 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Application of NMR crystallography to the determination of the mechanism of charge-balancing in organocation-templated AlPO STA-2†
Valerie R. Seymour,
Eike C. V. Eschenroeder,
Maria Castro,
Paul A. Wright and
Sharon E. Ashbrook*
School of Chemistry and EaStCHEM, University of St Andrews, North Haugh, St Andrews KY16 9ST, UK. E-mail: sema@st-andrews.ac.uk
First published on 9th July 2013
Abstract
The combination of solid-state MAS NMR spectroscopy and first-principles calculations is used to elucidate the structure of an as-prepared microporous AlPO (STA-2), in which the organocation template (bis-diazabicyclooctane-butane) is charge balanced by hydroxyl groups coordinated to framework aluminium species. 27Al MAS NMR spectra show Al exists in both tetrahedral and five-fold coordination, with the latter directly coordinated to hydroxyl O atoms as well as framework O atoms. The relative intensities of these resonances can be used to determine that the hydroxyls bridge between the two types of Al. Calculation of NMR parameters using a periodic density functional theory approach are able to suggest assignments for the resonances in both 27Al and 31P NMR spectra. 31P chemical shifts are shown to depend not only on the topologically-distinct sites in the SAT framework but also on whether or not the P atoms form part of a six-membered P(OAl)2OH ring, where OH is a bridging hydroxyl. By diffraction six possible sites for the charge-balancing hydroxyls have been identified, but all are refined with an average occupancy of 0.33. However, predicted peak positions in two-dimensional J-HETCOR NMR spectra indicate that only one hydroxyl is found in each cancrinite cage, and suggest that the most likely arrangements are those that avoid the close approach of bridging hydroxyl groups in adjacent cages. We show that the use of a combination of NMR spectroscopy and calculation to elucidate structure, often referred to as “NMR Crystallography”, offers a promising route for the future study of as-made and substituted microporous materials.
Introduction
The wide range of porous structures exhibited by zeotypic materials has resulted in a variety of potential applications, including gas storage and separation, drug delivery and in catalysis. Aluminophosphates (AlPOs), first reported in 1982,1 are the most extensive group of zeotypes, forming many structures isomorphous with those of the common silica zeolites, but others with unique topologies. In order to avoid the formation of dense phases, synthesis of AlPOs typically requires an organic amine base (usually referred to as a structure-directing agent (SDA) or template), enabling open frameworks to be produced. As the SDA is usually positively charged, a charge-balancing mechanism is required in the “as-made” materials to maintain the inherently neutral tetrahedral AlPO framework. This can be achieved by the partial substitution of divalent metal cations for Al, or of Si for P, but in many cases the coordination of anions from the synthesis solution, typically OH− or F−, to the aluminium cations of the framework is observed, creating five- and/or six-coordinate Al species. Where hydroxyls are present, different coordination modes can be observed; the hydroxyls may be terminal (i.e., connected to a single Al species), as in STA-15,2 or they may be bridging (i.e., connected to two Al species simultaneously), as in AlPO-173 and AlPO-18.4 In most cases the SDA, charge-balancing anions and any water within the pores of the AlPO can be removed by calcination to produce a neutral (and purely tetrahedral) framework.NMR spectroscopy is an ideal tool for the investigation of phosphate frameworks, owing to its sensitivity to the local structural environment, and its ability to provide information in the absence of long-range periodicity. For aluminophosphates, NMR can be used to study the basic components of the framework, as both 27Al (I = 5/2) and 31P (I = 1/2) have 100% natural abundance. More recently, using ionothermal synthesis, cost-efficient low-level (i.e., 4–10%) 17O enrichment of AlPO frameworks has been demonstrated, overcoming the low natural abundance (∼0.037%) of this isotope, and offering an interesting new approach to studying host–guest interactions.5 For as-made AlPOs, further structural information can be obtained as both the charge-balancing anions and SDA molecules contain a range of NMR-active nuclides (e.g., 13C, 14/15N, 1/2H, 19F). Although the AlPO framework exhibits a periodic, repeating structure, the substitution of other elements into this framework, and the disordered arrangements of many charge-balancing anions and SDAs can pose a challenge for diffraction-based methods, and the combination of multi-dimensional high-resolution NMR alongside more conventional crystallographic methods can provide a more complete and more detailed picture.2,3,6–14 For example, multinuclear NMR spectroscopy was recently applied to provide information on the distribution of OH−/F− anions in AlPO-CJ2,9 the non-periodic F subnetwork in AlPO cloverite,10 and the in situ reversible transformation of AlPO-53 to JDF-2.7 NMR spectroscopy is also sensitive to dynamics over a wide range of timescales, and has been applied to investigate the μs-timescale motion of different SDAs (isopropylamine and piperidine) in AlPO-14.13
In recent years, experimental NMR investigation has been complemented by an increasing use of first-principles calculations, facilitating spectral interpretation and assignment in simple systems, but providing insight into local structure, disorder and dynamics in more complex materials. The recent introduction of the GIPAW approach, which exploits the inherent periodicity of the solid state,15 has led to a resurgence of interest in the calculation of NMR parameters in solids, and applications in areas as wide ranging as biomaterials, minerals, energy materials and ceramics.16–19 Although applied relatively rarely to AlPO frameworks,2,7,11,12 this approach has been used to assign spectra and to understand phase transformations and dehydration. Notably, NMR parameters calculated from published crystal structures can be in poor agreement with experiment in some cases, unless optimization of the geometry is performed.12,18 This suggests that a combination of NMR and first-principles calculations, often termed “NMR crystallography”, will be a useful tool in the structural refinement of microporous materials in the future, particularly for structures that have been derived from powder diffraction experiments.
Here we use an NMR crystallography approach to investigate the as-made microporous aluminophosphate framework STA-2. STA-2 was first reported in 1997, using 1,4-bis-N-quinuclidiniumbutane (BQNB) as the SDA, in the magnesium aluminophosphate (MgAPO) form (framework composition Mg0.15Al0.85PO4).20 In subsequent work, a pure AlPO form was prepared using either BQNB or bis-diazabicyclooctane-butane (BDAB) as an SDA, with the latter a potentially cheaper alternative to the former.21 In both cases, charge balance was achieved though the coordination of OH− species to the framework, and although refinement of synchrotron X-ray powder diffraction suggested locations for some of these hydroxyl groups they were neither fully nor unambiguously located in the structure. Although NMR spectra of the as-made forms were not assigned in this work, the similarity in the spectra observed for the two SDAs led to the conclusion that (i) BDAB was also divalent when incorporated into the material (this SDA has the potential for its charge to vary from 2+ to 4+) and (ii) that the structures of the two as-made forms were very similar. In this work, we combine multinuclear NMR spectroscopy and first-principles calculations to investigate the distribution of hydroxyls in as-made STA-2(BDAB). A variety of one- and two-dimensional NMR experiments are used, alongside GIPAW calculations performed for a range of simple structural models with a variety of hydroxyl positions, in an attempt to assign and interpret the NMR spectra obtained and to gain insight into the atomic-scale structure of STA-2.
Experimental details
Synthesis
AlPO STA-2 materials were prepared according to the method of Castro et al.21 The SDA (BDAB) was prepared in the bromide form by the Menschutkin reaction of diazabicyclooctane with dibromobutane. Before use in the AlPO synthesis, the bromide salt of BDAB was converted to a solution of the hydroxide form either by reaction with excess Ag2O or by stirring with a large excess of the Amberlite® IRN78 resin (fully OH− exchanged) for 12 h at room temperature. AlPO STA-2 was crystallised by hydrothermal treatment of a gel of composition R(OH)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Al(OH)3:H3PO4:H2O = 0.44:1:1:40 in a Teflon-lined stainless steel autoclave at 190 °C for 48 h. After slow cooling, the product was suspended in water, sonicated to remove uncrystallised material in suspension and the crystalline solid was filtered off (see ESI† for further information). The NMR spectra described below were measured on AlPO samples prepared via Ag2O treatment whereas crystals suitable for single-crystal X-ray diffraction studies (SXRD) were prepared via anion exchange over the resin.
:Al(OH)3:H3PO4:H2O = 0.44:1:1:40 in a Teflon-lined stainless steel autoclave at 190 °C for 48 h. After slow cooling, the product was suspended in water, sonicated to remove uncrystallised material in suspension and the crystalline solid was filtered off (see ESI† for further information). The NMR spectra described below were measured on AlPO samples prepared via Ag2O treatment whereas crystals suitable for single-crystal X-ray diffraction studies (SXRD) were prepared via anion exchange over the resin.
The samples were confirmed as STA-2 via laboratory PXRD on a Stoe STAD i/p powder diffractometer using Cu Kα1 radiation. The particle morphology was determined by scanning electron microscopy using a JEOL JSM-5600 at 5 kV acceleration voltage and a working distance of 21 mm. The spot size was set to 25 nm.
Diffraction
Single crystals of as-prepared AlPO STA-2 of suitable size (10–20 μm, see ESI†) were examined on a Rigaku Saturn92 diffractometer with a CCD detector using Cu Kα1 radiation (rotating anode). The structure was solved and refined employing SHELX-97,22 (see Table 1 and crystallographic file in ESI†). The framework was solved with alternating Al and P in tetrahedral cation positions and remaining extra-framework electron density in the large cage was assigned to the template. Electron density found in the smaller cancrinite cage was identified as charge-balancing hydroxide groups in the cages. The occupancy of the hydroxide O atoms (represented as O100 in the refinement of STA-2 in R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ) was allowed to vary, giving an average occupancy of 0.33. Lowering the occupancy gave rise to physically unrealistic thermal parameters for this species.
) was allowed to vary, giving an average occupancy of 0.33. Lowering the occupancy gave rise to physically unrealistic thermal parameters for this species.
| Chemical formula unit | (C8H12N2)2 Al12P12O50 |
| Z | 3 |
| Unit cell weight | 5301.69 |
| Calculated density/g cm−3 | 2.029 |
| Temperature/K | 173 |
| Space group | R |
| X-ray source | Rotating anode Cu Kα1 |
| Wavelength/Å | 1.54187 |
| Crystal size/mm | 0.01 × 0.01 × 0.02 |
| Unit cell dimensions/Å | a = 13.0310(6) |
| b =13.0310(6) | |
| c = 29.4980(16) | |
| α = 90° β = 90° γ = 120° | |
| Unit cell volume/Å3 | 4337.89(4) |
| Rp (all data) | 0.1121 |
| Rp (I > 2σI) | 0.1689 |
| Max and min residual e− density/e− Å−3 | −0.77, 1.21 |
NMR spectroscopy
MAS NMR spectra were acquired using Bruker Avance spectrometers at B0 field strengths of either 9.4, 14.1, 18.8 or 20.0 T, at Larmor frequencies of 104.3, 156.4, 208.4 and 221.5 MHz for 27Al, and 162.0, 242.9, 323.9 and 344.1 MHz for 31P, at the University of St Andrews (9.4 and 14.1 T), L'Universite de Lille, France (18.8 T) and the UK 850 MHz Solid-State NMR Facility (20.0 T). Powdered samples were packed into conventional 4 or 3.2 mm rotors and rotated at MAS rates between 12.5 and 20 kHz. Typical recycle intervals were 1–3 s for 27Al and 10–30 s for 31P. Chemical shifts are reported (in ppm) relative to 85% H3PO4 for 31P and 1 M Al(NO3)3 (aq) for 27Al. Triple-quantum MAS NMR experiments were carried out using a phase-modulated split-t1 shifted-echo pulse sequence,23 or a z-filtered pulse sequence.24 In each case, the final pulse was chosen to be selective for the central transition. The scale in the indirect dimension is referenced according to the convention in ref. 25. Two-dimensional heteronuclear correlation experiments were performed using transfer through either the J coupling (with an INEPT transfer from 27Al to 31P)26,27 or the dipolar coupling (using a D-HMQC28 experiment). In each case, for 27Al the pulses applied were selective for the central transition.DFT calculations
Calculations of total energies and NMR parameters were carried out using the CASTEP density functional theory (DFT) code (version 5),29 employing the GIPAW algorithm,15 to reconstruct the all-electron wave function in the presence of a magnetic field. Calculations were performed using the GGA PBE functional,30 with core–valence interactions described by ultrasoft pseudopotentials.31 A planewave energy cutoff of 50 Ry was used, and integrals over the Brillouin zone were performed using a Monkhurst–Pack grid with k-point spacing of 0.04 Å−1. Calculations were performed on a 198-node (2376 core) Intel Westmere cluster with 2 GB memory per core and QDR Infiniband interconnect at the University of St Andrews.The initial STA-2 structure was taken from diffraction measurements, with protons added manually to the SDA. Hydroxyl groups were also added manually as discussed in detail in later sections. In a preliminary step the positions of all protons in the structure were optimised (with the coordinates of all other atoms and the unit cell size and shape fixed). Subsequently, geometry optimisation was performed (using a planewave energy cutoff of 50 Ry and a k-point spacing of 0.04 Å−1) with the positions of all atoms, and the unit cell size and shape, allowed to vary. After 30 iterations of optimisation (5–6 days calculation time) the forces on the atoms had reduced to less than 0.1 eV Å−1, although the optimization was not converged (within the criteria specified: 1 × 10−4 eV per atom, 0.05 eV Å−1 and 1 × 10−3 Å for total energy, ionic force and ionic displacement, respectively), at this point. This could be seen to be due to motion of the template within the pores, rather than any change in the position of the framework atoms. Geometry optimization for one structural model was continued until completion (110 steps, 24 days). However, no significant change was observed in the position of the framework atoms. Owing to the lengthy calculation times, for all other models structures optimised for 30 iterations were deemed sufficient.
Calculations generate the absolute shielding tensor (σ) and the electric field gradient (EFG) tensor (V) in the crystal frame. In each case, diagonalisation yields the three principal components of the tensor (σXX, σYY and σZZ for the shielding tensor and VXX, VYY and VZZ for the EFG tensor). The isotropic shielding, σiso = (1/3) Tr{σ}, and the isotropic chemical shift, δiso, is given by −(σiso − σref), where σref is a reference shielding. This was determined to be 552.3 ppm for 27Al and 278.9 ppm for 31P, from calculations carried out on calcined STA-2 (see ESI† for further details). The magnitude of the quadrupolar coupling constant is given by CQ = eQVZZ/h, where Q is the nuclear quadrupole moment (for which a value of 146.6 mB was used for 27Al).32 The asymmetry parameter is given by ηQ = (VXX − VYY)/VZZ.
Results and discussion
NMR spectroscopy
The STA-2 structure (framework type SAT), shown in Fig. 1a, can be described in terms of the stacking of its secondary building units (SBUs), with six-membered rings (6MRs) stacked at different positions in the xy plane of a hexagonal unit cell, connected via four-membered rings (4MRs).20,21 As shown in Fig. 1b, the resulting framework contains single and double six-membered rings (S6Rs and D6Rs, respectively), 4MRs and both cancrinite (can) and elongated cages. In the calcined framework there are two distinct Al and two distinct P sites (shown also in Fig. 1b). Al2 and P1 are located in D6R units, and Al1 and P2 are located in S6R units. These join to form cancrinite cages that share a face of a 4MR. Therefore, via oxygen, Al1 is bonded to one P1 and three P2, and Al2 is bonded to one P2 and three P1. The 27Al and 31P NMR spectra for calcined (dehydrated) STA-2(BDAB) have been shown in previous work,21 and are also shown in Fig. 2a. The 31P MAS NMR spectrum shows two distinct resonances at −29.6 and −34.8 ppm, corresponding to P1 (D6R) and P2 (S6R), respectively. The 27Al spectrum also showed two resonances, albeit overlapping, which could be resolved by MQMAS, with δiso of 36.0 and 42.0 ppm, corresponding to Al1 (S6R) and Al2 (D6R), respectively. NMR parameters for calcined STA-2 are summarised in Table 2. The spectral assignment was supported by DFT calculations and two-dimensional heteronuclear correlation experiments.21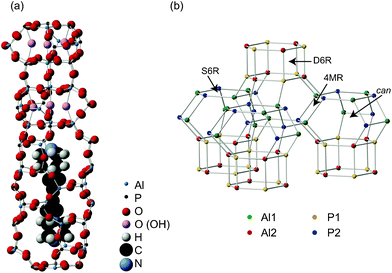 | ||
| Fig. 1 (a) Part of the structure of STA-2(BDAB), showing the BDAB template within the pores. The six possible hydroxyl positions suggested by diffraction measurements are also shown (as pink spheres). (b) Layers of cancrinite cages in the STA-2 framework structure, with Al1, Al2, P1 and P2 shown by green, red, yellow and blue spheres, respectively. Single and double six-membered rings (S6Rs and D6Rs, respectively), four membered rings (4MRs) and cancrinite (can) cages are shown. | ||
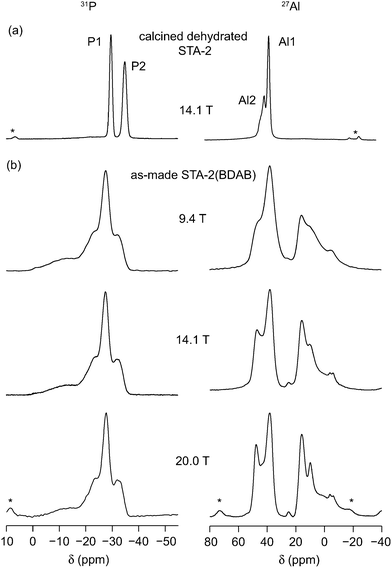 | ||
| Fig. 2 31P (left) and 27Al (right) MAS NMR spectra of (a) calcined dehydrated STA-2 and (b) as-made STA-2(BDAB) at varying B0 field strengths. The MAS rate was between 12.5 and 14 kHz. Spinning sidebands are denoted by *. | ||
| Species | SBU | δiso (ppm) | CQ/MHz | ηQ | PQ/MHz |
|---|---|---|---|---|---|
| Al1 | S6R | 36.0(5) | 2.0(2) | 0.7(2) | 2.1(2) |
| Al2 | D6R | 42.0(5) | 3.5(1) | 0.9(5) | 3.9(2) |
| P1 | D6R | −29.6 | |||
| P2 | S6R | −34.8 |
The as-made AlPO contains the SDA (BDAB), encapsulated within the pores, and charge-balancing hydroxyls coordinated to Al atoms in the framework. The local environment of each Al and P species is now modified, with the presence of both four- and five-coordinated Al sites, while each P species now has a potentially different next-nearest neighbour (NNN) coordination, P(OAlIV)4−n(OAlV)n, where the superscript indicates the Al coordination number. Fig. 2b shows 31P and 27Al MAS NMR spectra of STA-2(BDAB) at variable external magnetic field strength. (MAS NMR spectra at B0 = 14.1 T were previously reported by Castro et al.,21 but were unassigned.) For both 27Al and 31P, the spectra are more complex than those for the calcined form, with a number of broad and overlapping lineshapes observed. For 31P, intense resonances are observed between −20 and −40 ppm, with a much broader, low intensity component at higher chemical shift. No difference in the spectrum is observed when using cross polarization, indicating all peaks result from Q4 P species and not from species with any directly-bonded hydroxyls. Two distinct sets of resonances are observed in the 27Al MAS NMR spectrum, corresponding to four-coordinate Al between 35 and 50 ppm and five-coordinate Al between 0 and 20 ppm.33 A broader resonance with low intensity is also observed at lower chemical shift, perhaps resulting from an amorphous or less crystalline impurity phase. As discussed previously,21 the relative ratio of the signal intensity for AlIV and AlV can be used to provide insight into the position of the charge-balancing hydroxyls. As demonstrated previously by comparison to STA-2(BQNB), the charge on the SDA is 2+, and so two hydroxyls are required to charge balance each template molecule.21 The basic formula therefore becomes Al12P12O48(OH)2·BDAB. If the hydroxyls are terminal ligands (i.e., coordinated only to one Al species) then two Al would be five coordinate and the remainder four coordinate, resulting in a 2:10 (or 1:5) AlV:AlIV ratio. If, however, the hydroxyls bridge between Al species there would be four five-coordinate and eight four-coordinate Al species, and a 4:8 (or 1:2) AlV:AlIV ratio. The latter case is in much better agreement with ratio observed experimentally (between 1:1.5 and 1:1.7 at the three fields), suggesting the hydroxyls in STA-2(BDAB) are bridging.
27Al (14.1 T) multiple-quantum (MQ) MAS NMR spectra (shown in ESI†) exhibit broadened resonances, despite the resolution enhancement expected from the removal of the second-order quadrupolar broadening, as a result of a distribution in both δiso and PQ. Spectra have been recorded using both phase-modulated shifted-echo23 and z-filtered24 experiments to ensure that any broadened resonances are acquired accurately and that no signal is lost as a result of T2 relaxation during the echo period. Two broadened resonances corresponding to four-coordinate Al are clearly resolved, and are presumed to arise from species that correspond to Al1 and Al2 in the calcined form (i.e., in S6Rs and D6Rs respectively), although these cannot be assigned simply from the spectrum. A small but distinct shoulder is observed between the two. Only one (broadened) resonance is observed in the five-coordinate region of the spectrum. NMR parameters (extracted using dmfit34 assuming a Czjzek distribution of quadrupolar products from spectra acquired at a range of field strengths) are given in Table 3. (Note that most resonances most likely result from the overlap of a number of signals.) The integrated intensities of the two major AlIV resonances is 1:1, leading to the suggestion that a similar number of bridging hydroxyls are attached to both Al1 and Al2.21 (Although MQMAS is formally a non-quantitative experiment, both species have a similar distribution of PQ, meaning that accurate relative intensities should be obtained.) The presence of one major resonance corresponding to AlV is, therefore, a little surprising as one might have expected two resonances for AlV in S6Rs and D6Rs (i.e., formally Al1 and Al2 species), casting some doubt on the previous spectral interpretation.
| Species | <δiso> (ppm) | FWHM (ppm)a | PQ/MHzb |
|---|---|---|---|
| a Assuming a Gaussian distribution.b Extracted using a Czjzek (GIM, d = 5) distribution of the quadrupolar product.34c Note that most resonances most likely result from the overlap of a number of signals. | |||
| Al(V) | 17(1) | 4 | 3.1(2) |
| Al(IV) | 39(1) | 4 | 2.4(2) |
| Al(IV) | 45(1) | 3 | 3.6(2) |
| Al(IV) | 49(1) | 3 | 3.1(2) |
Fig. 3 shows two-dimensional 27Al–31P heteronuclear correlation (J-HETCOR) spectra26,27 of STA-2(BDAB), acquired at B0 = 20.0 T. Several cross peaks are observed, with varying intensity, although notably the 31P resonance at −23 ppm is not connected to the AlIV species at 49 ppm. The shoulder at δ1 ≈ 45 ppm (observed in the MQMAS spectra) is clearly visible, and exhibits a connectivity similar to that of the Al species at δ1 ≈ 49 ppm, suggesting it has a more similar environment to this Al species than that with δ1 ≈ 39 ppm. No significant difference was observed in the spectra when the evolution interval, τ, was increased, suggesting all Al–O–P J couplings are of similar magnitude. MQ-J-HETCOR experiments27 were also performed, but owing to the disordered nature of the sample there was little increase in resolution, and a significant loss of sensitivity. A D-HMQC spectrum28 (exploiting transfer via the dipolar coupling) was also acquired (at B0 = 18.8 T), and is shown in the ESI.† In general, similar cross peaks are observed (note the reversal of the nuclear dimensions), with increased intensity of the cross peak between the resonances (now) at −22 ppm (31P) and 47 ppm (AlIV), which is absent in the experiment probing through-bond connectivity, confirming these species are close in space but not linked through covalent bonds. However, a broad, low intensity cross peak is observed between the broad resonances observed in both 27Al and 31P spectra, confirming these are present in the same chemical phase. Notably, no correlations are observed for either of these resonances with any of the sharper, more intense peaks, confirming the suggestion that they result from a less crystalline impurity phase.
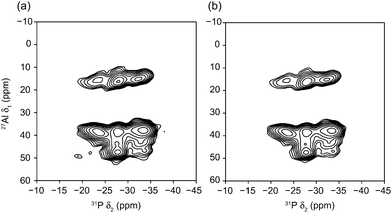 | ||
| Fig. 3 27Al–31P (20.0 T) two-dimensional J-HETCOR spectra of STA-2(BDAB), acquired using an INEPT transfer from 27Al to 31P, with τ of (a) 1.6 ms and (b) 2.4 ms. The MAS rate was 12.5 kHz. | ||
STA-2 is known to absorb water in its as-made form.20,21 However, there were no significant changes in the 27Al and 31P spectra of a sample of STA-2(BDAB) dried at 250 °C for 3 hours (shown in ESI†), suggesting that the water is present only within the pores and is not coordinated to the framework Al species. A comparison of the 1H MAS NMR spectra (also shown in ESI†) reveals a decrease in intensity of the resonance at ∼5 ppm upon drying, confirming water was present in the pores initially. (The residual intensity at 5 ppm in the spectrum of the dried material probably results from the hydroxyl protons.) A two-dimensional 27Al/31P J-HETCOR spectrum (see ESI†) of the dried material, acquired at 14.1 T, shows no differences to that of the hydrated as-made material, confirming that the framework structure (and, therefore, the NMR parameters) are unaffected by the presence of water.
Diffraction
Although there are two crystallographically-distinct Al and P sites in calcined STA-2,20 the presence of the SDA and the associated hydroxyls must lower the local symmetry from rhombohedral to triclinic. This makes each of the twelve Al and P species in the primitive P1 triclinic cell formally different (six for each of the two topologically-distinct sites). The six Al located in S6Rs (i.e., formally Al1 in the calcined material) are denoted in this work Al11–Al16, whereas those in D6Rs (i.e., formally Al2 in the calcined material) are denoted Al21–Al26. Similarly P11–P16 and P21–P26 refer to P derived from P1 and P2, respectively, in the calcined material. In previous work synchrotron powder X-ray diffraction data for dehydrated STA-2(BDAB) was refined starting from an initial structural model with the position of the template determined by molecular modelling.21 Difference Fourier maps showed scattering within the cancrinite cages, thought to be due to the hydroxyls. Using information from the 27Al MAS NMR spectra (which demonstrate the hydroxyls are bridging between two different Al species), six potential positions for hydroxyls within the cancrinite cages (i.e., three within each cage in the unit cell) could be refined, and are shown in Fig. 4a, numbered O91 to O96. More detailed refinement determined one of the two hydroxyls within the structure was definitely located within one of the cancrinite cages (on O91 or O92, with fractional occupancies of 0.25 and 0.75, respectively). The second hydroxyl could not be located unambiguously, but probably also lay within a cancrinite cage.21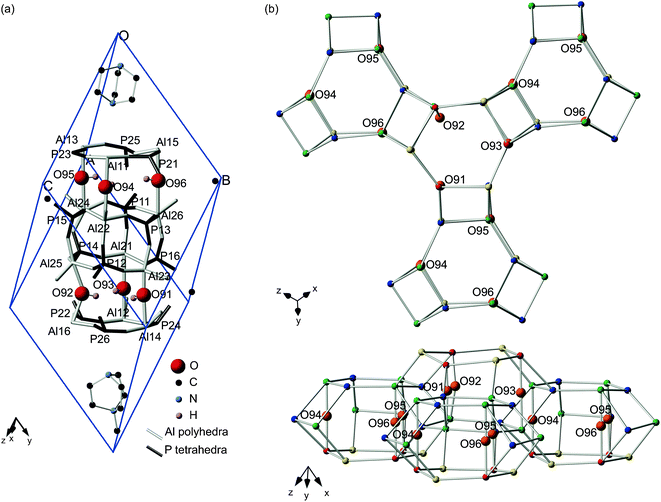 | ||
| Fig. 4 (a) Unit cell corresponding to dehydrated STA-2(BDAB), showing the distinct Al and P species, and the six possible hydroxyl locations. (b) Part of framework structure of dehydrated STA-2(BDAB), shown as a wireframe model, indicating the proximity of the hydroxyl groups. Atoms in the BDAB template are not shown for clarity. | ||
During the course of the current work it was possible to prepare single crystals suitable for structural analysis in the as-prepared form by SXRD (structural details given in Table 1). The data was indexed in the rhombohedral space group R and structure solution confirmed the STA framework (average Al–O framework bond lengths (disregarding coordinated OH−), Al1–O = 1.72(2) Å; Al2–O = 1.74(1) Å; average P–O bond lengths, P1–O = 1.51(1) Å; P2–O = 1.51(1) Å), and located the intact BDAB template. Furthermore, and consistent with 27Al MAS NMR, it was possible to locate O atoms in bridging positions between Al1 and Al2 cations in the top and bottom 6MRs of the cancrinite cages, with an average occupancy of 0.33, corresponding to one bridging OH− per cage, or two per BDAB SDA, as expected for complete charge balance of the template. The Al–O distances (Al1–O100 = 2.28(2) Å; Al2–O100 = 2.35(2) Å) are longer than expected, but note that this can be attributed to the fractional occupancy of the hydroxyl group, such that the Al positions are weighted averages of coordinated and uncoordinated environments. It was not possible to determine the positions of the OH− groups relative to one another owing to the statistical disorder in the R symmetry.
First-principles calculations
A series of nine possible structural models of STA-2(BDAB) were created, assuming both hydroxyls are located in cancrinite cages, on two of the six hydroxyl sites located by diffraction, and are denoted subsequently as OXOY, where X and Y refer to the position of the hydroxyls as shown in Fig. 4a. In the models chosen, either O91 or O92 were occupied, with the second hydroxyl placed on one of the other (O93 to O96) possible positions. No water was included in any of the models, as this was not located by diffraction – however, as shown in the ESI,† there are no significant changes in the 27Al and 31P NMR spectra for hydrated or dried samples. The models can be divided into four separate groups, according to the structural relationship between the two occupied hydroxyl sites, i.e., whether they share a cancrinite cage or a 4MR, and their spatial proximity. This is summarised in Table 4, and shown schematically in Fig. 4b. All structures were optimised as described in the Experimental section. Fig. 5 shows an overlay of part of one of the structural models (O92O94) both prior to and post DFT optimisation. There are some significant changes in the framework geometry after optimisation, as the coordinates determined by diffraction are the weighted averages of atomic positions when the hydroxyl group is present or absent. Furthermore, the Al–OH distances are between 2.15 and 2.3 Å prior to optimisation, but between 1.95 and 1.98 Å post optimisation. The initial distances (too long for typical Al–O bonds) result, as described above, from the weighted average of the Al atomic coordinates.21 The distances are considerably shorter after optimisation as there is a significant change in the position of the framework Al.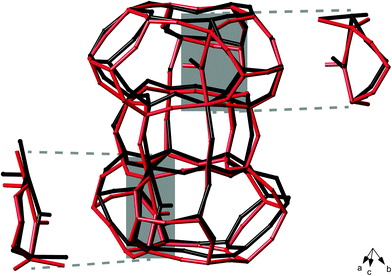 | ||
| Fig. 5 Overlay of part of the framework (shown as a wireframe model) in the O92O94 structural model of dehydrated STA-2(BDAB), both pre (black) and post (red) optimisation of the geometry using DFT calculations. Atoms in the BDAB template are not shown for clarity. The significant changes in the position of the AlV species once the neighbouring hydroxyl site is occupied are highlighted. | ||
| Model | Relationship of hydroxyl groups |
|---|---|
| O91O92, O91O93, O92O93 | Located in same can cage |
| O91O94, O92O95 | Located in different can cages with closest O–O distance of 5.97 Å |
| O91O95, O92O96 | Located in different can cages but share a 4MR |
| O91O96, O92O94 | Located in different can cages with closest O–O distance of 6.19 Å |
After optimisation, 31P and 27Al NMR parameters were calculated for each of the structural models. These are given in full in the ESI.† The 31P calculated isotropic chemical shifts are plotted in Fig. 6a. For P in D6Rs (P11–P16) a range of shifts are observed centred on −26 ppm, reflecting the variation of the local geometry around P in the disordered structure. P11–16 are found with three different NNN coordination environments in the models, i.e., P(OAlIV)4, P(OAlIV)3(OAlV) and P(OAlIV)2(OAlV)2, as shown in Fig. 6a. However, there appears to be no significant differences in the calculated chemical shifts for these species, with resulting spectral resonances expected to have considerable overlap in the MAS spectrum. However, there is a significant difference in the 31P calculated isotropic chemical shifts for P in S6Rs (P21–P26), with two separate ranges, centred on −33 and −22 ppm, respectively. The P2 sites have a number of different possible environments, as also shown in Fig. 6a, with 0, 1 or 2 AlV NNN. However, the large difference in calculated chemical shift results not from the number of AlV NNN, but from whether the AlV are coordinated to the same hydroxyl group, i.e., whether the two Al species the hydroxyl group bridges are attached to the same P atom. These environments are denoted S6R* in Fig. 6, and result in an increase in the calculated chemical shift of 8–14 ppm. It has been shown previously in the literature35 that for PO4 species there is a relationship between the chemical shift and the average Al–O–P angle, with a decrease in shift corresponding to an increased average Al–O–P angle. Examination of the average P–O–Al bond angles for the structural models used here reveals a decrease from 150°–154° for P21–26 in S6R environments to 142°–147° for S6R* environments, with the latter much more similar to those for P11–P16 (of 140°–148°), in agreement with the results of Müller.35 For each of the different models used the range of calculated shifts (shown in Fig. 6b) is very similar, and it is not possible to distinguish between them purely on this basis, although those where the hydroxyls share a cancrinite cage (O91O92, O91O93 and O92O93) perhaps show slightly poorer agreement with experiment. The three major resonances in the 31P MAS NMR spectrum may, therefore, be assigned to P in S6R, D6R and S6R* environments in order of increasing chemical shift.
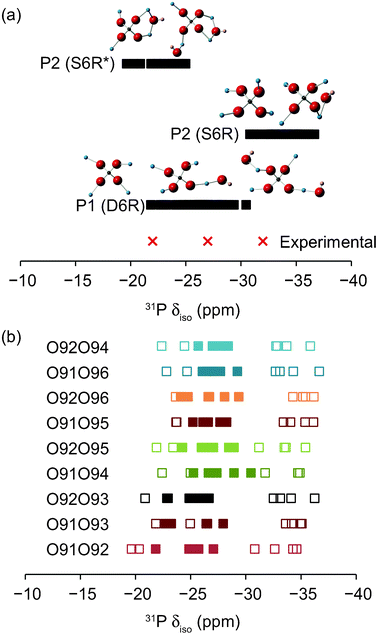 | ||
| Fig. 6 Calculated 31P isotropic chemical shifts, δiso, for the structural models of STA-2(BDAB), described in the text. Experimental values are shown for comparison. Also shown are the possible local coordination environments for each P. In (a) all calculated shifts are shown together, while in (b) data for each individual model are shown separately. In (b), filled and empty squares denote P in D6Rs and S6Rs, respectively. | ||
The 27Al calculated isotropic chemical shifts are plotted in Fig. 7a, and compared to the average shifts extracted from the MQMAS experiments shown in the ESI† for the resonances in STA-2(BDAB). The shifts predicted for AlIV in D6Rs (i.e., Al21–26) are generally higher than those for S6R AlIV (i.e., Al11–Al16). The distribution of local environments produces a range of calculated CQ values. In general, those for AlIV in D6Rs are generally higher (between 3 and 6 MHz) than those for S6R (between 1 and 4 MHz). This prediction agrees with the appearance of the resonances in the 27Al MQMAS spectra shown in the ESI,† where the resonance at higher chemical shift is broader than that at lower shift, although in both cases the lineshapes suggest a distribution of CQ. It should also be noted that the CQ values observed for Al1 and Al2 in calcined STA-2 are 2.0 and 3.5 MHz, respectively. For AlV species, those in D6R and S6R have similar ranges of chemical shifts, between 15 and 30 ppm, and so are unlikely to be separated in the experimental MAS spectrum, supporting (and explaining) the observation of a single broad resonance in this region. A broad range of CQ values are predicted for these species, from 2 to 9 MHz, though the majority lie between 3.6 and 8.4 MHz. As also observed for 31P, the shifts calculated for each of the models are very similar, with only those containing hydroxyls in the same cancrinite cage showing some differences to the rest, and these are generally in poorer agreement with experiment. In contrast to 31P, the position of a resonance for 27Al depends not just on the isotropic chemical shift, but also on the isotropic second-order quadrupolar shift, δQ (given by −(16/15) ((νQPAS)2/ν0) (1 + ηQ2/3) for an I = 5/2 nucleus).36 The presence of the quadrupolar interaction will, therefore, have a small effect upon the resonance position when CQ is small (e.g., ∼2.2 ppm for a spin I = 5/2 nucleus with a CQ of 3 MHz at 14.1 T) but an increasingly important effect as the quadrupolar interaction is larger.
 | ||
| Fig. 7 Calculated 27Al isotropic chemical shifts, δiso, for structural models of STA-2(BDAB), described. Experimental values (extracted from MQMAS NMR spectra) are shown for comparison. In (a) all calculated shifts are shown together, while in (b) data for each individual model are shown separately. In (b), filled and empty squares denote Al in D6Rs and S6Rs, respectively. | ||
It is also possible to consider the agreement between experimental 27Al MQMAS NMR spectra and the calculated NMR parameters by plotting the predicted centre-of-gravity (δ1, δ2) of the resonance associated with each Al species in the calculated models of STA-2(BDAB). The δ2 position is simply a sum of the isotropic chemical and second-order quadrupolar shifts (as described for the MAS spectrum above). The position in δ1 is given (for a sheared triple-quantum MAS spectrum) by δ1 = (17/31) δiso + (32/93) δQ (for I = 5/2, using the scaling convention of ref. 25). Fig. S7.1 in the ESI† shows the predicted centre-of-gravity for each of the Al species in the structural models considered. When all models are considered together, there is good agreement between the resonance positions for calculated and experimental data for AlIV species. The agreement for AlV species is less good, perhaps reflecting the disorder and possible dynamics of the coordinated hydroxyl groups. The calculated data for hydroxyl groups found in the same cancrinite cage (O91O92/O91O93/O92O93) is in poorest agreement with experiment, with a particularly large spread for the AlV species. The differences between the three other groups of models are much smaller, with all in reasonable agreement with experiment.
Fig. 8a shows the predicted position of cross peaks in a 27Al/31P (20.0 T) J-HETCOR spectrum for each of the structural models of STA-2(BDAB). These plots were generated by correlating the isotropic chemical shifts for 31P with the 27Al resonance position (a sum of both isotropic chemical shifts and isotropic quadrupolar shifts as described above) for all species joined by a single Al–O–P linkage. The Al and P species linked by two bonds are shown in Table 5. In general, and as noted previously for the MQMAS spectra, the agreement between calculated results and experiment is better for AlIV than for AlV. Particularly good agreement is observed for the two sets of models where the hydroxyl groups are located in different cancrinite cages, with the obvious absence of a cross peak between the 31P at ∼−22 ppm and the 27Al at ∼48 ppm. For the set of models where the hydroxyl groups are in the same cancrinite cage the agreement with experiment is poor, with predicted cross peaks observed where the signal is absent experimentally, and with more generally a wider spread of intensity, particularly for cross peaks involving AlV. The models where hydroxyls are in separate cancrinite cages but sharing a 4MR result in correlation plots that do not completely agree with experiment, with only cross peaks observed at higher 31P shift, i.e., few or no cross peaks observed at 31P shifts lower than −24 ppm. However, this does not rule out the presence of this type of hydroxyl ordering in STA-2(BDAB), but shows that if it is present it must correspond to a relatively minor component of the structure, co-existing with significantly more local environments where the hydroxyls are more remote in order to generate the correct cross peaks with 31P at lower chemical shift. The ESI† shows a similar set of plots to those in Fig. 8, but where only the 31P and 27Al isotropic chemical shifts are correlated, i.e., the isotropic quadrupolar shift for 27Al is not included. Whilst, in principle, this should provide less accurate results, the agreement with experiment is perhaps better than that seen in Fig. 8 – a somewhat surprising result. However, while it has been shown that good agreement between experiment and calculation is usually obtained when calculating chemical shifts, the calculation of quadrupolar NMR parameters is often not quite as accurate, and a small but systematic overestimation of CQ has been observed for a number of nuclei including 25Mg, 71Ga, 93Nb, 17O and 27Al.7,17,37–41 This error ultimately leads to errors in the isotropic quadrupolar shifts, and it appears in this case these are more significant than the small errors associated with the calculation of δiso. In the latter case, of course, there may be a cancellation of any systematic errors within the referencing process, whereas this is clearly not possible when calculating quadrupolar shifts.
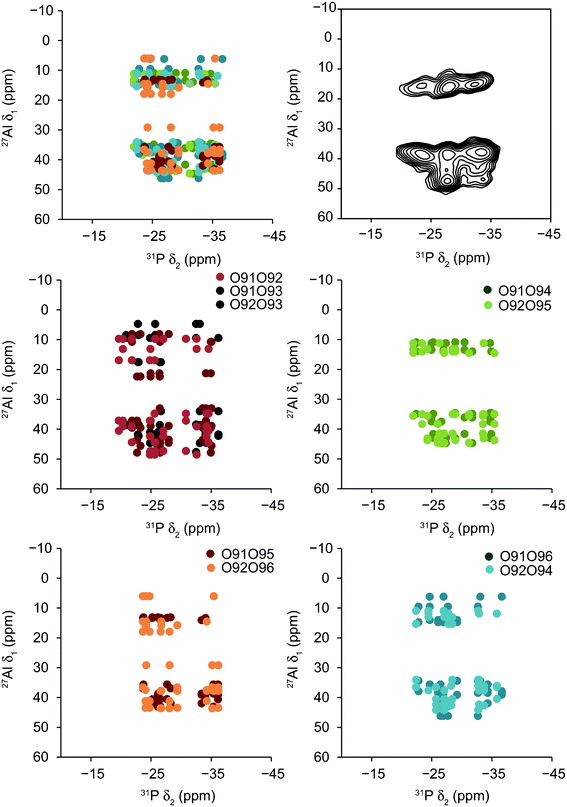 | ||
| Fig. 8 Calculated cross peak positions in 27Al/31P (20.0 T) J-HETCOR spectra for each of the structural models of STA-2(BDAB), generated by correlating the 31P isotropic chemical shift with the 27Al resonance position (a sum of both isotropic chemical shifts and isotropic quadrupolar shifts as described above) for species joined by an Al–O–P linkage, as shown in Table 5. Also shown is the experimental spectrum (20.0 T) of STA-2(BDAB) for comparison. | ||
| P1 in calcined dehydrated structure (D6R) | P2 in calcined dehydrated structure (S6R) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Al/P in templated structure | 11 | 12 | 13 | 14 | 15 | 16 | 21 | 22 | 23 | 24 | 25 | 26 | |
| Al1 in calcined dehydrated structure (S6R) | 11 | X | X | X | X | ||||||||
| 12 | X | X | X | X | |||||||||
| 13 | X | X | X | X | |||||||||
| 14 | X | X | X | X | |||||||||
| 15 | X | X | X | X | |||||||||
| 16 | X | X | X | X | |||||||||
| Al2 in calcined dehydrated structure (D6R) | 21 | X | X | X | X | ||||||||
| 22 | X | X | X | X | |||||||||
| 23 | X | X | X | X | |||||||||
| 24 | X | X | X | X | |||||||||
| 25 | X | X | X | X | |||||||||
| 26 | X | X | X | X | |||||||||
Although attention has focussed on the calculation of 27Al and 31P NMR parameters in order to understand the location of the charge-balancing hydroxyls in STA-2(BDAB), DFT calculations also provide information on the NMR parameters for the template molecule (i.e., for 13C and 15N in BDAB), and these are shown in full in the ESI.† Very similar results are obtained for all of the sets of models used, suggesting little interaction between the SDA and the framework itself. For 15N, the calculations confirm the assignment of the apical and quaternary nitrogens in the previous work,21 although the difference between the calculated values (∼52 ppm) is slightly greater than that observed experimentally (∼43 ppm). However, it should also be noted that the previous experimental work was carried out on hydrated samples of STA-2(BDAB), and the presence of water (while having almost no effect upon the framework structure) may have a greater effect on the template molecules with which is shares the pore space. The DFT calculations also confirm the previous assignment of the 13C CP MAS NMR spectrum of STA-2(BDAB).
It should be noted that significant template dynamics have been observed in AlPO frameworks (often evidenced by changes to the NMR spectra of the framework atoms).13 Furthermore, significant template motion was also observed upon geometry optimization in the calculations in this work, indicating that dynamics may also play a role in this material. Dynamics may affect the experimental NMR parameters and, ultimately, the agreement between experiment and the calculated results. A further source of disagreement between calculation and experiment is the underestimation of dispersion forces in the DFT methods used (which may be of more significance for these inorganic/organic frameworks than for simple rigid solids). Although not used in this work, recently semi-empirical dispersion correction schemes have become available in periodic codes to attempt to account for this problem, and this approach does offer promise for future work. However, given the complex nature of the systems DFT calculations have been used here to aid spectral assignment and interpretation and to offer overall insight into the local structure, rather than providing precise agreement with experimental values, although both dynamics and dispersion forces may need to be taken into account when considering other materials.
Conclusions
Insight into the detailed structure of the pure aluminophosphate form of STA-2 was obtained by using a combination of diffraction, multinuclear solid-state NMR spectroscopy and first-principles calculations. Although previous work had shown that STA-2 could be prepared using bis-diazabicyclooctane-butane (BDAB) as an alternative SDA to the 1,4-bis-N-quinuclidiniumbutane (BQNB) used in the initial synthesis, the NMR spectra of the as-made form had not been assigned, and the positions of the charge-balancing hydroxyls not been located. The similarity of NMR spectra for STA-2(BDAB) and STA-2(BQNB) led to the suggestion in the earlier work that BDAB was also divalent when incorporated into the material and that the structures of the two as-made forms were very similar. Hence, the conclusions of the investigation in the current work are expected to be equally applicable to STA-2(BQNB).From the ratio of AlIV–AlV species in 27Al MAS NMR spectra it is possible to determine that the charge-balancing hydroxyls are bridging rather than terminal in the structure. The similar relative intensities of the two major AlIV resonances suggests that the hydroxyl groups bridge formal “Al1” and “Al2” species (i.e., those in S6R and D6R, respectively), although the presence of only one resonance in the region of the spectrum corresponding to AlV is, therefore, somewhat of a surprise. However, DFT calculations carried out using a series of structural models where hydroxyls were located on two of six possible sites within the cancrinite cages identified by diffraction, predicted distinctly different isotropic chemical shifts for Al11–16 and Al21–26 when four coordinate, but showed that the shifts were very similar for five-coordinate species in both cases, enabling the experimental spectrum to be assigned. A range of CQ values were predicted for all types of Al species, but those for AlIV in D6R are generally higher (3–6 MHz), than those for AlIV in S6R (1–4 MHz), in agreement with the shape and width of the resonances observed in two-dimensional 27Al MQMAS spectra. DFT calculations were also able to assign the three resonances in the 31P spectrum, with that at −27 ppm resulting from P in D6R (i.e., P11–P16), while the two less intense resonances at −23 and −33 ppm result from P in S6R (i.e., P21–P26). Little difference in the 31P chemical shift was observed as the number of AlV NNN increased, but a significant change in shift was observed for S6R species when the hydroxyl group bridged between two of the NNN Al species (S6R*). This was shown to result from a significant change in the Al–O–P bond angles (from 150°–154° for S6R to 142°–147° in S6R* environments).
From a consideration of simply 27Al and 31P calculated isotropic shifts it is difficult to rule out completely any particular type of structural model, as all show reasonable agreement with experiment. However, a comparison of experimental and calculated 27Al MQMAS spectra, and in particular 27Al/31P J-HETCOR spectra (created using the expected Al–O–P connectivities in the STA-2 framework structure), revealed some differences between the model types. In both cases, the Al resonance position was considered (i.e., a sum of isotropic chemical and quadrupolar shifts); however, it was shown that the (systematic) errors associated with the calculation of the quadrupolar coupling constants (and therefore the quadrupolar shifts) resulted in a greater spread of the calculated results compared to those seen in experiment, particularly for AlV species. In general, models where the hydroxyls were located in the same cancrinite cage were shown to be in less good agreement, ruling out this possibility in the real material. Generally poorer agreement was also shown between experimental and calculated data when the hydroxyl groups were in different cancrinite cages but shared a 4MR. However, this generally resulted in the absence of cross peaks in J-HETCOR spectra, rather than the presence of cross peaks that were not observed experimentally, leading to the conclusion that while most hydroxyls must be located in different cancrinite cages and also separated spatially, the presence of a small number of hydroxyls that shared 4MR could not be ruled out completely. As described in the experimental section, owing to template movement it was difficult to ensure the geometry optimisations were “converged”, making it difficult to compare the relative energies of the structural models. However, in general, for the optimized structures used, most of the models where the hydroxyls shared the same cancrinite cage were slightly higher in energy (0.3 eV per unit cell of STA-2(BDAB), or 28.9 kJ mol−1 of STA-2(BDAB)), tentatively supporting the conclusions from the NMR calculations.
Although it is difficult to refine a “structural model” for disordered materials using diffraction-based methods, if the information obtained is combined with that from NMR spectroscopy and from DFT calculations of NMR parameters a much more detailed understanding of both long- and short-range structure can be obtained. In many cases, the local structure gives important details on the synthesis mechanisms of these solids, and furthermore plays a very important role in determining the physical and chemical properties of the bulk material. The approach of combining diffraction and NMR spectroscopy and simulation to elucidate structure, often referred to as NMR Crystallography, offers a promising route for the future study of as-made and substituted microporous materials, and an understanding of the structure–property relationships that will ultimately determine their use and applications.
Acknowledgements
We would like to thank EPSRC (EP/E041825/1, EP/J501542/1, and EP/F018096/1) the European Commission FP6 Marie Curie Research Training Network “INDENS” (MRTN-CT-2004-005503), Johnson Matthey and the Leverhulme Trust (F/00 268/BJ) for support. We thank EaStCHEM for computational support through the EaStCHEM Research Computing Facility. The UK 850 MHz solid-state NMR Facility used in this research was funded by EPSRC and BBSRC, as well as the University of Warwick including via part funding through Birmingham Science City Advanced Materials Projects 1 and 2 supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF). We would also like to thank Dr Julien Trebosc and Dr Dinu Iuga for their help at the facilities in Lille and Warwick, respectively. Professor Alex M. Z. Slawin (University of St. Andrews) is thanked for collection of single crystal diffraction data and helpful discussions.References
- S. T. Wilson, B. M. Lok, C. A. Messina, T. R. Cannan and E. M. Flanigen, J. Am. Chem. Soc., 1982, 104, 1146 CrossRef CAS.
- Z. Han, A. L. Picone, A. M. Z. Slawin, V. R. Seymour, S. E. Ashbrook, W. Zhou, S. P. Thompson, J. E. Parker and P. A. Wright, Chem. Mater., 2010, 22, 338 CrossRef CAS.
- A. Tuel, C. Lorentz, V. Gramlich and C. Baerlocher, C. R. Chim., 2005, 8, 531 CrossRef CAS.
- A. Simmen, L. B. McCusker, Ch. Baerlocher and W. M. Meier, Zeolites, 1991, 11, 654 CrossRef CAS.
- J. M. Griffin, L. Clark, V. R. Seymour, D. W. Aldous, D. M. Dawson, D. Iuga, R. E. Morris and S. E. Ashbrook, Chem. Sci., 2012, 3, 2293 RSC.
- S. P. Brown, S. E. Ashbrook and S. Wimperis, J. Phys. Chem. B, 1999, 103, 812 CrossRef CAS.
- S. E. Ashbrook, M. Cutajar, J. M. Griffin, Z. A. D. Lethbridge, R. I. Walton and S. Wimperis, J. Phys. Chem. C, 2009, 113, 10780 CAS.
- C. M. Morais, V. Montouillout, M. Deschamps, D. Iuga, F. Fayon, F. A. A. Paz, J. Rocha, C. Fernandez and D. Massiot, Magn. Reson. Chem., 2009, 47, 942 CrossRef CAS.
- C. Martineau, C. Mellot-Draznieks and F. Taulelle, Phys. Chem. Chem. Phys., 2001, 13, 18078 RSC.
- C. Martineau, B. Bouchevreau, Z. J. Tian, S. J. Lohmeier, P. Behrens and F. Taulelle, Chem. Mater., 2001, 23, 4799 CrossRef.
- P. J. Byrne, J. E. Warren, R. E. Morris and S. E. Ashbrook, Solid State Sci., 2009, 11, 1001 CrossRef CAS.
- S. E. Ashbrook, M. Cutajar, C. J. Pickard, R. I. Walton and S. Wimperis, Phys. Chem. Chem. Phys., 2008, 10, 5754 RSC.
- S. Antonijevic, S. E. Ashbrook, S. Biedasek, R. I. Walton, S. Wimperis and H. X. Yang, J. Am. Chem. Soc., 2006, 128, 8054 CrossRef CAS.
- A. Tuel, S. Calarelli, A. Meden, L. B. McCusker, C. Baerlocher, A. Ristic, N. Rajic, G. Mali and V. Kaucic, J. Phys. Chem. B, 2000, 104, 5697 CrossRef CAS.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 245101 CrossRef.
- T. Charpentier, Solid State Nucl. Magn. Reson., 2011, 40, 1 CrossRef CAS.
- C. Bonhomme, C. Gervais, F. Babonneau, C. Coelho, F. Pourpoint, T. Azais, S. E. Ashbrook, J. M. Griffin, J. R. Yates, F. Mauri and C. J. Pickard, Chem. Rev., 2012, 112, 5733 CrossRef CAS.
- S. E. Ashbrook and D. M. Dawson, Acc. Chem. Res., 2013 DOI:10.1021/ar300303w.
- J. Cuny, S. Messaoudi, V. Alonzo, E. Furet, J. F. Halet, E. Le Fur, S. E. Ashbrook, C. J. Pickard, R. Gautier and L. Le Polles, J. Comb. Chem., 2008, 13, 2279 Search PubMed.
- G. W. Noble, P. A. Wright and A. Kvick, J. Chem. Soc., Dalton Trans., 1997, 4485 RSC.
- M. Castro, V. R. Seymour, D. Carnevale, J. M. Griffin, S. E. Ashbrook, P. A. Wright, D. C. Apperley, J. E. Parker, S. P. Thompson, A. Fecant and N. Bats, J. Phys. Chem., 2010, 114, 12698 CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112 CrossRef.
- S. P. Brown and S. Wimperis, J. Magn. Reson., 1997, 128, 42 CrossRef CAS.
- J. P. Amoureux, C. Fernandez and S. Steuernagel, J. Magn. Reson., Ser. A, 1996, 123, 116 CrossRef CAS.
- K. J. Pike, R. Malde, S. E. Ashbrook, J. McManus and S. Wimperis, Solid State Nucl. Magn. Reson., 2000, 16, 203 CrossRef CAS.
- C. A. Fyfe, K. C. Wongmoon, Y. Huang and H. Grondey, J. Am. Chem. Soc., 1995, 117, 10397 CrossRef CAS.
- J. P. Amoureux, J. Trebosc, J. Wiench and M. Pruski, J. Magn. Reson., 2007, 184, 1 CrossRef CAS.
- G. Tricot, O. Lafon, J. Trebosc, L. Delevoye, F. Mear, L. Montagne and J. P. Amoureux, Phys. Chem. Chem. Phys., 2011, 13, 16786 RSC.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. J. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- J. R. Yates, C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 024401 CrossRef.
- P. Pyykko, Mol. Phys., 2008, 106, 1965 CrossRef CAS.
- K. J. D. MacKenzie and M. E. Smith, Multinuclear Solid-State NMR of Inorganic Materials, Pergamon Press, Oxford, 2002, ch. 5 Search PubMed.
- D. R. Neuville, L. Cormier and D. Massiot, Geochim. Cosmochim. Acta, 2004, 68, 5071 CrossRef CAS.
- D. Muller, E. Jahn, G. Ladwig and U. Haubenreisser, Chem. Phys. Lett., 1984, 109, 332 CrossRef.
- A. J. Vega, Quadrupolar Nuclei in Solids, in eMagRes, 2010 Search PubMed.
- J. M. Griffin, A. J. Berry and S. E. Ashbrook, Solid State Nucl. Magn. Reson., 2011, 40, 91 CrossRef CAS.
- P. J. Pallister, I. L. Moudrakovski and J. A. Ripmeester, Phys. Chem. Chem. Phys., 2009, 11, 11487 RSC.
- J. M. Griffin, S. Wimperis, A. J. Berry, C. J. Pickard and S. E. Ashbrook, J. Phys. Chem. C, 2009, 113, 465 CAS.
- K. E. Johnston, J. M. Griffin, R. I. Walton, D. M. Dawson, P. Lightfoot and S. E. Ashbrook, Phys. Chem. Chem. Phys., 2010, 13, 7565 RSC.
- F. Blanc, D. S. Middlemiss, L. Buannic, J. L. Palumbo, I. Farnan and C. P. Grey, Solid State Nucl. Magn. Reson., 2012, 42, 87 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further information on the synthesis of AlPO STA-2, referencing of the DFT calculations, 27Al MQMAS spectra of STA-2(BDAB), D-HMQC spectra of STA-2(BDAB), NMR spectra of dehydrated STA-2(BDAB), calculated NMR parameters for structural models of STA-2(BDAB), comparison of DFT calculations and MQMAS spectra for STA-2(BDAB), DFT calculations of J-HETCOR spectra and calculated 13C/15N NMR parameters of the template in STA-2(BDAB). The crystallographic information file is also provided. See DOI: 10.1039/c3ce40965a |
| This journal is © The Royal Society of Chemistry 2013 |