 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of sequence controlled acrylate oligomers via consecutive RAFT monomer additions†
Joke
Vandenbergh
a,
Gunter
Reekmans
b,
Peter
Adriaensens
b and
Tanja
Junkers
*a
aPolymer Reaction Design Group Institute for Materials Research (IMO-IMOMEC), Universiteit Hasselt, Agoralaan Building D, B-3590 Diepenbeek, Belgium. E-mail: tanja.junkers@uhasselt.be; Web: http://www.polymatter.net Fax: +32 (11)268399; Tel: +32 (11)268318
bNuclear Magnetic Resonance Spectroscopy Group, Institute for Materials Research (IMO-IMOMEC), Universiteit Hasselt, Agoralaan Building D, B-3590 Diepenbeek, Belgium
First published on 17th September 2013
Abstract
An effective procedure to synthesize sequence controlled oligoacrylates with up to four monomer insertions via the RAFT technique is presented. Two different sets of oligomers with a preferred monomer order were synthesized. Recycling SEC allowed for facile and automated purification of the monodisperse sequence-controlled materials.
During the last decade, the synthesis and design of polymer-based materials have undergone a tremendous evolution due to the development of the controlled/living as well as ionic polymerization techniques1,2 in combination with the advent of very efficient conjugation reactions, generalized under the concept of click chemistry.3 Due to the elaboration of these techniques, polymer materials can nowadays be designed and synthesized virtually without limitations with regard to chain length, dispersity, composition, functionality and overall architecture.4,5 However, processes are still limited when it comes to monomer sequence control, which still separates up-to-date polymer synthesis from molecularly encoded materials produced by nature. Sequences of biological materials such as proteins or polynucleotides are perfectly defined, giving these materials the ability to fold into tertiary structures, capable of performing various advanced tasks such as selective transport, catalytic reactions or signal transduction.6 Despite these advantages, the development of methods to control the specific sequence order of monomers in synthetic polymer materials is yet one of the last major challenges in modern polymer science.7 The development of reliable techniques that allow for synthesis of even only short oligomeric chains with precisely tuneable order of chain units would already mark a very significant advance in the field and give rise to many applications. Sequence controlled oligomers could be built in larger structures for example as molecular recognition elements or anchor points for chain folding.
A highly valuable contribution to the development of macromolecular structures with defined monomer sequences consists of the Merrifield-synthesis of artificial peptides on a solid support.8 However, this method is only successful for relatively short peptides and due to consecutive protection–deprotection reactions and purification steps it is very time consuming and far from being quantitative. Another study describes the synthesis of sequence-controlled oligomers via living cationic polymerization of vinyl ethers and styrene derivatives.9 In yet another attempt to control monomer sequences, controlled radical polymerization techniques such as atom transfer radical polymerization (ATRP)10 and nitroxide mediated polymerization (NMP)11 were applied to synthesize AB alternating monomer sequences, by using the strong cross-polymerization behaviour of styrene and N-substituted maleimides. By adding small amounts of maleimides to an ATRP reaction of styrene at specific time intervals, very short segments of maleimide (1 or 2 units) could be built in the main chain of polymers at precisely located positions on the backbone, allowing for sequence control, but not removing the statistical nature of the entire chain.12–14 Furthermore, ATRP was also used as an approach to synthesize copolymers with defined sequence order of styrene, acrylate and vinyl chloride starting from defined oligomers.15 In a very recent approach reported by Tsanaktsidis et al.,16 reversible addition fragmentation radical transfer (RAFT)17 was used to sequentially insert two single monomer units into a RAFT agent. Using this approach, insertion of a single styrene unit followed by insertion of a single N-isopropyl acrylamide (NIPAM) monomer into a trithiocarbonate RAFT agent was successful, demonstrating the proof-of-concept for the generation of monodisperse material from the RAFT process. After each insertion step, the resulting macro-RAFT agent was purified via classical column chromatography, however, the reaction had to be stopped after the dimer formation due to separation issues.
In the current study, we adopted the core idea of the single monomer insertion strategy via the RAFT approach and used it for true oligomer sequence control synthesis. Two independent sets of monodisperse oligoacrylates consisting of up to four monomer units in precisely tuned order have been synthesized. The novelty of our approach lies in the fact that no cumbersome and time consuming classical column chromatography is used to purify the materials, but an automated preparative recycling size-exclusion chromatography (SEC) technique.18 In such a set-up, crude reaction products are passed over columns with size exclusion limits of 1000 and 5000 Da. To overcome the problem of insufficient separation, samples are repeatedly recycled by returning the elution flow back to the column entry. This procedure is repeated until molecules with different sizes are perfectly separated, allowing for facile purification of materials which would otherwise be inseparable. The product separation is followed via UV and RI signal detection and the entire process and collection of fractions is computer controlled, making the purification substantially less time consuming and more accurate than any classical column chromatography.
In a first step, 2-cyano-2-propyl dodecyl trithiocarbonate (CPD-TTC) was used as a chain transfer agent for the RAFT polymerization of n-butyl acrylate (nBuA) (Scheme 1). Recently, we have shown that in microreactions such RAFT agents can be used to approximate sequence controlled materials by synthesizing narrowly-distributed pentablock copolymers.19 However, since the aim was not to produce a polymer, but to insert only one single nBuA monomer into the RAFT agent, only 10 equiv. of nBuA were used and the reaction proceeded at 100 °C for a very short reaction time of 10 min. Optimization of the reaction showed that such an approach is more efficient than using equimolar amounts. Premature stopping of the reaction allows for a higher trithiocarbonate endgroup retention, which is of highest importance for the next insertion step. After quenching of the reaction, excess of solvent and monomer is evaporated to yield the crude product mixture. The success of the reaction was studied using soft electrospray ionization mass spectrometry (ESI-MS). Fig. 1a depicts the ESI-MS spectrum obtained for the crude nBuA macro-RAFT agent 1 mixture. The three different mass-to-charge (m/z) signals present in the spectrum can be assigned to macro-RAFT agents with single, double and triple nBuA insertions, respectively (intensities do not reflect concentration ratios). To isolate the single adduct from the side products with multiple insertions, the crude mixture was purified using recycling SEC as described above. Fig. 2a shows the UV signal recorded during recycling SEC of crude mixture 1. Already after two cycles, the different reaction products were separated successfully. The purity of oligo-RAFT agent 1 was verified with 1H NMR (see ESI†) and ESI-MS (Fig. 1b), which reveals only one mass-to-charge signal, corresponding to pure nBuA oligo-RAFT agent 1.
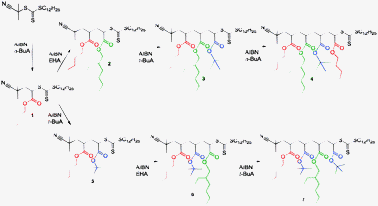 | ||
| Scheme 1 Sequence controlled insertion of four single monomer units forming 2 sets of oligoacrylate RAFT agents. | ||
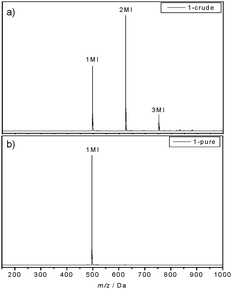 | ||
| Fig. 1 ESI-MS spectra of nBuA oligo-RAFT agent 1, (a) before and (b) after purification with recycling SEC. | ||
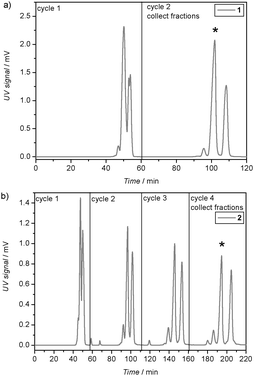 | ||
| Fig. 2 Recycling SEC trace recorded during consecutive purification cycles of (a) nBuA oligo-RAFT agent 1 and (b) nBuA-EHA macro-RAFT agent 2. The desired oligomer is marked with an asterisk. | ||
In a second step, the purified nBuA oligo-RAFT agent 1 was used as a chain transfer agent for a single RAFT insertion of either 2-ethylhexyl acrylate (EHA) or tert-butyl acrylate (tBuA), yielding macro-RAFT agents 2 and 5, respectively (Scheme 1). Generally, acrylates have been used as model compounds since they allow for facile incorporation of functionality into the products without significantly affecting the reactivity. Hence, they are ideal candidates to later truly encode chemical functionalities in the sequence. It is well-known from RAFT polymerization that the insertion of subsequent monomer units occurs significantly faster than the insertion of the very first monomer unit into a RAFT agent.20,21 Therefore, the reaction was optimized for best yields and only one equivalent of either EHA or tBuA was used for the second insertion reaction. ESI-MS of the resulting crude reaction products revealed again a distribution of single, double and triple insertion products (ESI†), which could be purified again without problems to obtain pure oligo-RAFT agents 2 and 5via recycling SEC, however, requiring higher cycle times (see Fig. 2b for purification of 2).
The same experimental conditions were repeated for the successful synthesis of macro-RAFT agents 3, 4, 6 and 7. The products were always purified by recycling SEC (purification time and final yields are provided in the ESI,† Table S23) and analysed using 1H-NMR, SEC and ESI-MS (ESI†). For all products synthesized, ESI-MS shows RAFT end group retentions of ≥99%. In the final stages of the synthesis procedure, small impurities in the product are observed, which stem from hydrolysis of the tert-butyl ester. It should be noted that such hydrolysis might be an analysis artefact, but even if present, does not pose a conceptual problem since the herein used esters are only placeholders and may be replaced virtually by any other functional ester. Generally, separation of products becomes more tedious when more monomers are added to the oligomer chain. Size exclusion chromatography separates according to hydrodynamic volume of species, which does not linearly increase with chain length. Thus, after each monomer addition, more recycling cycles are required to achieve separation, which is, however, limited by band broadening effects and thus reducing the overall yields after each addition. The present case represents chains with six following chain units and a molar mass of 914 Da (considering the R and Z groups of the RAFT agent separately, since these may also be functionalized and/or replaced in click-reactions). Regarding yields and efficiency (yields of 10–15% are reached in the last step as compared to 55% in the first addition step), access to longer oligomers is still somewhat limited and more chain additions might be desirable. It should, however, be stressed that the present method does not rely on solid support systems and hence 6 consecutive functionalities represent already a very efficient process.
While SEC and MS analyses unambiguously confirm the monodisperse growth of the chains, the exact chain structure (and thus the sequence) remains unknown. When using acrylates the possible formation of so-called mid chain radicals via backbiting—a process well-known for acrylate polymerization—must be considered.22 This process is likely to happen as soon as three consecutive monomers are inserted and hence might influence the structure of the product from the forth addition on. Backbiting occurs via 6-membered ring transition states, in which the secondary radical at the end of the chain can migrate from the chain end to the position of the first inserted monomer, thereby forming a more stable tertiary radical (see ESI† for structures). The consequence would be a branched product, destroying the chain sequence information.
Since both the linear as well as the branched structure of the oligo-RAFT agents 4 or 7 would exhibit exactly the same mass-to-charge signal in ESI-MS, other methods need to be applied to distinguish product purity. Therefore, the purified oligo-RAFT agents 4 and 7 were studied using Attached Proton Test 13C-NMR (APT) (ESI†). No resonances typical for branched structures23 are present in the spectrum and all peaks can be neatly assigned to the linear structure of either macro-RAFT agent 4 or 7. It can thus safely be assumed that backbiting did not occur during the RAFT insertion reactions.
In summary, an effective way of synthesizing sequence controlled acrylate oligo-RAFT agents was established. Two different sets of sequence controlled polymer structures could be synthesized in which the order of monomers was chosen as preferred. Purification of the obtained reaction mixtures could be executed effectively via automated recycling SEC giving access to these materials in a non-tedious procedure. SEC, ESI MS and APT 13C NMR have proven that the products are of high structural integrity. The demonstrated ability to use the RAFT mechanism to deliberately control the insertion and order of monomers in a polymer chain is truly a step forward towards the development of sequence controlled materials in future. Further developments will focus on the optimization to reach higher chain lengths and to prepare materials with defined functionality. Even though only oligomers are accessible to this point, high prospects can be associated with these materials. Embedding such oligomers into other materials will allow for development of precise molecular recognition materials ranging from self-folding biomimic materials towards new materials for biosensing.
The authors are grateful for the award of the Odysseus project “Precise Polymer Design for the Development of New Materials” by the Fund for Scientific Research-Flanders. (FWO). T.J. acknowledges the European Science Foundation – Precision Polymer Materials (P2M) program.
Notes and references
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93 CrossRef CAS PubMed.
- K. Matyjaszewski, Prog. Polym. Sci., 2005, 30, 858 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- O. W. Webster, Science, 1991, 251, 887 CrossRef CAS PubMed.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369 RSC.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, W. H. Freeman, New York, 6th edn, 2006 Search PubMed.
- (a) J.-F. Lutz, Polym. Chem., 2010, 1, 55 RSC; (b) M. Ouchi, N. Badi, J.-F. Lutz and M. Sawamoto, Nat. Chem., 2011, 3, 917 CrossRef CAS PubMed; (c) S. Ida, T. Terashima, M. Ouchi and M. Sawamoto, J. Am. Chem. Soc., 2009, 131, 10808 CrossRef CAS PubMed; (d) J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 628 CrossRef CAS PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149 CrossRef CAS.
- M. Minoda, M. Sawamoto and T. Higashimura, Macromolecules, 1990, 23, 4889 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS PubMed.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 301, 3661 CrossRef PubMed.
- S. Pfeifer and J.-F. Lutz, J. Am. Chem. Soc., 2007, 129, 9542 CrossRef CAS PubMed.
- R. Kakuchi, M. Zamfir, J.-F. Lutz and P. Theato, Macromol. Rapid. Commun., 2012, 33, 54 CrossRef CAS PubMed.
- M. Zamfir and J.-F. Lutz, Nat. Commun., 2012, 3, 1138 CrossRef.
- K. Satoh, S. Ozawa, M. Mizutani, K. Nagai and M. Kamigaito, Nat. Commun., 2010, 1, 6 CrossRef PubMed.
- S. Houshyar, D. J. Keddie, G. Moad, R. J. Mulder, S. Saubern and J. Tsanaktsidis, Polym. Chem., 2012, 3, 1879 RSC.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- L. Zeng, L. Burton, K. Yung, B. Shushan and D. B. Kassel, J. Chromatogr., A., 1998, 794, 3 CrossRef CAS.
- J. Vandenbergh, T. M. Ogawa and T. Junkers, J. Polym. Sci., Polym. Chem. Ed., 2013, 51, 2366–2374 CrossRef CAS.
- E. T. A. van den Dungen, H. Matahwa, J. B. McLeary, R. D. Sanderson and B. Klumperman, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 2500 CrossRef CAS.
- G. Moad, Y. K. Chong, R. Mulder, E. Rizzardo and S. H. Thang, ACS Symp. Ser., 2009, 1024, 3 CrossRef CAS PubMed.
- T. Junkers and C. Barner-Kowollik, J. Polym. Sci., Polym. Chem. Ed., 2008, 46, 7585 CrossRef CAS.
- L. J. Mathias, Solid State NMR of Polymers, Plenum Press, New York, 1991, p. 134 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental section and characterization of products. See DOI: 10.1039/c3cc45994b |
| This journal is © The Royal Society of Chemistry 2013 |