fig. 7(b) and 7(c)) illustrate the true utility and power of the DOR technique when disorder and chemical shift dispersion are not present within the structure. The chemically inequivalent O positions defining both systems (four in LiHD and two in KHD) are clearly resolved. The δiso and PQ results show some inconsistencies with the MAS derived and NMR-CASTEP calculated results, however, given the limited rotational frequencies achievable with DOR and the large 17O CSA contributions experienced in organic systems,48 and the inability to effectively 1H decouple in the presence of H motion, the overall agreement is acceptable. In contrast, the 17O DOR NMR result for the RbHD systems represents no resolution enhancement and a complete inability to average the quadrupole dominated lineshape. In fact, the resultant DOR lineshape is a factor of ~2.5 times broader than the MAS averaged lineshape at 14.1 T implying that the DOR experiment is recoupling elements of the quadrupolar and dipolar interactions caused by fluctuations in the proton position and its specific frequency of motion. This situation is somewhat retrieved for the CsHD case as both inequivalent O positions are now resolved, however the residual linewidths are not as narrow as those observed in the BZA, LiHD and KHD cases and the apparent resolution is reduced.
An SSNMR analysis of each central Group I alkali metal was performed to characterise the positions within each structure and rationalise the GIPAW DFT calculations of the isotropic chemical shifts (δiso) and EFG parameters (CQ and ηQ) against the changing geometry and H bond strength characterising these arrangements. The 7Li, 39K, 87Rb and 133Cs SSNMR data are shown in Fig. 9(a)–9(d), respectively, and the experimentally measured data and the GIPAW DFT calculated data are summarised in Table 4. As observed in Fig. 9(a) and 9(d), the small 7Li and 133Cs quadrupole moments (Q) afford measurement to be undertaken with the MAS NMR technique. The 7Li MAS NMR spectrum of Fig. 9(a) shows one resonance at δiso 3.38 ppm in agreement with the proposed LiHD structure from the X-ray studies (see above), and the δiso calculated with NMR-CASTEP exhibits a reasonable agreement with this value. Similarly, the 133Cs MAS NMR data of Fig. 9(d) is also characterised by narrow resonances, however in this case both the monoclinic (δiso −188.2 ppm) and orthorhombic (δiso −195.3 ppm) forms are observed simultaneously; this is corroborated by excellent agreement with the NMR-CASTEP values given in Table 4. In both the 7Li and 133Cs cases, the very small CQ values characterising these nuclei induce featureless resonances and the experimental CQ values are likely to be smaller than what is predicted by DFT methods. In particular, for the 133Cs case the spin of I = 7/2 scales the width of the quadrupole interaction accordingly, making evidence of a quadrupole interaction more difficult to observe. Thus, the lack of sensitivity to B0 variation and the obvious absence of satellite transitions sidebands precludes the measurement of the EFG parameters.
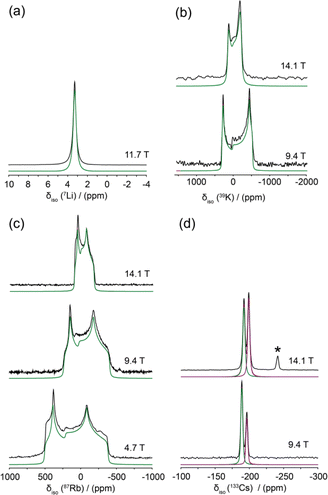 | ||
| Fig. 9 The solid state NMR data of the Group I alkali metals, (a) 7Li MAS (νr = 27.5 kHz), (b) multiple field static 39K, (c) multiple field static 87Rb and (d) multiple field 133Cs MAS (νr = 10 kHz). The resultant simulations are shown below each spectrum in green and purple. The asterisk indicates the presence of Cs benzoate in this compound. | ||
| Sample | Nucleus | δiso(MAS)/δiso (static)a ppm | CQ MHz | ηQ | δiso(MAS) CASTEP | CQ CASTEP | ηQ CASTEP |
|---|---|---|---|---|---|---|---|
| a Measured from a 39K static lineshape.b Measured from a 87Rb static lineshape.6 6-Coordinate.8 8-Coordinate variants.m Monoclinic.o Orthorhombic variants. | |||||||
| LiHD | 7Li | 3.4 | 2.0 | 0.39 | 0.04 | ||
| KHD | 39Ka | 9.8 | 1.38 | 0.09 | −6.2 | 1.30 | 0.15 |
| RbHD | 87Rb6b | −82.08 | 10.12 | 0.34 | −173.8 | 9.41 | 0.69 |
| 87Rb8 | −175.4 | 9.27 | 0.76 | ||||
| CsHD | 133Csm | −188.2 | −184.5 | 0.34 | 0.79 | ||
| 133Cso | −195.3 | −196.7 | 0.34 | 0.79 | |||
In contrast, the 39K and 87Rb measurements were performed with multiple field static (solid echo) NMR experiments which afforded a more complete analysis of the δiso and EFG parameters characterising each position. The 39K values (CQ = 1.38 MHz, ηQ = 0.09) and 87Rb values (CQ = 10.12 MHz, ηQ = 0.34, six-coordinate polymorph only) display a good agreement with the calculated quadrupole parameter values. The 87Rb static lineshape also possesses a small CSA contribution which is very difficult to quantify accurately; if this could be incorporated further improvement in the agreement with the 87Rb calculations could be achieved. Furthermore, the 39K δiso value also exhibits a good agreement with its calculated counterpart, considering that previously established 39K chemical shifts for diagmagnetic materials and complexes span a range of δiso ~ +100 – −100 ppm.49–54 In addition to the 1H MAS NMR data discussed above, this general corroboration between the measured and chemical shift and quadrupolar tensor parameters for these alkali metals also suggests that the reported H crystallographic positions defining each H bonding arrangement are reliable despite the intrinsic uncertainty in these positions from the X-ray data. These results are consistently reliable despite the significantly changing dimensions in the M2O2 rings defining each system. The X-ray measured M–O distances (within each M2O2 ring) summarised in Tables 1 and 5, and the corresponding M–O distances obtained from the geometry optimised isostructural series KHD, RbHD(6) and CsHD(m) (see Table 5 and supplementary data S6†) both indicate that KHD clearly exhibits the shortest M–O distance (2.690 Ǻ), which progressively lengthens to 2.821 and 3.063 Ǻ for RbHD and CsHD, respectively. This dimension is consistent with the significantly increasing ionic radius in the Group I alkali metals.
| Sample | δiso(MAS)1Ha O⋯1HO ppm/±0.25 | δiso(MAS)13Cb −13COOH ppm/±0.5 | δ2213Cc −13COOH ppm/±2.0 | IR stretching frequencyd CO, cm−1 | O–H–O distancee Å | M2O2 rings, M–O distance X-raye Å | M2O2 rings M–O distance DFTf Å | O–H–O anglee degrees | O–C–O anglee degrees |
|---|---|---|---|---|---|---|---|---|---|
| a Directly measured from the experimental 1H MAS NMR data.b Directly measured from the experimental 13C CPMAS NMR data.c The δ22 term of the 13C chemical shift tensor derived from slow MAS.d As measured from powder IR.e As measured from single crystal X-ray data.f Determined from DFT energy minimisation and geometry optimisation.g 20 K, major component, from Wilson et al., see Ref. 38.m Monoclinic variant.o Orthorhombic variant.NM No alkali metal present. | |||||||||
| BZA | 13.2 | 172.2 | 187.08 | 1678.0 | 2.608g | NM | NM | 174.7 | 123.7 |
| LiHD | 13.0 | 173.5 | 189.2 | 1677.3 | 2.613 | 1.919 | 1.915 | 171.0 | 123.0 |
| KHD | 20.8 | 176.3 | 179.3 | 1689.9 | 2.456 | 2.690 | 2.810 | 174.0 | 110.8 |
| RbHD | 20.2 | 176.0 | 178.4 | 1675.6 | 2.475 | 2.820 | 2.930 | 180.0 | 113.6 |
| CsHD | 20.9 | 173.2 | 168.6 | 1633.7 | 2.457 | 3.063 | 3.095 | 168.0m/170.1o | 114.8m/114.3o |
Table 5 summarizes the SSNMR parameters and other indicators from diffraction studies and vibrational spectroscopy that have traditionally helped to define H bond strengths across series of organic and organometallic complexes. The 1H MAS NMR method is the most obvious technique to have recourse to when a characterisation of the H bond strength is required. The 1H δiso values from Tables 2 and 5 clearly show that a stepwise change is exhibited, with the BZA and LiHD systems representing ‘weaker’ H bonding arrangements and the KHD, RbHD and CsHD complexes containing the larger alkali cations signifying ‘stronger’ H bonding situations. This step in the bonding strength is echoed by the 13C δiso values, although the CsHD gives an anomalous result. However, the δ22 element of the 13C CSA (which is aligned along the CO bond) is directly affected by the strength of the H bond interaction, yet it displays a gradual monotonic decrease upon progression through the series from BZA to CsHD. This conflicting behaviour suggests that these indicators describe individual characteristics of what is a more complex process, especially when H motion is involved.
The IR stretching frequency of the carbonyl group is typically used to describe the H bonding interaction.15 It usually gives a broad resonance caused by the variation in the H bonding. The frequencies shown in Table 5 have very limited correlation with the SSNMR data. Other literature methods resort to measuring the O⋯O distance across the hydrogen bond.55 This parameter exhibits a stepwise decrease in length from 2.608 and 2.613 Ǻ for BZA and LiHD, respectively (i.e. the weaker H bonds in this series), to 2.456, 2.455 and 2.457 Ǻ for the isostructural KHD, RbHD and CsHD, respectively (i.e. the stronger H bonds). This behaviour mirrors the clear 1H δiso trend, and also correlates with a less-defined stepwise trend that can also be associated with the δ22 element of the 13C CSA tensor. In contrast, the O–H–O and O–C–O angles show no discernible trend. This suggests that the true complexity of H bonding is not easily rationalised solely by geometric parameters and that SSNMR tensorial parameters play a critical role in these description of this phenomenon.
Conclusions
This work has demonstrated that a NMR crystallography approach involving SSNMR, single crystal X-ray structure determinations and GIPAW DFT calculation of the NMR parameters places realistic constraints on the description of H bonding in organic crystals. XRD methods often struggle to locate H positions accurately, however with the aid of SSNMR and ab initio GIPAW DFT calculations the level of constraint is greatly increased and the overall accuracy of the structure determination can rely on the relationship displayed by these accompanying techniques. The multinuclear SSNMR/GIPAW DFT approach described here has sought to accurately analyse all aspects of the H bonding in alkali metal containing HD systems by investigating nuclei such as 1H, 13C, 17O and 7Li/39K/87Rb/133Cs that are relevant to each of the BZA, LiHD, KHD, RbHD and CsHD H bonded complexes.The important aspects of this work highlight the ability of 1H and 13C MAS NMR to focus on H bonding character in organic systems bearing Group I alkali metal centres. Traditionally, the 1H δiso has been used to describe the H bonding nature of the carboxyl proton, and in this study a stepwise change in H bonding character was observed to separate BZA and LiHD (which are identified as weakly H bonded systems) from KHD, RbHD and CsHD (which are identified as strongly H bonded systems). This trend was precisely predicted by the GIPAW DFT calculation of the 1H δiso values which agree unequivocally with the measured values. Both the 13C δiso value and the δ22 CSA tensor value exhibit a smoother and monotonic progression with increasing alkali cation size, although the 13C δiso value for CsHD is anomalous. GIPAW DFT calculations of these 13C parameters are also of high precision and thus corroborate the different reported trends. The close relationship between the measurement and calculations for both the 1H and 13C parameters suggest that these atoms play quite separate roles in the mechanism of a H bond.
Subsequent 17O MAS and DOR NMR, and associated DFT calculations emphasise the greater difficulties connected with acquiring and treating experimental 17O data, and correlating that data with DFT calculated parameters. The 17O DOR experiment proved a very powerful tool to establish the exact number of inequivalent O sites in each HD system with very high resolution data being acquired for all systems except RbHD. Both 17O MAS and DOR NMR techniques were used to elucidate the δiso and quadrupole parameters such as CQ and ηQ, however the correlation with the DFT calculations was not as precise. The complex data generated by the presence of quadruple and CSA contributions, convoluted with the effects generated by H motion, has compromised the quality of the 17O data simulation and hence the relationship between the experimental and calculated δiso, CQ and ηQ values. Nevertheless, useful correlations were able to be established and it was shown that hydroxyl O tends to have a smaller δiso and CQ value when compared to the carboxyl O species.
Finally, the alkali metal SSNMR studies involving 7Li, 39K, 87Rb and 133Cs nuclei and the associated GIPAW DFT calculation of their chemical shift and quadrupole tensor parameters highlighted an excellent agreement between these approaches. In particular, the ability to accurately predict the 7Li, 39K and 133Cs δiso values and the 39K and 87Rb quadrupole parameters served as a useful addition to the understanding of these H bonded systems. This study promotes the further possibilities for the incorporation of guest nuclei such as monovalent Group I alkali metal ions as a probe of H bond geometry and strength in other organic/organometallic systems, and stresses the value of an NMR crystallographic approach as an effective method of characterisation.
Experimental Section
General
IR spectra were recorded on a Perkin Elmer Spectrum 100 FT-IR Spectrometer using Attenuated Total Reflection sampling unless otherwise stated, and are reported in cm−1. Mass spectra were recorded at the EPSRC Mass Spectrometry Centre at the University of Swansea. GCMS was carried out at NTU using Agilent Technologies 6890N GC equipped with a 5975 mass detector. Chemical analysis data were obtained from Mr Stephen Boyer, London Metropolitan University. Microwave synthesis was performed using a CEM Discovery apparatus and all 17O labelled water was purchased from Cortecnet.17O-Enriched benzoic acid (BZA)
α,α,α-Trichlorotoluene (660 mg, 3.38 mmol) and 70% 17O-enriched water (Cortecnet, 193 mg, 10.1 mmol) were reacted together at 160 °C for 15 min in a 10 ml vial at a pressure of 16 bar in a microwave reactor. The vessel was allowed to cool to room temperature, and then the excess pressure (HCl vapour) released. The mixture was again reacted at 160 °C for 45 min at 16 kbar, and then cooled to room temperature and the pressure released. Depending on the degree of conversion to benzoic acid (as determined by GC-MS analysis), a further amount of 17O-labelled water added (typically 50–100 μL), and the mixture was treated under the same microwave conditions (45 mins). This was repeated, if necessary, until GC-MS showed almost complete conversion to benzoic acid. The crude product was recrystallized from heptane, typical isolated yield 80%. An ampoule of 70% 17O-enriched water gave benzoic acid containing 40.5% PhC(17O)2H, 45.7% PhC(17O)(16O)H and 13.8% PhCO2H by mass spectral analysis (64% enrichment); elemental analysis calcd (%) for C7H6O2: C 68.16, H 4.90; found: C 68.25, H 4.82; 90% 17O-enriched water gave benzoic acid containing 74.2% PhC(17O)2H, 23.6% PhC(17O)(16O)H and 2.2% PhCO2H by mass spectral analysis (86% enrichment), m.p. 121–122 °C, IR (ATR): ν˜ = 3073, 2982, 2815, 2658, 2540, 1664 (CO), 1603, 1582, 1454, 1419, 1323, 1285 and 1276 (C–O), 1187, 1180, 1121, 1073, 1027, 931, 810, 798, 703, 684, 661 cm−1 (cf. non-enriched benzoic acid, from heptane: IR (ATR): ν˜ = 3072, 2831, 2668, 2554, 1678, 1602, 1583, 1454, 1421, 1324, 1289 (C–O), 1280 sh, 1187, 1180, 1128, 1073, 1027, 932, 805, 704, 684, 666 cm−1).Preparation of hydrogen dibenzoates and tribenzoate
The hydrogen dibenzoate (HD) salts were prepared by the reaction of benzoic acid with half an equivalent of the appropriate metal hydroxide in hot ethanol/water mixtures (KHD, RbHD, CsHD) or methanol/water mixtures (LiHD) and subsequent cooling. For CsHD, the use of THF and methanol mixtures for the reaction led to a different polymorph (orthorhombic) of the product. The unlabelled NaDT sample was prepared by slow evaporation of 2:1 mixture of BZA and NaOH prepared in ethanol/water. Further details are given in the ESI.†17O labelled HD samples were prepared in a similar way, but using 17O enriched BZA.X-ray crystallography
X-ray diffraction datasets for most crystals were measured with MoKα radiation (λ = 0.71073 Å) on a Bruker-Nonius FR591 diffractometer equipped with a rotating anode and a Bruker-Nonius Roper CCD camera, and the X-ray data for the 6-coordinate RbHD polymorph were collected on a Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn 724+ detector mounted at the window of an FR–E+ Superbright MoKα rotating anode generator with HF Varimax optics56 (National Crystallography Service, Southampton University, UK). The diffraction data for the orthorhombic polymorph of CsHD were measured on an Oxford Diffraction Xcalibur diffractometer equipped with a Sapphire CCD detector at NTU. Crystal cooling was provided by an Oxford Cryosystems Cryostream low temperature system. Diffraction data was collected by φ and ω scans, and Lorentz polarisation correction and an absorption correction were applied. Crystal structure solution and refinement (on F2) were carried out with SHELXS and SHELXL programs57 using the XSEED interface.58 Non-hydrogen atoms were assigned anisotropic displacement parameters. All hydrogen atom positions were determined geometrically and refined with a riding model, except as follows: LiHD and NaDT: coordinates of the (O)H atoms refined; KHD: two (O)H positions, related by a centre of symmetry in the middle of the O–O atomic vector, refined with half occupancies based on observations of the difference electron density; the six-coordinate RbHD polymorph was refined with the (O)H atom located at a centre of symmetry in the middle of the O–O atomic vector based on observation of the difference electron density map; the eight-coordinate RbHD polymorph and both polymorphs of CsHD: electron density corresponding to the (O)H atom located on a two-fold axis and equidistant from two O atoms (O–O: (Rb) 2.455 Å, (Cs monoclinic) 2.457 Å, (Cs orthorhombic) 2.457 Å) and only the y coordinate refined in each case. Difference electron density maps59 from the structural models without the (O)H atom found two centrosymmetrically related peaks of electron density between the two O atoms for the K salt, but only one such peak positions equidistant from the two O atoms for the Rb and Cs cases. Molecular illustrations were made with Mercury60 and POVRAY.61Crystal data
Lithium hydrogen dibenzoate: C14H11O4Li, Mr = 250.17, triclinic, a = 5.3600(3), b = 8.5693(7), c = 13.4999(11) Å, α = 98.830(4)°, β = 93.800(5)°, γ = 95.524(5)°, V = 607.80(8) Å3, Z = 2, P−1, crystal: 0.16 × 0.08 × 0.05 mm, Dcalc. = 1.37 g cm−3, μ = 0.099 mm−1, T = 120 K, 11772 measured reflections, max. 2Θ = 27.57°, 2763 unique reflections, 1833 with F2 > 2σ, R(F, F2 > 2σ) = 0.054, Rw(F2, all data) = 0.13. max. and min. residual e density: 0.23 and −0.27 eÅ−3.Sodium dihydrogentribenzoate: C21H17O6Na, Mr = 388.34, monoclinic, a = 5.8077(1), b = 14.4331(4), c = 22.0139(6) Å, β = 97.047(2)°, V = 1831.33(8) Å3, Z = 4, P21/c, crystal: 0.50 × 0.26 × 0.05 mm, Dcalc. = 1.41 g cm−3, μ = 0.123 mm−1, T = 120 K, 21737 measured reflections, max. 2Θ = 27.51°, 4185 unique reflections, 3434 reflections with F2 > 2σ, R(F, F2 > 2σ) = 0.041, Rw(F2, all data) = 0.10, max. and min. residual e density: 0.27 and −0.29 eÅ−3.
Potassium hydrogen dibenzoate: C14H11O4K, Mr = 282.33, monoclinic, a = 29.586(2), b = 3.7964(3), c = 11.1214(8) Å, β = 97.096(9)°, V = 1239.59(16) Å3, Z = 4, C2/c, crystal: 0.50 × 0.40 × 0.03 mm, Dcalc = 1.51 g cm−3, μ = 0.435 mm−1, T = 120 K, 12549 measured reflections, max. 2Θ = 27.65°, 1428 unique reflections, 1203 with F2 > 2σ, R(F, F2 > 2σ) = 0.043, Rw(F2, all data) = 0.11, max. and min. residual e density: 0.37 and −0.37 eÅ−3; original measurement.62
Rubidium hydrogen dibenzoate, six coordinate polymorph: C14H11O4Rb, Mr = 328.70, monoclinic, a = 29.347(4), b = 3.8697(5), c = 11.3973(16) Å, β = 95.478(7)°, V = 1288.4(3) Å3, Z = 4, C2/c, crystal: 0.34 × 0.31 × 0.05 mm, Dcalc. = 1.69 g cm−3, μ = 3.85 mm−1, T = 100 K, 4341 measured reflections, max. 2Θ = 27.42°, 1423 unique reflections, 1404 with F2 > 2σ, R(F, F2 > 2σ) = 0.025, Rw(F2, all data) = 0.069, max. and min. residual e density: 0.59 and −0.90 eÅ−3.
Rubidium hydrogen dibenzoate, eight coordinate polymorph: C14H11O4Rb, Mr = 328.70, monoclinic, a = 29.0315(9), b = 6.9630(2), c = 6.6156(2) Å, β = 99.297(2)°, V = 1319.75(7) Å3, Z = 4, C2/c, crystal: 0.28 × 0.20 × 0.08 mm, Dcalc. = 1.65 g cm−3, μ = 3.76 mm−1, T = 120 K, 8615 measured reflections, max. 2Θ = 27.49°, 1510 unique reflections, 1439 with F2 > 2σ, R(F, F2 > 2σ) = 0.028, Rw(F2, all data) = 0.070, max. and min. residual e density: 0.34 and −0.61 eÅ−3.
Cesium hydrogen dibenzoate, monoclinic polymorph: C14H11O4Cs, Mr = 376.14, monoclinic, a = 28.9095(8), b = 7.0509(2), c = 6.7779(2) Å, β = 99.970(2)°, V = 1360.73(7) Å3, Z = 4, C2/c, crystal: 0.55 × 0.45 × 0.04 mm, Dcalc. = 1.84 g cm−3, μ = 2.73 mm−1, T = 120 K, 11461 measured reflections, max. 2Θ = 27.57°, 1565 unique reflections, 1529 with F2 > 2σ, R(F, F2 > 2σ) = 0.019, Rw(F2, all data) = 0.046, max. and min. residual e density: 0.61 and −0.80 eÅ−3.
Cesium hydrogen dibenzoate, orthorhombic polymorph: C14H11O4Cs, Mr = 376.14, orthorhombic, a = 28.3393(19), b = 7.0215(3), c = 6.9066(4) Å, V = 1374.31(14) Å3, Z = 4, Pbcn, crystal: 0.35 × 0.33 × 0.12 mm, Dcalc. = 1.82 g cm−3, μ = 2.70 mm−1, T = 150 K, 7535 measured reflections, max. 2Θ = 29.13°, 1664 unique reflections, 1389 with F2 > 2σ, R(F, F2 > 2σ) = 0.031, Rw(F2, all data) = 0.050, max. and min. residual e density: 0.66 and −0.45 eÅ−3.
Solid state NMR
All solid state 1H MAS NMR measurements were undertaken at 11.7 T using a Bruker Avance III-500 spectrometer operating at a 1H frequency of 500.1 MHz. These data were acquired using a Bruker 2.5 mm double resonance probe spinning each sample at 27.5 kHz, and with single pulse experiments of 1.5 μs pulse duration (flip angle of π/4) and recycle delays of 15 s. Each 1H MAS NMR spectrum was referenced to the primary standard of tetramethylsilane (TMS, δiso = 0 ppm).63 The corresponding 13C MAS NMR data were acquired at 9.4 T on a Bruker DSX-400 spectrometer operating at a 13C frequency of 100.3 MHz. The 1H–13C cross-polarisation MAS (CPMAS) technique was implemented using a Bruker 3.2 mm double resonance probe; a variable amplitude CPMAS experiment was employed which used an initial 1H π/2 pulse of 3 μs, a contact period of 1 ms, a recycle delay of 2 s and a 1H decoupling field of ~95 kHz during acquisition. For each system 13C CPMAS data were acquired at a range of MAS frequencies (νr = 0.8, 1.5 and 12 kHz) to allow a detailed sideband analysis to be undertaken thus enabling an accurate determination of the 13C chemical shift anisotropy (CSA). Spinning sideband manifold simulations were completed on all 13C CPMAS data using the spectral analysis function available in TOPSPIN 2.1. Each 13C CPMAS NMR spectrum was referenced to the primary standard of tetramethylsilane (TMS, δiso = 0 ppm) via a secondary solid reference of alanine (δiso(CH3) = 20.5 ppm, δiso(CH) = 50.5 ppm).6317O (I = 5/2) MAS NMR studies were performed at 9.4 and 14.1 T using Bruker DSX-400 and Bruker Avance II-600 spectrometers operating at 17O frequencies of 52.23 and 81.34 MHz, respectively. A Bruker 2.5 mm double resonance probe was used at each field which facilitated a MAS frequency of 27.5 kHz for each measurement. All 17O MAS NMR data were acquired using a rotor-synchronised Hahn echo experiment (θ–τ–2θ–τ–acquire); a ‘non-selective’ (solution) π/2 pulse time of 6 μs was calibrated using 10% H217O, from which a ‘selective’ (solid) θ pulse of 1 μs was used which represented a ~π/4 flip angle. A recycle delay of 2–3 s was used throughout; this was checked against acquisitions using recycle delays of up to 60 s. All 17O apparent shifts (centre-of-gravity, δcg) and isotropic chemical shifts (δiso) are reported against a primary reference of H217O (δiso = 0 ppm). Two dimensional 17O triple quantum MAS (2D 3QMAS) measurements were undertaken at 14.1 T using a Bruker Avance II-600 spectrometer and a double resonance Bruker 2.5 mm probe which delivered an MAS frequency of 27.5 kHz. 3QMAS data were acquired using a three pulse Z-filter pulse experiment which consisted of a ‘non-selective’ 5.0 μs 3 π/2 excitation pulse, a 1.5 μs π/2 conversion pulse, and followed by two ‘selective’ 40.00 μs π/2 pulses which comprised the Z-filter. The recycle delay was 2 s. 17O double rotation (DOR) experiments using odd order sideband suppression64 were undertaken at 14.1 and 20.0 T using Bruker Avance II-600 and Avance III-850, respectively, operating at the 17O frequencies of 81.34 and 115.25 MHz. The measurements at each field were performed using Samoson designed DOR probes which functioned with an external rotor spinning frequency of 1.2–1.8 kHz and an internal rotor spinning frequency of 5.8–8.8 kHz.
The 7Li (I = 3/2) MAS NMR measurements were performed at 11.7 T using a Bruker Avance III-500 spectrometer operating at a 7Li frequency of 194.37 MHz, and a Bruker double resonance 3.2 mm probe enabling a MAS frequency of 20 kHz. The single pulse experiment consisted of a ‘selective’ (solid) π/2 pulse of 1.50 μs duration (i.e. ‘non-selective’ (solution) π/2 pulse of 3 μs)and a recycle delay of 60 s. This pulse time was calibrated on a 9.6 M LiCl (aq) solution (δiso = 0 ppm) which also acted as the primary chemical shift reference. Static (broadline) 39K (I = 3/2) NMR measurements were performed at 9.4 and 14.1 T using Bruker DSX-400 and Avance II-600 specrometers operating at 39K frequencies of 18.66 and 27.97 MHz, respectively. These experiments were implemented using a single channel Bruker 10 mm static probe, and the data was acquired using a solid echo (θ–τ–θ–τ–acquire) experiment. A ‘selective’ (solid) pulse time of 5 μs (i.e. ‘non-selective’ (solution) π/2 pulse of 10 μs) duration was calibrated on a 0.1 M KCl (aq) solution (δiso = 0 ppm) which also acted as the primary chemical shift reference, and recycle delay was 10 s. Similar static 87Rb (I = 3/2) NMR measurements were undertaken at 4.7, 9.4 and 14.1 T using Chemagnetics CMX-200, Bruker DSX-400 and Bruker Avance II-600 spectrometers operating at 87Rb frequencies of 65.25, 130.50 and 196.13 MHz, respectively. The data at all three fields was acquired using the same Bruker 10 mm probe and solid echo experiment configuration. A ‘selective’ (solid) π/2 pulse duration of 1.0 μs (i.e. ‘non-selective’ (solution) π/2 pulse of 2 μs) was calibrated using cubic RbF (s) which also acted as a secondary chemical shift reference (δiso = 19.1 ppm) with respect to the primary reference of 0.01 M RbCl (aq) solution (δiso = 0 ppm). The recycle delay was 1 s. Lastly, 133Cs MAS NMR data were obtained at 9.4 and 14.1 T using Bruker DSX-400 and Avance II-600 spectrometers operating at 133Cs frequencies of 52.32 and 78.62 MHz, respectively. These data were acquired using a Bruker 4 mm double resonance probe which delivered a MAS frequency of 10 kHz. All measurements were undertaken with single pulse experiments which used a ‘selective’ (solid) π/2 pulse of 1.5 μs (i.e. ‘non-selective’ (solution) π/2 pulse of 6 μs) duration which was calibrated on 1 M CsNO3(aq) solution. The recycle delay was 5 s. This also served as the primary chemical shift reference (δiso = 0 ppm).
All quadrupole dominated MAS and static NMR spectra were simulated with the DMFit simulation package,65 while some additional calculated of MAS NMR lineshapes under specific experimental conditions were produced with Simpson.41
DFT calculations
All density functional theory (DFT) calculations were performed using the CASTEP 6.01 code which employs Kohn-Sham DFT methodology using periodic plane-waves under the ultrasoftpseudopotential approximation.66 The generalized gradient approximation for the exchange correlation energy was employed using the Perdew–Burke–Ernzerhof (PBE) functional.67,68 Pseudopotentials were generated ‘on-the-fly’ using the standard Accelrys Software Materials Studio pseudo-atom definitions.69 Each calculation was converged with respect to basis-set size and Brillouin zone k point sampling to at least an accuracy of 0.4 mH (~0.000002% of total energy) per atom for each of the systems under investigation. To confirm this level of energy convergence was sufficient to produce accurate ionic forces, energy minimisation with respect to ionic positions was repeated with increasing plane wave cut off energy and density of k points within the Monkhorst–Pack Brillouin zone grid. This level of convergence was achieved using a plane wave cut-off energy of 900 eV for the Li, Cs and Rb systems and 950 eV for the K system, and by invoking k point Monkhorst-Pack grids of 1 × 4 × 2, 3 × 2 × 2, for the K and Li systems respectively, and a 1 × 2 × 2 grid for the Cs and Rb systems.Geometry optimisation and hydrogen position optimisation calculations were performed on all systems using the over-converged values described previously to ensure accurate forces. For these structural optimisations the lattice parameters were kept fixed whilst the atomic positions were optimised to a force tolerance of 0.05 eV/Å, a maximum ionic displacement of 1 × 10−3 Å and a total energy change of 2 × 10−5 eV per atom. NMR parameter calculation of the chemical shift and electric field gradient tensors invoked the gauge-included projector augmented-wave (GIPAW)-DFT method which extended the pseudopotential (valence electron approximation) approximation to recover all electron charge densities.70–72 This approach demonstrated that the NMR parameters depend more sensitively upon the density than the total energy, so each NMR parameter will be more sensitive to basis set truncation errors than total energies. This is compensated for by the over-convergence of the basis set to a level which experience suggests will produce fully converged NMR parameters. Consequently, these calculations were completed using the plane wave cut-off and k-point Monkhorst–Pack grids described earlier. In order to calculate isotropic chemical shifts for each nucleus the relationship δiso = −[σ − σref] is employed, where σ is the calculated isotopic shielding calculated against a bare atom, and σref is the reference isotropic shielding against a bare atom. This necessitated GIPAW-DFT calculations be performed on a set of references including benzoic acid (1H), alanine (13C), LiCl (7Li), KCl (39K), RbCl (87Rb) and CsCl (133Cs). These calculations used the same basis set convergence (0.4 mH) as the original calculations in order to minimise the propagation of errors.
The XRD structure determination of KHB obtained at a low temperature (~120 K) has identified two possible sites of equal probability and equal spacing about the middle of the O–H–O moiety supporting the H bonded proton (see S3 ESI†). This form of partial occupancy signifies that the H bonding is undergoing thermal fluctuations on the timescale of the XRD experiment (i.e. on the order of ms–s), thus manifesting itself as two (equally) possible average atomic positions in the XRD data. Since all of the NMR measurements were undertaken at a higher temperature of ~293 K, and the timescale of the NMR experiment is very much faster (i.e. on the order of μs) then this proton position will be observed as an average nuclear environment. In this case the average H position will reside in the middle of the O–H–O arrangement. Hence, a rationalisation of the difference in the timescale of measurement (and different measurement temperatures) has alleviated the need to treat partial occupancies (and particularly geometry optimisations) with CASTEP in this case. This assumption is justified by the excellent agreement displayed between the δiso values measured by 1H MAS NMR and calculated NMR-CASTEP.
Acknowledgements
J. V. H., J. D. W. and M. E. S. would like to thank the EPSRC for grant (EP/E018203/1) from the Physical Organic Chemistry Initiative, the EPSRC Mass Spectrometry Service for data and the Chemical Database Service73 for access to the Cambridge Structural Database. J. V. H acknowledges the continued funding of the 400, 500 and 600 MHz solid state NMR instrumentation at Warwick used in this research facilitated by EPSRC, the University of Warwick and the Birmingham Science City Advanced Materials Projects 1 and 2, which are supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF). The UK 850 MHz High Field Solid State NMR Facility used in this research was funded by EPSRC, BBSRC, the University of Warwick with partial funding through Birmingham Science City Advanced Materials Projects 1 and 2, which are supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF). J. D. W. would also like to acknowledge the support from Nottingham Trent University for diffraction facilities. J. V. H. and D. Q. acknowledge the facilities of HECToR, the UK's national high-performance computing service, which is provided by UoE HPCx Ltd. at the University of Edinburgh, Cray Inc. and NAG Ltd., and funded by the Office of Science and Technology through EPSRC's High End Computing Programme.Notes and references
- C. M. Gowda, F. Vasconcelos, E. Schwartz, E. R. H. van Eck, M. Marsman, J. J. L. M. Cornelissen, A. E. Rowan, G. A. de Wijs and A. P. M. Kentgens, Phys. Chem. Chem. Phys., 2011, 13, 13082–13095 RSC.
- J. W. Traer, J. F. Britten and G. R. Goward, J. Phys. Chem. B, 2007, 111, 5602–5609 CrossRef CAS PubMed.
- Z. T. Gu, C. F. Ridenour, C. E. Bronnimann, T. Iwashita and A. McDermott, J. Am. Chem. Soc., 1996, 118, 822–829 CrossRef CAS.
- Z. T. Gu, R. Zambrano and A. McDermott, J. Am. Chem. Soc., 1994, 116, 6368–6372 CrossRef CAS.
- N. Asakawa, S. Kuroki, H. Kurosu, I. Ando, A. Shoji and T. Ozaki, J. Am. Chem. Soc., 1992, 114, 3261–3265 CrossRef CAS.
- S. Kuroki, S. Ando, I. Ando, A. Shoji, T. Ozaki and G. A. Webb, J. Mol. Struct., 1990, 240, 19–29 CrossRef CAS.
- J. Sponer, J. Leszczynski and P. Hobza, J. Biomol. Struct. Dyn., 1996, 14, 117–135 CAS.
- G. A. Jeffrey and W. Saenger, Hydrogen bonding in biological structures, Springer, Berlin, 1991 Search PubMed.
- G. A. Jeffrey, An introduction to hydrogen bonding, Oxford University Press, New York, Oxford, 1997 Search PubMed.
- E. D. Arunan, G. R. Klein, R. A. Sadlej, J. Scheiner, S. Alkorta, I. Clary, D. C. Crabtree, R. H. Dannenberg, J. J. Hobaza, P. Kjaergaard, H. G. Legon, A. C. Mennucci, B. Nesbitt and D. J. , Pure Appl. Chem., 2011, 83, 1637–1641 CrossRef CAS.
- J. M. Cole, G. J. McIntyre, M. S. Lehmann, D. A. A. Myles, C. Wilkinson and J. A. K. Howard, Acta Crystallogr., Sect. A: Found. Crystallogr., 2001, 57, 429–434 CAS.
- B. Slootmaekers and H. O. Desseyn, Appl. Spectrosc., 1991, 45, 118–120 CrossRef CAS.
- G. Gemmecker, Angew. Chem., Int. Ed., 2000, 39, 1224 CrossRef CAS.
- A. K. Cheetham and A. P. Wilkinson, Angew. Chem., Int. Ed. Engl., 1992, 31, 1557–1570 CrossRef.
- M. Gorman, J. Chem. Educ., 1957, 34, 304 CrossRef CAS.
- A. Witkowski and M. Wójcik, Chem. Phys., 1973, 1, 9–16 CrossRef CAS.
- S. Grabowski, Hydrogen Bonding - New Insights, London, 2006 Search PubMed.
- Z. T. Gu and A. McDermott, J. Am. Chem. Soc., 1993, 115, 4282–4285 CrossRef CAS.
- Y. F. Wei, A. C. de Dios and A. E. McDermott, J. Am. Chem. Soc., 1999, 121, 10389–10394 CrossRef CAS.
- A. Wong, K. J. Pike, R. Jenkins, G. J. Clarkson, T. Anupold, A. P. Howes, D. H. G. Crout, A. Samoson, R. Dupree and M. E. Smith, J. Phys. Chem. A, 2006, 110, 1824–1835 CrossRef CAS PubMed.
- S. E. Ashbrook and M. E. Smith, Chem. Soc. Rev., 2006, 35, 718–735 RSC.
- T. J. Bastow and S. N. Stuart, Chem. Phys., 1990, 143, 459–467 CrossRef CAS.
- T. J. Bastow, P. J. Dirken, M. E. Smith and H. J. Whitfield, J. Phys. Chem., 1996, 100, 18539–18545 CrossRef CAS.
- G. Wu and K. Yamada, Solid State Nucl. Magn. Reson., 2003, 24, 196–208 CrossRef CAS.
- G. Wu, Prog. Nucl. Magn. Reson. Spectrosc., 2008, 52, 118–169 CrossRef CAS PubMed.
- E. Oldfield, M. A. Keniry, S. Shinoda, S. Schramm, T. L. Brown and H. S. Gutowsky, J. Chem. Soc., Chem. Commun., 1985, 791–793 RSC.
- P. M. Panchmatia, A. Orera, G. J. Rees, M. E. Smith, J. V. Hanna, P. R. Slater and M. S. Islam, Angew. Chem., Int. Ed., 2011, 50, 9328–9333 CrossRef CAS PubMed.
- E. D. Glowacki, M. Irimia-Vladu, S. Bauer and N. S. Sariciftci, J. Mater. Chem. B, 2013 Search PubMed.
- C. G. Cannon, Spectrochim. Acta, 1958, 10, 341–368 CrossRef CAS.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- R. K. Harris, P. Jackson, L. H. Merwin, B. J. Say and G. Hagele, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 3649–3672 RSC.
- R. Gobetto, C. Nervi, E. Valfre, M. R. Chierotti, D. Braga, L. Maini, F. Grepioni, R. K. Harris and P. Y. Ghi, Chem. Mater., 2005, 17, 1457–1466 CrossRef CAS.
- J. S. Waugh, L. M. Huber and U. Haeberlen, Phys. Rev. Lett., 1968, 20, 180–180 CrossRef CAS.
- U. Haeberlen and J. S. Waugh, Phys. Rev., 1968, 175, 453–453 CrossRef CAS.
- A. Wong, I. Hung, A. P. Howes, T. Anupold, J. Past, A. Samoson, S. P. Brown, M. E. Smith and R. Dupree, Magn. Reson. Chem., 2007, 45, S68–S72 CrossRef CAS PubMed.
- J. C. Speakman, The hydrogen bond and other intermolecular forces, Chemical Society, London, 1975 Search PubMed.
- P. Gilli, V. Bertolasi, L. Pretto, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2004, 126, 3845–3855 CrossRef CAS PubMed.
- C. C. Wilson, N. Shankland and A. J. Florence, Chem. Phys. Lett., 1996, 253, 103–107 CrossRef CAS.
- F. Fillaux, A. Cousson, J. F. R. Archilla and J. Tomkinson, J. Chem. Phys., 2008, 128, 204502 CrossRef PubMed.
- E. W. Hagaman, B. H. Chen, J. Jiao and W. Parsons, Solid State Nucl. Magn. Reson., 2012, 41, 60–67 CrossRef CAS PubMed.
- T. Vosegaard, Z. Tošner and N. C. Nielsen, in eMagRes, John Wiley & Sons, Ltd, 2007 Search PubMed.
- J. M. Griffin, L. Clark, V. R. Seymour, D. W. Aldous, D. M. Dawson, D. Iuga, R. E. Morris and S. E. Ashbrook, Chem. Sci., 2012, 3, 2293–2300 RSC.
- A. Wong, A. P. Howes, J. R. Yates, A. Watts, T. Anupold, J. Past, A. Samoson, R. Dupree and M. E. Smith, Phys. Chem. Chem. Phys., 2011, 13, 12213–12224 RSC.
- A. Samoson and E. Lippmaa, J. Magn. Reson., 1989, 84, 410–416 CAS.
- T. Anupold, A. Reinhold, P. Sarv and A. Samoson, Solid State Nucl. Magn. Reson., 1998, 13, 87–91 CrossRef CAS.
- A. P. Howes, T. Anupold, V. Lemaitre, A. Kukol, A. Watts, A. Samoson, M. E. Smith and R. Dupree, Chem. Phys. Lett., 2006, 421, 42–46 CrossRef CAS PubMed.
- R. Dupree, in eMagRess, Wiley, 2011 Search PubMed.
- I. Hung, A. Wong, A. P. Howes, l. T. Anupõ, J. Past, A. Samoson, X. Mo, G. Wu, M. E. Smith, S. P. Brown and R. Dupree, J. Magn. Reson., 2007, 188, 246–259 CrossRef CAS PubMed.
- F. J. Milford and W. B. Gager, Phys. Rev., 1961, 121, 716–720 CrossRef CAS.
- P. K. Lee, R. P. Chapman, L. Zhang, J. Hu, L. J. Barbour, E. K. Elliott, G. W. Gokel and D. L. Bryce, J. Phys. Chem. A, 2007, 111, 12859–12863 CrossRef CAS PubMed.
- I. L. Moudrakovski and J. A. Ripmeester, J. Phys. Chem. B, 2007, 111, 491–495 CrossRef CAS PubMed.
- K. Shimoda, A. Yamane, T. Ichikawa and Y. Kojima, J. Phys. Chem. C, 2012, 116, 20666–20672 CAS.
- J. Kim, J. L. Eglin, A. S. Ellaboudy, L. E. H. McMills, S. Huang and J. L. Dye, J. Phys. Chem., 1996, 100, 2885–2891 CrossRef CAS.
- G. Wu, Z. Gan, I. C. M. Kwan, J. C. Fettinger and J. T. Davis, J. Am. Chem. Soc., 2011, 133, 19570–19573 CrossRef CAS PubMed.
- K. R. Hirsch and W. B. Holzapfel, Phys. Lett. A, 1984, 101, 142–144 CrossRef.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- L. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van De Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- D. Buck and A. Collins, Persistence of Vision Pty. Ltd. (2004). Persistence of Vision (TM) Raytrace Persistence of Vision Pty. Ltd., Williamstown, Victoria, Australia, http://www.povray.com/.
- J. M. Skinner, G. M. D. Stewart and J. C. Speakman, J. Chem. Soc., 1954, 180–184 RSC.
- R. K. Harris, E. D. Becker, S. M. C. de Menezes, R. Goodfellow and P. Granger, Solid State Nucl. Magn. Reson., 2002, 22, 458–483 CrossRef CAS PubMed.
- A. Samoson and J. Tegenfeldt, J. Magn. Reson., Ser. A, 1994, 110, 238–244 CrossRef CAS.
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calve, B. Alonso, J. O. Durand, B. Bujoli, Z. H. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567–570 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter, 1992, 46, 6671–6687 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter, 1993, 48, 4978–4978 CrossRef CAS.
- Accelrys, 10188 Telesis Court, Suite 100 San Diego, CA 92121, USA., 2013.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter, 2001, 63, 245101 CrossRef.
- M. Profeta, F. Mauri and C. J. Pickard, J. Am. Chem. Soc., 2002, 125, 541–548 CrossRef PubMed.
- J. R. Yates, C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 024401 CrossRef.
- D. A. Fletcher, R. F. McMeeking and D. Parkin, J. Chem. Inf. Model., 1996, 36, 746–749 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC numbers 940110–940116. For crystallographic data in CIF or other electronic format. see DOI: 10.1039/c3ce41258j |
| This journal is © The Royal Society of Chemistry 2013 |