 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient asymmetric synthesis of lamivudine via enzymatic dynamic kinetic resolution†
Lei
Hu
,
Fredrik
Schaufelberger
,
Yan
Zhang
and
Olof
Ramström
*
Royal Institute of Technology, Department of Chemistry, Teknikringen 30, Stockholm, Sweden. E-mail: ramstrom@kth.se; Fax: +46 8 7912333
First published on 10th September 2013
Abstract
The anti-HIV nucleoside lamivudine was asymmetrically synthesized in only three steps via a novel surfactant-treated subtilisin Carlsberg-catalyzed dynamic kinetic resolution protocol. The enantiomer of lamivudine could also be accessed using the same protocol catalyzed by Candida antarctica lipase B.
In the last decade, lamivudine (3TC, 1a) (Fig. 1) has proven to be one of the most successful agents for the treatment of HIV as well as chronic Hepatitis B.1 The compound inhibits both type 1 and type 2 of the human HIV reverse transcriptase and also the reverse transcriptase of hepatitis B in vitro.2–6 As a permanent cure for HIV has remained elusive to date, efficient access to bulk quantities of lamivudine is synthetically very valuable as continuous demand is expected. There are several reported methods to synthesize isomerically pure lamivudine. Most of the previous reports introduced the chiral 1,3-oxathiolane motif by either crystallizing the correct isomer from a racemic mixture,7,8 or by enzymatic hydrolysis/acetylation of the other stereoisomers.9–11 For example, Liotta and Koszalka developed an efficient six-step pathway to racemic nucleosides and utilized a late-stage enzymatic kinetic resolution with a series of lipases to esterify the undesired enantiomer. Lamivudine was then obtained in good enantiomeric excess but moderate yield.10 The major disadvantage of these methods is the great loss of yield due to the nature of the kinetic resolution process, as no more than 50% yield could be theoretically achieved while maintaining high enantiomeric purity. In 2005, Goodyear et al. employed a crystallization-induced dynamic kinetic resolution (DKR) method for the lamivudine synthesis,12 however requiring not only seven steps of synthesis, and undesirable reagents such as SOCl2, but also the use of chiral auxiliary-derived starting materials.
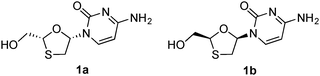 | ||
| Fig. 1 Lamivudine (1a) and its enantiomer (1b). | ||
Compared with other commonly used catalysts for asymmetric synthesis, such as chiral transition-metal complexes and organocatalysts, biocatalysis is now becoming a highly potent alternative in both academia and the pharmaceutical industry.13–16 This is in part due to the high stereoselectivities that can be obtained, the potential to modify/optimize the performances through directed evolution protocols, the inherent environmentally benign (green) nature of the catalysts, and the ease of recycling processes.17–25 In this study, we report a highly efficient three-step asymmetric synthesis of lamivudine and its enantiomer through a novel enzyme-catalyzed DKR protocol based on reversible formation of the intermediate stereoisomers. The key enantioenriched 1,3-oxathiolane structure was obtained in good yield and enantiopurity from two achiral starting materials through an enzyme-catalyzed cascade addition–cyclization–acetylation reaction, leading directly to a suitable substrate for the subsequent Vorbrüggen coupling reaction that is most frequently used for nucleoside synthesis. The advantage of this strategy is obvious, as the formation of the hemiacetal and its transformation into a better leaving group for the subsequent coupling are performed in one pot. In addition, by using different types of enzymes, the stereochemical configuration of the oxathiolane intermediate could be easily controlled, dynamically yielding the precursor of lamivudine or its enantiomer (Fig. 1), respectively. This sets up a useful example for the construction of highly enantioenriched oxathiolane-based nucleosides with access to both enantiomers in a short synthetic route. The method represents an improvement to the previous lamivudine syntheses with respect to both efficiency and environmental friendliness.
Taking advantage of the dynamic formation of hemithioacetals,26,27 we recently reported that Candida antarctica lipase B (CAL B) catalyzes the cyclization of the intermediate generated from the reversible nucleophilic addition of sulfanylacetate 2 to aldehyde 3, asymmetrically forming 1,3-oxathiolan-5-one derivative 4 (Scheme 1).19 Since the core structures of compounds 1 and 4 are very similar, the possibility of applying the same strategy to the enzyme-catalyzed asymmetric synthesis of lamivudine appeared to be natural.
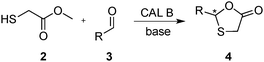 | ||
| Scheme 1 CAL B-catalyzed synthesis of 1,3-oxathiolan-5-ones. | ||
Initially, compounds 5 and 6 were mixed with triethylamine (TEA) as the base and phenyl acetate as the acyl donor in the presence of CAL B in toluene (Scheme 2). The intention was that compounds 5 and 6 would react to form compound 7b (2R) (Scheme 3) in a one-pot cyclization–diol acetylation reaction. According to the 1H NMR and chiral HPLC analyses, an enantioenriched intermediate was obtained in 88% yield, with 6.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, and 83% ee after two days. To elucidate the absolute configuration of precursor 7, the asymmetric nucleoside was synthesized through a modified Vorbrüggen coupling and deacylation procedure.28–30 The overall yield for this three-step asymmetric synthesis was 37%. Chiral HPLC was then used to identify the selectivity of CAL B by analyzing samples from both the CAL B-catalyzed nucleoside synthesis and commercial lamivudine. The result showed that the amplified isomer was compound 1b; therefore it could be deduced that CAL B selectively enriched the undesired intermediate stereoisomer 7a (2S). Nine different lipases were subsequently screened: the lipases from Candida rugosa, Rhizopus niveus, Rhizopus arrhizus, porcine pancreas, Penicillium camemberti, Aspergillus niger, Pseudomonas fluorescens, Burkholderia (Pseudomonas) cepacia, and Candida antarctica lipase A. Of these, the lipases from Burkholderia cepacia and Pseudomonas fluorescens were also able to catalyze the formation of the cyclized intermediate, showing similar stereochemical preferences to CAL B.
:1 dr, and 83% ee after two days. To elucidate the absolute configuration of precursor 7, the asymmetric nucleoside was synthesized through a modified Vorbrüggen coupling and deacylation procedure.28–30 The overall yield for this three-step asymmetric synthesis was 37%. Chiral HPLC was then used to identify the selectivity of CAL B by analyzing samples from both the CAL B-catalyzed nucleoside synthesis and commercial lamivudine. The result showed that the amplified isomer was compound 1b; therefore it could be deduced that CAL B selectively enriched the undesired intermediate stereoisomer 7a (2S). Nine different lipases were subsequently screened: the lipases from Candida rugosa, Rhizopus niveus, Rhizopus arrhizus, porcine pancreas, Penicillium camemberti, Aspergillus niger, Pseudomonas fluorescens, Burkholderia (Pseudomonas) cepacia, and Candida antarctica lipase A. Of these, the lipases from Burkholderia cepacia and Pseudomonas fluorescens were also able to catalyze the formation of the cyclized intermediate, showing similar stereochemical preferences to CAL B.
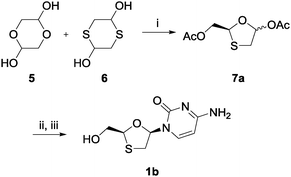 | ||
| Scheme 2 Synthesis of ent-lamivudine (1b) using CAL B; (i) phenyl acetate, CAL B, TEA, toluene, rt, 92%; (ii) silylated N4-acetylcytosine, TMSI, MeCN, 0 °C; (iii) K2CO3, MeOH, rt, 40% for two steps. | ||
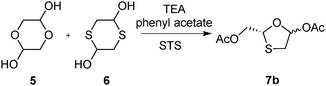 | ||
| Scheme 3 Synthesis of intermediate 7b with STS; rt, 87%. | ||
In order to obtain the correct stereoisomer, an enzyme with different stereoselectivity was required. The protease subtilisin Carlsberg is known to have the opposite selectivity for acyclic secondary alcohols compared to the selectivities of many lipases.31–34 The commercially available subtilisin Carlsberg was thus treated with octyl β-D-glucopyranoside and Brij 56 to enhance its activity and stability in organic solvents.35 The surfactant-treated subtilisin Carlsberg (STS) was applied to the same reaction conditions as in Scheme 2. Upon comparing the intermediate with compound 7a, it is clear that STS selected enantiomer 7b (Scheme 3), which would lead to the asymmetric synthesis of lamivudine. However, unsatisfactory enantiomeric purities (up to 64% ee) were obtained, despite extensive screening of the reaction conditions.
In order to obtain higher ees from the enzymatic DKR reaction, benzoyl protected aldehyde 8 was chosen instead of glycolaldehyde dimer 5 to react with 1,4-dithiane-2,5-diol 6 in the presence of STS (Scheme 4), assuming that the larger structure would fit the enzyme active site more rigidly. This substrate was subjected to similar reaction conditions as depicted in Scheme 2 for two days. The isolated intermediate 9a was obtained with an enantiomeric purity of 45% ee, which was promising for the further optimization. After Vorbrüggen coupling and deprotection, the final product (40% overall yield for three steps) was again compared with the standard sample using chiral HPLC, and the result showed that lamivudine (1a) was still amplified over its enantiomer under these conditions.
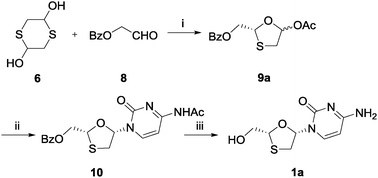 | ||
| Scheme 4 Synthesis of lamivudine (1a) using STS: (i) phenyl acetate, STS, TEA, THF, 4 °C, 89%; (ii) silylated N4-acetylcytosine, TMSI, MeCN, 0 °C, 51%; (iii) K2CO3, MeOH, rt, 89%. | ||
With the correct isomer in hand, several parameters were screened to improve the STS-mediated DKR protocol (Table 1). TBME and THF were tested as enzyme-tolerable solvents besides toluene according to the previous studies.17,19–21,23,27 The results in TBME were very similar to those obtained in toluene, whereas increased dr and ee from 2:1 and 45% to 4.3:1 and 65%, respectively, were obtained in THF, indicating that STS favors more polar solvents. Varying the amount of base and enzyme loading did not significantly affect the yield and selectivity. The performances of STS at different temperatures were also addressed. The highest yield was obtained at 40 °C but with very low stereoselectivity. Both the dr and ee could however be well improved by lowering the temperature from 25 °C to 4 °C without significant loss of efficiency. Lowering the temperature further gave even higher selectivities, although resulting in lower STS activity, where the highest enantiomeric purity (85% ee) could be recorded at –18 °C.
| Entry | Solvent | T [°C] | STS [mg] | drb | eec [%] | Yieldd [%] |
|---|---|---|---|---|---|---|
| a Reaction conditions: 0.1 mmol 8, 0.06 mmol 6, 0.1 mmol TEA, 0.3 mmol phenyl acetate; 2 d. b Estimated by 1H NMR spectroscopy from the isolated racemic mixture. c Analyzed by chiral HPLC (Chiralpak OJ column, λ = 254 nm) using 10% of 2-propanol in hexane. d Isolated yield. | ||||||
| 1 | Toluene | 25 | 20 | 2.0:1 |
45 | 74 |
| 2 | TBME | 25 | 20 | 2.5:1 |
47 | 69 |
| 3 | THF | 25 | 20 | 4.3:1 |
67 | 92 |
| 4 | THF | 25 | 10 | 4.9:1 |
67 | 81 |
| 5 | THF | 25 | 30 | 4.5:1 |
68 | 92 |
| 6 | THF | 4 | 20 | 4.3:1 |
82 | 89 |
| 7 | THF | –18 | 20 | 4.3:1 |
85 | 23 |
To verify the selectivity pattern of CAL B for the same substrates, the reaction was carried out under the optimized conditions for two days. Chiral HPLC analysis revealed that CAL B selectively formed isomer 9b (84% ee) (Scheme 5), the enantiomer of the STS-catalyzed intermediate 9a, indicating the retained selectivity of CAL B in spite of different starting materials.
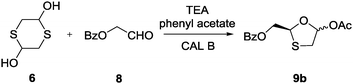 | ||
| Scheme 5 Synthesis of intermediate 9b with CAL B; rt, 83%. | ||
In summary, we have developed a three-step asymmetric synthesis of lamivudine using surfactant-treated subtilisin Carlsberg as a green catalyst. This strategy represents a first entry to efficient high-enantiopurity nucleoside analog synthesis, whereby application of enzyme optimization techniques, such as rational redesign or directed evolution protocols, could lead to enhanced selectivities. We have also described that the stereochemistry of the target molecules could be well controlled, as different isomers of the nucleoside intermediates were selectively obtained using different enzymes. This study thus offers a valuable methodology for the asymmetric synthesis of lamivudine as well as other nucleosides such as emtricitabine and apricitabine, where specific stereoisomers are desired.
This work was in part supported by the Swedish Research Council and the Royal Institute of Technology. LH and YZ thank the China Scholarship Council for special scholarship awards.
Notes and references
- G. Romeo, U. Chiacchio, A. Corsaro and P. Merino, Chem. Rev., 2010, 110, 3337 CrossRef CAS PubMed.
- J. W. Beach, L. S. Jeong, A. J. Alves, D. Pohl, H. O. Kim, C. N. Chang, S. L. Doong, R. F. Schinazi, Y. C. Cheng and C. K. Chu, J. Org. Chem., 1992, 57, 2217 CrossRef CAS.
- C. N. Chang, S. L. Doong, J. H. Zhou, J. W. Beach, L. S. Jeong, C. K. Chu, C. H. Tsai, Y. C. Cheng, D. Liotta and R. Schinazi, J. Biol. Chem., 1992, 267, 13938 CAS.
- J. A. Coates, N. Cammack, H. J. Jenkinson, A. J. Jowett, M. I. Jowett, B. A. Pearson, C. R. Penn, P. L. Rouse, K. C. Viner and J. M. Cameron, Antimicrob. Agents Chemother., 1992, 36, 733 CrossRef CAS.
- L. S. Jeong, R. F. Schinazi, J. W. Beach, H. O. Kim, S. Nampalli, K. Shanmuganathan, A. J. Alves, A. McMillan, C. K. Chu and R. Mathis, J. Med. Chem., 1993, 36, 181 CrossRef CAS.
- S. L. Doong, C. H. Tsai, R. F. Schinazi, D. C. Liotta and Y. C. Cheng, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 8495 CrossRef CAS.
- J.-Z. Li, L.-X. Gao and M.-X. Ding, Synth. Commun., 2002, 32, 2355 CrossRef CAS PubMed.
- B. N. Roy, G. P. Singh, D. Srivastava, H. S. Jadhav, M. B. Saini and U. P. Aher, Org. Process Res. Dev., 2009, 13, 450 CrossRef CAS.
- J. Milton, S. Brand, M. F. Jones and C. M. Rayner, Tetrahedron Lett., 1995, 36, 6961 CAS.
- L. K. Hoong, L. E. Strange, D. C. Liotta, G. W. Koszalka, C. L. Burns and R. F. Schinazi, J. Org. Chem., 1992, 57, 5563 CrossRef CAS.
- R. P. C. Cousins, M. Mahmoudian and P. M. Youds, Tetrahedron: Asymmetry, 1995, 6, 393 CrossRef CAS.
- M. D. Goodyear, M. L. Hill, J. P. West and A. J. Whitehead, Tetrahedron Lett., 2005, 46, 8535 CrossRef CAS PubMed.
- M. T. Reetz, in Adv. Catal., ed. C. G. Bruce and K. z. Helmut, Academic Press, 2006, vol. 49, p. 1 Search PubMed.
- M. Ikunaka, Chem.–Eur. J., 2003, 9, 378 CrossRef CAS PubMed.
- M. T. Reetz, Angew. Chem., Int. Ed., 2002, 41, 1335 CrossRef CAS.
- E. M. Anderson, K. M. Larsson and O. Kirk, Biocatal. Biotransform., 1998, 16, 181 CrossRef CAS.
- P. Vongvilai, R. Larsson and O. Ramström, Adv. Synth. Catal., 2008, 350, 448 CrossRef CAS.
- H. E. Schoemaker, D. Mink and M. G. Wubbolts, Science, 2003, 299, 1694 CrossRef CAS PubMed.
- M. Sakulsombat, Y. Zhang and O. Ramström, Chem.–Eur. J., 2012, 18, 6129 CrossRef CAS PubMed.
- P. Vongvilai and O. Ramström, J. Am. Chem. Soc., 2009, 131, 14419 CrossRef CAS PubMed.
- P. Vongvilai, M. Angelin, R. Larsson and O. Ramström, Angew. Chem., Int. Ed., 2007, 46, 948 CrossRef CAS PubMed.
- R. Larsson and O. Ramström, Eur. J. Org. Chem., 2006, 285 CrossRef CAS.
- M. Sakulsombat, P. Vongvilai and O. Ramström, Org. Biomol. Chem., 2011, 9, 1112 CAS.
- M. Breuer, K. Ditrich, T. Habicher, B. Hauer, M. Keßeler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788 CrossRef CAS PubMed.
- A. Houde, A. Kademi and D. Leblanc, Appl. Biochem. Biotechnol., 2004, 118, 155 CrossRef CAS.
- R. Caraballo, H. Dong, J. P. Ribeiro, J. Jiménez-Barbero and O. Ramström, Angew. Chem., Int. Ed., 2010, 49, 589 CrossRef CAS PubMed.
- M. Sakulsombat, M. Angelin and O. Ramström, Tetrahedron Lett., 2010, 51, 75 CrossRef CAS PubMed.
- H. Jin, M. A. Siddiqui, C. A. Evans, H. L. A. Tse, T. S. Mansour, M. D. Goodyear, P. Ravenscroft and C. D. Beels, J. Org. Chem., 1995, 60, 2621 CrossRef CAS.
- W. B. Choi, L. J. Wilson, S. Yeola, D. C. Liotta and R. F. Schinazi, J. Am. Chem. Soc., 1991, 113, 9377 CrossRef CAS.
- H. Vorbrüggen and C. Ruh-Pohlenz, Organic Reactions, John Wiley & Sons, Inc., 2004 Search PubMed.
- R. J. Kazlauskas, A. N. E. Weissfloch, A. T. Rappaport and L. A. Cuccia, J. Org. Chem., 1991, 56, 2656 CrossRef CAS.
- M.-J. Kim, Y. I. Chung, Y. K. Choi, H. K. Lee, D. Kim and J. Park, J. Am. Chem. Soc., 2003, 125, 11494 CrossRef CAS PubMed.
- L. Borén, B. Martín-Matute, Y. Xu, A. Córdova and J.-E. Bäckvall, Chem.–Eur. J., 2006, 12, 225 CrossRef PubMed.
- R. J. Kazlauskas and A. N. E. Weissfloch, J. Mol. Catal. B: Enzym., 1997, 3, 65 CrossRef CAS.
- L. K. Thalén, A. Sumic, K. Bogár, J. Norinder, A. K. Ö. Persson and J.-E. Bäckvall, J. Org. Chem., 2010, 75, 6842 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cc45551c |
| This journal is © The Royal Society of Chemistry 2013 |