 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in stereoselective bromofunctionalization of alkenes using N-bromoamide reagents
Chong Kiat
Tan
and
Ying-Yeung
Yeung
*
Department of Chemistry, NUS, 3 Science Drive 3, 11754, Singapore. E-mail: chmyyy@nus.edu.sg; Fax: +65-6779-1691; Tel: +65-6516-7760
First published on 3rd July 2013
Abstract
Bromination reactions have been made a lot more convenient since the invention of N-bromoamide reagents. These reagents are more easily handled when compared to molecular bromine. In comparison to other halogens, brominating reagents sit in between chlorine and iodine on the reactivity scale, giving them an advantage in some cases. Recently, several important advances in enantioselective bromofunctionalization of alkenes using such reagents have been reported. This article will highlight the challenges and methods to surmount these problems. In addition, this article will also show the use of N-bromoamide reagents in expanding the scope of diastereoselective bromofunctionalization of alkenes. Examples include bromine initiated cyclic ether cascades and novel multicomponent reactions (MCRs).
 Chong Kiat Tan | Chong Kiat Tan is a final year PhD candidate at the Department of Chemistry at the National University of Singapore. He obtained his BSc (Hons) from the same university. His research interests include organic synthesis and the development of asymmetric organocatalysis. |
 Ying-Yeung Yeung | Ying Yeung Yeung received his BSc (2001) at The Chinese University of Hong Kong. He continued his graduate research in the same university under the supervision of Prof. Tony K. M. Shing. After four years research dedicated to natural product synthesis, Dr Yeung moved to USA to conduct postdoctoral research with Prof. E. J. Corey at Harvard University. In 2008, he joined National University of Singapore, Department of Chemistry, as an assistant professor. His research interests include asymmetric catalysis, green oxidation, and methodology development. |
1. Introduction
Bromination reactions have been a workhorse in organic synthesis. However the use of bromine as a reagent itself is not entirely convenient due to its high reactivity, volatility and toxicity. The invention of N-bromoamide reagents like N-bromosuccinimide (NBS)1 (Fig. 1) for example has greatly enhanced the utility of bromination reactions due to its safe and easy handling. In addition to NBS, a number of commercially available N-bromoamide reagents exist including N-bromophthalimide (NBP), N-bromoacetamide (NBA) and the more reactive 1,3-dibromo-5,5-dimethydantoin (DBDMH). In terms of halogen reactivity, bromine is more reactive than chlorine but less reactive than iodine towards electrophilic halogenation of alkenes.2 In terms of the leaving group ability of the halogens in the products, alkyl bromides offer higher reactivity towards SN2 displacement than alkyl chlorides. In addition, alkyl bromides are also more stable towards unwanted nucleophilic displacement when compared to alkyl iodides.3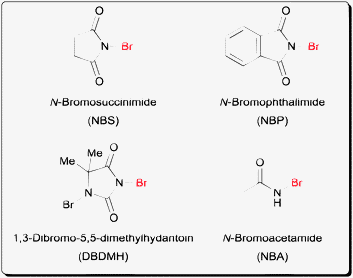 | ||
| Fig. 1 Examples of N-bromoamide reagents. | ||
Reactions ranging from the radical bromination of allylic4 and benzylic5 positions to electrophilic bromination of carbonyl compounds,6 aromatic7 and heterocyclic8 compounds are just some examples of the bromination reactions that are possible with such reagents. However, this feature article will only review selected examples from the recent advances of the electrophilic bromofunctionalization of alkenes starting from 2005.
The generally accepted mechanism of such reactions begins with the formation of the bromonium ion once the electrophilic bromine source comes into contact with the alkene. Such bromonium ions were observed in liquid SO2 in a series of NMR studies performed by Olah et al.9
Subsequently, this reactive bromonium ion can be further captured by a compatible nucleophile (Scheme 1). Since products that contain an anti-relationship between the bromide and nucleophile are generally obtained, the nucleophile is believed to open the bromonium ring in an SN2 fashion.10
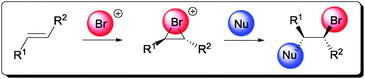 | ||
| Scheme 1 General mechanism of the electrophilic bromofunctionalization of alkene. | ||
Bromofunctionalization of alkenes generally affords products of high diastereoselectivity, as suggested by the proposed mechanism in Scheme 1.
In recent years a renewed focus has been cast on the long standing challenge of enantioselective bromofunctionalization of alkenes. More broadly, the solving of this long standing problem should be addressed under the umbrella of enantioselective halofunctionalization of alkenes. Several reviews on this topic are already in the literature.11
This article will only limit its coverage to the electrophilic bromofunctionalization of alkenes using N-bromoamide reagents. The article consists of two parts: part one will review the recent progress in the catalytic enantioselective bromofunctionalization of alkenes using N-bromoamide reagents, and the second part will address the other significant progress in diastereoselective bromofunctionalization of alkenes. Efforts from our own laboratory in these two fields will be described as well.
2. Part I – recent progress in enantioselective bromofunctionalization of alkenes
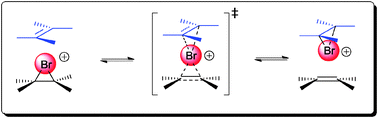
The enantioselective bromofunctionalization or more broadly halofunctionalization of alkenes has been an area of intensive research in recent years. Many of the difficulties in achieving bromofunctionalization with a useful level of enantioselectivity have been worked out mechanistically by Brown et al. as well as Denmark et al.Brown and co-workers were the first to demonstrate that a rapid and degenerative olefin-to-olefin transfer exists between the bromonium ion and free alkenes in NMR studies (Scheme 2).12 The rate of this olefin-to-olefin transfer process was also found to be competitive with the nucleophilic capture of the bromonium ion. With the existence of such a rapid bromonium transfer mechanism, one can rationalize the possibility of racemization of an enantioenriched bromonium ion. This possible racemization mechanism thus makes the development of the enantioselective bromofunctionalization of alkene a challenging task.
The next important mechanistic rationalization of the problem of enantioselective bromofunctionalization was demonstrated in a series of acetolysis experiments conducted by Denmark and co-workers.13 The experiments simulate the environment where the configurational stability of both bromonium and chloronium ions can be determined in the presence of both a nucleophile and excess olefin. The β-halo sulfonates 1a and 1b can generate the respective bromonium and chloronium ions in strongly ionizing media. The rates of racemization by excess (E)-4-octene (via olefin-to-olefin transfer, Scheme 2) can thus be compared to the rate of intermolecular nucleophilic capture of the halonium ion.
They discovered that the addition of 4-octene into a solution of bromotosylate 1a resulted in the erosion of enantiospecificity in the highly polar hexafluoroisopropyl alcohol (HFIP) solvent medium (Scheme 3). The main factors affecting the erosion of enantiospecificity were identified to be the concentration of the olefin (4-octene) and the counter cation of the acetate. The observation for bromotosylate stands in complete contrast to the analogous case of chlorotriflate 1b, where enantiospecificity was preserved even in the presence of 4-octene. These results established the relative stability of the chloronium ion compared to the bromonium ion.
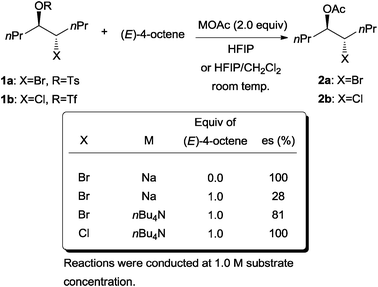 | ||
| Scheme 3 Effect of halogen, counter cation, and concentration of alkene on the erosion of enantiospecificity. | ||
The above two observations suggest the inherent difficulty of forming a stable and chiral bromonium ion due to the presence of a rapid and degenerate olefin-to-olefin transfer mechanism. Nonetheless, some solutions to this problem have been unravelled in recent years and will be described in the following sections. It is noteworthy that breakthroughs in chloro-14 and iodofunctionalization15 of alkenes were also achieved concurrently in recent years.
2.1 Enantioselective intramolecular bromofunctionalization of alkenes
One successful report of the enantioselective bromofunctionalization of alkenes was disclosed by the laboratory of Tang and co-workers.16 Their solution to this problem was the use of the quinine derived tosyl urea 3 as the chiral organocatalyst for the bromolactonization of Z-1,3-enynes (Scheme 4).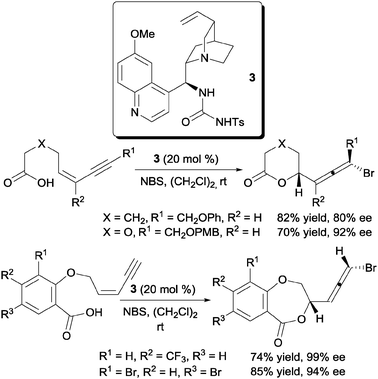 | ||
| Scheme 4 Enantioselective bromolactonization of Z-1,3-enynes. | ||
The design of catalyst 3 was preceded by a previous work from their laboratory which demonstrated the competence of 1,4-diazabicyclo-[2.2.2]octane (DABCO) as a bromolactonization catalyst.17 In this report they proposed a dual activation mode where NBS is activated via a hydrogen bonding with the NH groups of the urea and the carboxylate anion is bound to the quinuclidine of the quinine scaffold. This proposed mechanism was also supported by the change in enantioselectivities of the bromolactonization with various halogen sources especially between NBS and tetrabromocyclohexadienone.16b
Another competent chiral organocatalyst came in the form of the C3-symmetric trisimidazoline 4 reported by Fujioka et al. (Scheme 5).18 Using their protocol, a variety of 5-aryl-substituted hexenoic acids are amendable to enantioselective bromolactonization. Their substrate scope includes 1,1-disubstituted, trisubstituted, and tetrasubstituted hexenoic acids. Mechanistically, they found the presence of the three imidazoline units to be crucial to achieving high enantioselectivity. The use of both the mono- and di-imidazoline variants of catalyst 4 resulted in much lower enantioselectivities. Additionally, NMR studies conducted by the group suggest the probable templating of three units of hexenoic acids to one unit of catalyst 4 as shown in model A (Scheme 5).
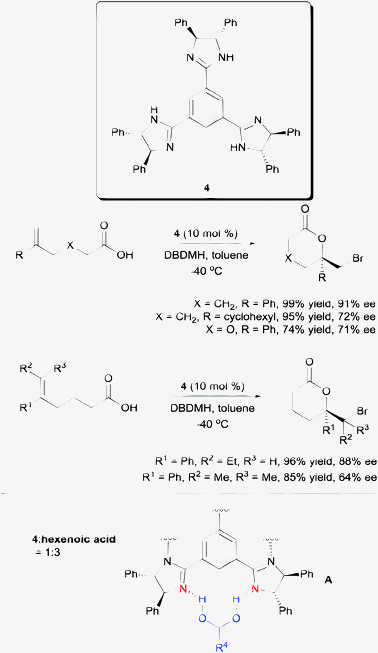 | ||
| Scheme 5 Enantioselective bromolactonization with C3-symmetric trisimidazoline. | ||
Another different solution was offered by our laboratory with a class of amino-thiocarbamate catalysts. The first of this class of amino-thiocarbamate came with a cinchona alkaloid scaffold (Scheme 6). After extensive studies, two catalyst tuning sites were discovered. These are the N-aryl substituent indicated as R1 and the O-alkoxy substituent at the quinoline unit indicated as R2. The tuning of these sites allowed a number of alkenoic acids to be accommodated into this protocol. These include 1,1-disubstituted pentenoic acid,19 both E and Z-pentenoic acid,20,21 and the styrene type carboxylic acid.22
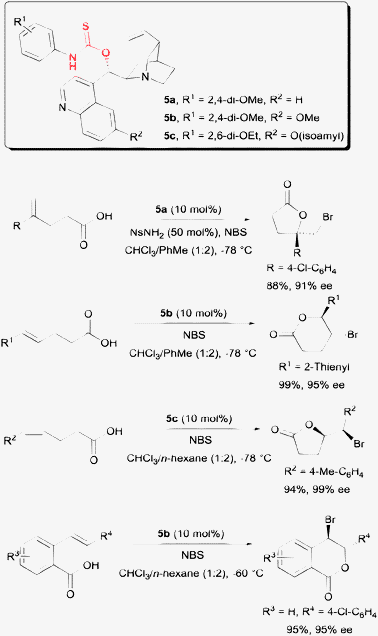 | ||
| Scheme 6 Enantioselective bromolactonization with amino-thiocarbamate catalysts. | ||
While the above example demonstrated the essentiality of using O-alkyl thiocarbamate on a cinchona alkaloid scaffold, subsequent work shown that S-alkyl thiocarbamates are also competent chiral organocatalysts on a proline backbone (Scheme 7).23 In one of our studies, S-alkyl thiocarbamate catalyst 6 was able to serve as a chiral catalyst for the bromolactonization of hexenoic acids. On the other hand, O-alkyl thiocarbamate 7 could catalyze the enantioselective bromolactonization of pentenoic acids. An additional motivation for carrying out the work on a proline scaffold is that both enantiomers of proline are available. In contrast, only pseudo-enantiomeric pairs of cinchona alkaloids are commercially available. The magnitudes of enantioselectivity of some bromolactones are not equal when a pseudo-enantiomeric pair of cinchona alkaloid derived thiocarbamates were used in some specific studies.21,22
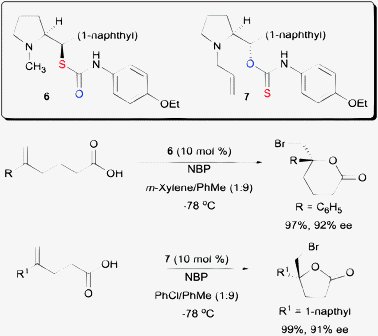 | ||
| Scheme 7 Enantioselective bromolactonization with both O- and S-alkyl amino-thiocarbamates. | ||
The enantioenriched bromolactones can be converted into valuable intermediates. In one example, we were able to perform the debromination of a bromolactone into (R)-(+)-boivinianin A (Scheme 8).24 The enantiomer of (R)-(+)-boivinianin A can readily be accessed by using the enantiomer of the proline based catalyst 7.
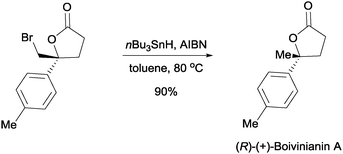 | ||
| Scheme 8 Synthesis of (R)-(+)-boivinianin A from chiral bromolactone. | ||
The amino-thiocarbamate catalysts were also found to be able to perform the enantioselective bromoaminocyclization of a variety of olefinic amides (Scheme 9). In the series of studies, amides 9, 11 and 13 were found to be possible substrates for achieving high enantioselectivities.25 One of the essential requirements for this reaction is the presence of the nosyl substituents. The NH of the 4-nosyl amide is then sufficiently acidic for the reaction to take place smoothly in an enantioselective fashion. The resulting enantioenriched bromo-pyrrolidine and piperidine products are useful building blocks of various pharmaceuticals.
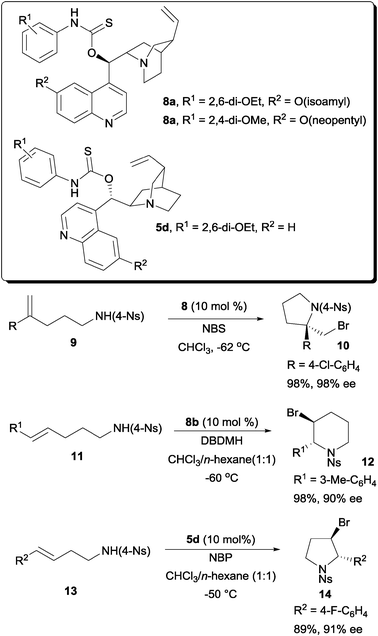 | ||
| Scheme 9 Enantioselective bromoaminocyclization with amino-thiocarbamates. | ||
Mechanistically, the above reactions catalyzed by amino-thiocarbamates were proposed to proceed via a dual activation mode. The sulfur atom in the catalyst is believed to act as a Lewis base activator of bromine,26 while the amine may interact with the nucleophile via an acid–base interaction (Model B, Scheme 10).19
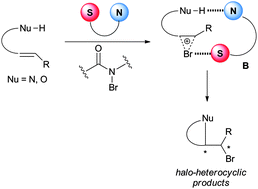 | ||
| Scheme 10 Proposed mechanism of the amino-thiocarbamate catalyzed enantioselective bromocyclization. | ||
Sulfur or more broadly chalcogens have been known to form stable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molecular adducts with bromine.27 Denmark et al. have also demonstrated the competence of sulfur-containing compounds as both bromolactonization and bromoetherification catalysts.28
:1 molecular adducts with bromine.27 Denmark et al. have also demonstrated the competence of sulfur-containing compounds as both bromolactonization and bromoetherification catalysts.28
In addition to the bifunctional amino-thiocarbamate catalyst, we later discovered that dialkyl cyclic selenides are also capable chiral catalyst for the enantioselective bromoaminocyclization of olefinic amides (Scheme 11). The motivation for designing such dialkyl selenides is to overcome the low reactivity of certain olefinic amides towards catalysis by amino-thiocarbamates. While higher reactivity was observed with amino-selenothiocarbamates, however, they were found to be unstable for long term storage as well as during the reaction.29
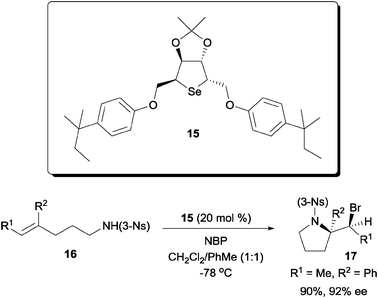 | ||
| Scheme 11 Enantioselective bromoaminocyclization with cyclic dialkyl selenide. | ||
Building up from d-mannitol, cyclic selenide catalyst 15 was capable of effecting the enantioselective cyclization of tri-substituted olefinic amides.30 Interestingly, in contrast to the cinchona alkaloid based amino-thiocarbamates, the 3-nosyl amide was found to be the optimal substituent for the substrate.
The above-mentioned examples show substrates containing both an olefin and a sufficiently acidic nucleophile. Enantioselective bromoetherifications were not reported by groups using the above catalytic systems. Instead, enantioselective bromoetherifications were reported by several groups using chiral phosphoric acid derivatives.
Hennecke and co-workers reported the desymmetrization of 1,8-oct-4-ene-diols with the chiral phosphate catalyst 18 (Scheme 12).31 While modest enantioselectivity was achieved with NBS, much higher enantioselectivity was reported with N-iodo-2-pyrrolidone.
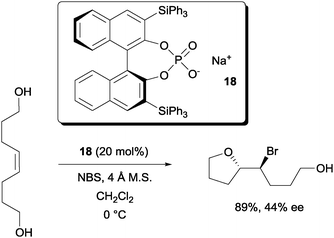 | ||
| Scheme 12 Desymmetrization of 1,8-oct-4-ene diols with a chiral phosphate. | ||
Later, more highly enantioselective bromoetherifications were reported independently by Denmark and Shi. In Denmark's case, the use of a co-catalyst triphenylphosphine sulfide was found to be an essential requirement (Scheme 13).32 A number of substrates including 5-aryl-4-pentenols are applicable to their protocol. In their study, they observed that the Z-pentenols gave an almost exclusive yield of the exo-cyclized bromoethers with high enantioselectivities. On the other hand, the E-configured pentenols gave a mixture of both exo and endo cyclized bromoethers. In terms of enantioselectivity, high ee was detected among the exo cyclized bromoethers but much reduced enantioselectivity was detected for the endo cyclized bromoethers.
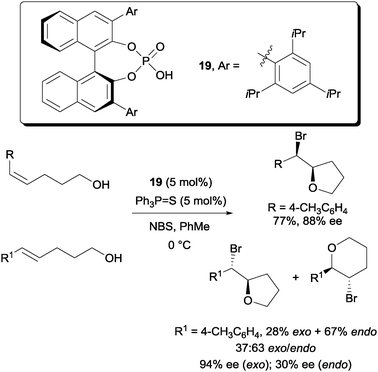 | ||
| Scheme 13 Enantioselective bromoetherification with chiral phosphoric acid and triphenylphosphine sulphide co-catalyst. | ||
Shi and co-workers utilized chiral phosphoric acid 19 as the sole catalyst (Scheme 14).33 5-Alkyl-4-pentenols were also applicable substrates with good enantioselectivity. In addition, they were also able to apply substrates with nitrogen nucleophile to the catalytic protocol.
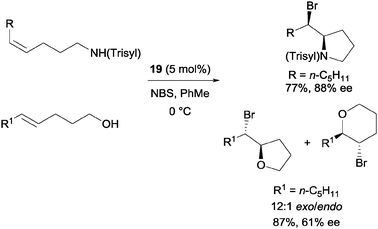 | ||
| Scheme 14 Enantioselective bromoetherification with chiral phosphoric acid catalyst. | ||
In another recent study, Toste et al. reported a highly enantioselective bromocyclization using chiral phosphoric acids together with a specialized DABCO derived tricationic brominating reagent.34
The final example of N-bromoamide induced enantioselective intramolecular bromofunctionalization in this section is the N-bromoacetamide induced semi-pinacol rearrangement of tertiary alcohol 20 to bromoketones 21. Using a combination of cinchona alkaloid based (DHQD)2Pyr and N-Boc-L-phenylglycine (NBLP) catalyst, they were able to afford (2R,3S)-21 with good yield and enantioselectivity (Scheme 15).35 The enantioenriched (2S,3R)-21 was obtainable by switching to (DHQ)2Pyr and N-Boc-D-phenylglycine (NBDP) catalyst.
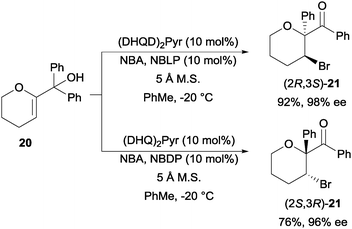 | ||
| Scheme 15 Enantioselective semi-pinacol rearrangement. | ||
2.2 Enantioselective intermolecular bromofunctionalization of alkenes
In comparison to enantioselective intramolecular bromofunctionalization of alkenes, the intermolecular format has proven to be a tougher problem to tackle. In a recent report, a promising enantioselective intermolecular bromoesterification was disclosed. Using chiral phosphoric acid catalyst 22, promising enantioselectivities could be obtained (Scheme 16).36 The yields were however low due to the catalyst acting as a competitive nucleophile in opening the bromonium intermediate.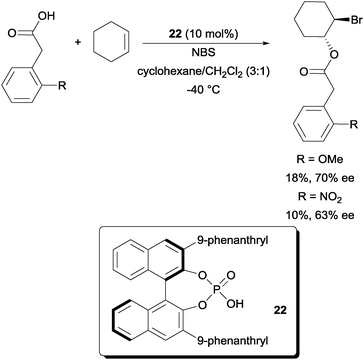 | ||
| Scheme 16 Enantioselective intermolecular bromoesterification with chiral phosphoric acid. | ||
Another organocatalytic approach to intermolecular bromoesterification was recently reported by Tang and co-workers. Using (DHQD)2PHAL with (+)-camphor sulfonic acid (CSA) as an additive, they were able to obtain the highly enantioselective bromoesterification of a range of allylic sulfonamides (Scheme 17).37
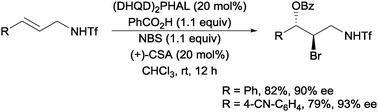 | ||
| Scheme 17 Enantioselective intermolecular bromoesterification with (DHQD)2PHAL. | ||
A highly enantioselective intermolecular bromofunctionalization was reported by Feng et al. with a scandium triflate–N,N′-dioxide metal complex catalyst (Scheme 18).38 Using just 0.05 mol% of the catalyst and ligand system, they were able to achieve the enantioselective bromoamination of chalcone with NBS and TsNH2. High yields and high diastereoselectivities were also achieved. It is also worth noting that successful chloro-39 and iodoamination40 of chalcones were reported by the same group.
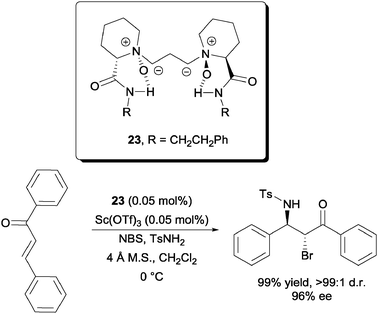 | ||
| Scheme 18 Enantioselective intermolecular bromoamination of chalcone. | ||
3. Part II – recent progress in diastereoselective bromofunctionalization of alkenes
Although it is mentioned in Scheme 1 that the opening of a rigid bromonium intermediate leads to an anti-relationship between the bromine and nucleophile, there has been much room for the development of novel diastereoselective bromofunctionalization of alkenes. In many cases, a Lewis acid or a Lewis base catalyst is needed to activate the brominating reagent towards such reactions. Nevertheless, recently discovered NBS-initiated cascades and multicomponent reactions (MCRs) have been found to proceed without the need for an additional catalyst. The Lewis acid catalyzed and catalyst-free MCRs will be addressed accordingly in this section.3.1 Diastereoselective bromofunctionalization of alkenes with a Lewis acid catalyst
In Corey's concise synthesis of oseltamivir phosphate (Tamiflu),41 they disclosed a novel Lewis acid catalyzed haloamidation of olefins in a Ritter type fashion. It was reported that a catalytic amount of SnCl4 could catalyze the haloamidation process with NBA as the brominating source (Scheme 19).42 In addition, a variety of nitriles and olefins could be subjected to this reaction.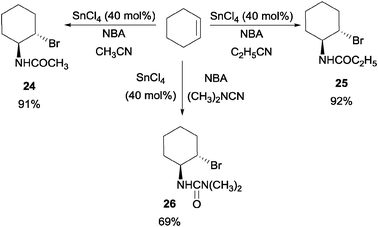 | ||
| Scheme 19 Tin(IV) chloride catalyzed haloamidation of olefin. | ||
The mechanism was proposed to proceed via a bromonium ion ring intermediate which was opened by acetonitrile to generate intermediate C (Scheme 20). Attack by water on the nitrile could result in intermediate D which could isomerize into the bromoamide product.
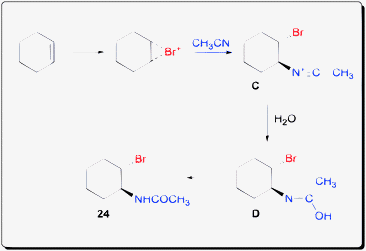 | ||
| Scheme 20 Proposed mechanism of haloamidation of olefin. | ||
Hajra et al. reported a series of metal triflate catalyzed bromofunctionalization of alkenes. In the first of their reports, they demonstrated that with Zn(OTf)2 as the catalyst, they could afford a series of 1,5-disubstituted tetrazoles 27 with an anti-relationship to the bromide (Scheme 21).43 Following the opening of the bromonium intermediate by the azide from TMSN3, assemblage into tetrazole ensued with the acetonitrile solvent. The reaction was found to be applicable to a range of olefins as well as a number of nitrile sources.
 | ||
| Scheme 21 NBS induced synthesis of 1,5-disubstituted tetrazoles from alkenes. | ||
With Yb(OTf)3 as the catalyst, Hajra and co-workers were able to achieve a highly diastereoselective bromoazidation of α,β-unsaturated carboxylic acid derivatives with the aid of chiral auxiliaries such as (2R)-bornane sultam and (2S,5S)-diphenyl pyrrolidine (Scheme 22).44
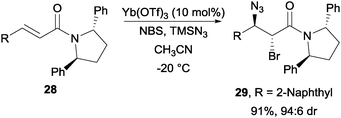 | ||
| Scheme 22 NBS induced bromoazidation of α,β-unsaturated carboxylic acid derivatives. | ||
Another interesting example was achieved via the use of Sm(OTf)3 as the catalyst for a NBS promoted Friedel–Crafts alkylation of alkenes (Scheme 23).45 With electron-rich aromatic rings including toluene, o-xylene, anisole, 1,2-dimethoxy benzene, and 1,2,3-trimethoxy benzene, they were able to achieve this transformation with a number of olefinic partners.
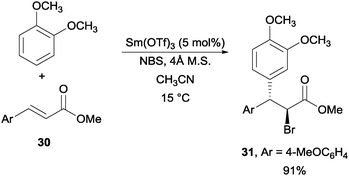 | ||
| Scheme 23 NBS promoted Friedel–Crafts alkylation of alkene. | ||
Lastly, with Cu(OTf)2 or Zn(OTf)2 Hajra and co-workers were able to achieve the stereoselective one-pot synthesis of oxazolines via a bromoamide intermediate (Scheme 24).46 In a process similar to Corey's haloamidation, the bromoamides were able to convert into oxazolines in the presence of NaHCO3 and water.
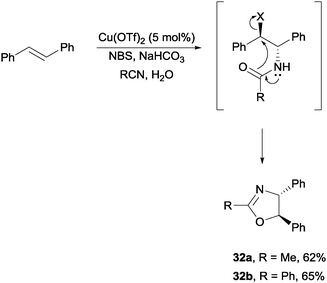 | ||
| Scheme 24 NBS promoted stereoselective one-pot synthesis of oxazolines. | ||
3.2 NBS initiated cyclic ether opening cascades
In Braddock and co-workers' study of model obtusallene systems, they discovered that NBS can be utilized to initiate a transannular oxonium ion formation–fragmentation reaction (Scheme 25).47 When the macrocyclic compound 33 was treated with NBS, the reaction proceeded in the presence of a nucleophile, for example acetic acid. Addition of 1 mol% of tetramethylguanidine (TMG) was found to be necessary. In addition, it was also found that bromine and water can also serve as the nucleophilic partners for this reaction.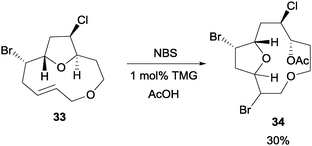 | ||
| Scheme 25 NBS initiated transannular oxonium ion formation–fragmentation. | ||
In a later study, Jamison et al. found that NBS can initiate an epoxide-opening cascade to afford products leading to the total synthesis of ent-dioxepandehydrothyrsiferol (Scheme 26).48 When the poly epoxide 35 with an OBoc terminal group was treated with NBS in a highly polar solvent hexafluoroisopropanol (HFIP), the desired brominated polycycle was afforded in 36% yield. The polycycle 36 was further elaborated to complete the total synthesis.
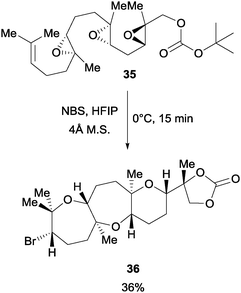 | ||
| Scheme 26 NBS initiated epoxide opening cascade. | ||
On simpler systems, Braddock and co-workers found that unlike Jamison's case48 and other earlier reports,49 the nucleophiles need not be tethered to the epoxide-containing molecules.50 Similar to their earlier study on the transannular oxonium ion formation–fragmentation, they found that treatment of epoxide 37 with NBS and acetic acid with 1 mol% of TMG afforded the isomeric bromoethers 38 and 39 in a 1:1 mixture with a combined yield of 43% (Scheme 27). While the yield of such NBS-initiated epoxide cascades was moderate, Snyder and co-workers have reported higher yielding epoxide cascades with the more reactive Et2SBr·SbBrCl5 (BDSB) reagent.51
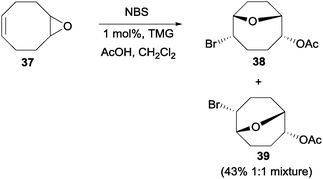 | ||
| Scheme 27 NBS initiated epoxide opening with an external nucleophile. | ||
3.3 Catalyst free NBS initiated MCRs
In contrast to the above-mentioned examples, our laboratory discovered a range of catalyst-free NBS-initiated multicomponent reactions (MCRs) with a variety of olefins. Such kinds of electrophilic MCRs are less reported, partly due to incompatibility of the reagents.52 In our initial conception, we envisaged the feasibility of activating olefins towards a nucleophilic attack by cyclic ethers yielding intermediate E (Scheme 28). The resulting oxonium intermediate F could then be further attacked by an amine source to yield the desired aminoalkoxylation product G.53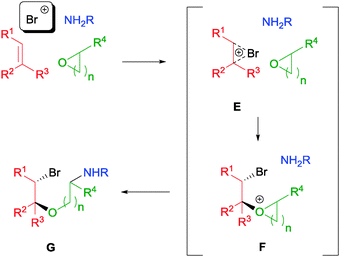 | ||
| Scheme 28 Proposed mechanism of NBS initiated aminoalkoxylation of olefins. | ||
The reaction generally works well with NsNH2 as the nucleophile and a variety of olefins and cyclic ethers were found to be tolerated. The products obtained had good positional- (Markonikov-type) as well as stereo-selectivities. In a demonstration of the usefulness of the reaction, the one-pot assembly of trans-1-phenyl-1-propene, NsNH2, ethylene oxide, and NBS resulted in the formation of the key intermediate 40 (Scheme 29). With further synthetic manipulation, (±)-phenmetrazine and BontrilTM were both obtained with good yields.54
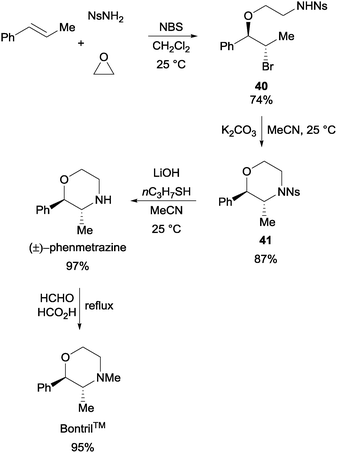 | ||
| Scheme 29 Synthesis of pharmaceutically relevant molecules via the NBS initiated MCR. | ||
In addition to using cyclic ethers, nitriles and cyanimides could be used in place to afford a range of imidazoline55 and guanidine,56 respectively. The MCRs proceed via the one-pot assembly of olefin, NBS, NsNH2, and nitriles or cyanimides to afford the respective bromoamides (Scheme 30). Subsequent intramolecular displacement of the bromides in the bromoamides and cyclization (either in situ or by further refluxing the bromoamides) led to the formation of imidazolines and guanidines.
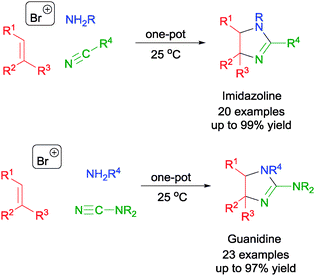 | ||
| Scheme 30 NBS initiated MCR in the synthesis of imidazolines and guanidines. | ||
Oxygen nucleophiles in the form of carboxylic acids were also found to be feasible reaction partners in this type of MCR. In a demonstration, the one-pot reaction of cyclohexene, NBS, acetic acid, and THF was found to yield the alkoxylether product 44 in good yield (Scheme 31).57 Likewise for this reaction, a variety of olefins, cyclic ethers as well as carboxylic acids were found to be applicable to this MCR protocol.
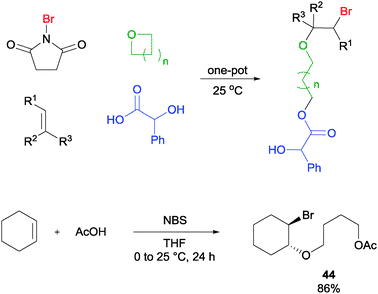 | ||
| Scheme 31 NBS initiated alkoxyetherification of olefin. | ||
A more challenging MCR involving mono-substituted epoxides was also tackled by our group (Scheme 32).58 In this reaction, after the formation of the bromo oxonium intermediate, the subsequent oxonium ring opening can proceed via the anti-Markovnikov (path 1) or the Markovnikov (path 2) pathways. In our investigation, we discovered that propylene oxide gave no regioselectivity (H:I = 1:1) despite the high reaction yield. Other bulky mono-substituted epoxides (e.g. R2 = OTBDPS) resulted in low regioselectivity as well. Nonetheless, it was found that when electron-deficient epoxides like epichlorohydrin and epibromohydrin were used, good regioselectivities were achieved.
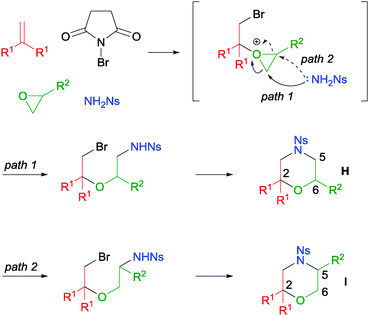 | ||
| Scheme 32 Proposed mechanism of MCR with mono-substituted epoxide. | ||
In a demonstration of the synthetic utility, enantiopure (R)-epichlorohydrin could assemble with olefin 45, NBS, and NsNH2 (Scheme 33). Upon treatment with base, enantiopure 2,2,6-trisubstituted morpholine 46 was obtained with good yield.
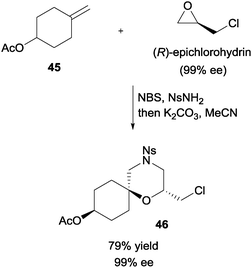 | ||
| Scheme 33 Enantioselective synthesis of 2,2,6-trisubstituted morpholine via NBS initiated MCR. | ||
More recently, it was also discovered that sufficiently acidic phenols could be used as a nucleophile in this MCR to generate a phenoxyetherification product like 47 (Scheme 34).59 Crucially, it was discovered that when relatively electron-rich phenols, including 4-methyl, 4-tert-butyl, and 2,6-dibromo-4-methyl phenols, were subjected to the reaction, no desired product was detected. This result is consistent with our previous observation in which the acidic proton plays a crucial role in the MCR, potentially acting as the NBS activator. Again, the MCR tolerates a range of olefinic partners as well as ether partners for a wider reaction scope. More interestingly, the dibromides on the aromatic ring can undergo a double Sonogashira coupling to generate compound 48, which may serve as ligands to self-assembly spherical complexes that are potentially applicable to the construction of high-density data-storage materials.60
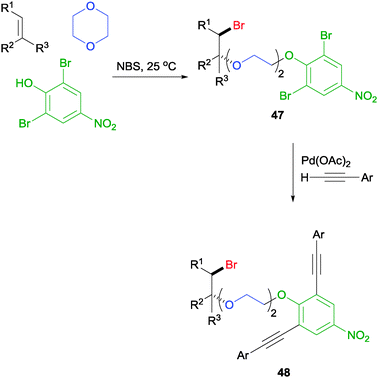 | ||
| Scheme 34 NBS initiated phenoxyetherification MCR and its application. | ||
4. Conclusion
This feature article has charted the recent advances of the use of N-bromoamide in the bromofunctionalization of alkenes. In the course of the article, the recent flurry of reports in the area of enantioselective bromofunctionalization of alkenes was highlighted. In these reports the previously noted difficulties of enantioselective bromofunctionalization of alkenes have been somewhat surmounted. However there is still room for expansion of reaction scope especially for the intermolecular enantioselective bromofunctionalization of alkenes. The arena of diastereoselective bromofunctionalization of alkenes still has much room for creative exploration. Examples of the use of brominating reagents for cyclic ether opening cascades and electrophilic multicomponent reactions are just some of the examples highlighted in this article. We believe other bromine initiated cascades may be explored in the future. Additionally, a clear mechanistic understanding of the described bromofunctionalization reactions has yet to emerge. Further investigation into the origin of the enantioselectivity for the asymmetric bromofunctionalization of alkenes will be especially useful for future development.We thank the financial support from National University of Singapore (Grant No. 143-000-509-112), ASTAR-Public Sector Funding (Grant no. 143-000-536-305), and GSK-EDB. C. K. Tan would like to acknowledge President's Graduate Fellowship for the sponsoring of his PhD scholarship.
Notes and references
- J. S. Pizey, Synthetic Reagents, Wiley, New York, 1974, vol. 2, p. 1 Search PubMed.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry Part A: Structure and Mechanisms, Springer, New York, 5th edn, 2007, pp. 476–482 Search PubMed.
- D. S. Noyce and J. A. Virgilio, J. Org. Chem., 1972, 37, 2643–2647 CrossRef CAS.
- (a) C. Djerassi, Chem. Rev., 1948, 43, 271–317 CrossRef CAS; (b) L. Horner and E. H. Winkelmann, Angew. Chem., 1959, 71, 349–365 CrossRef CAS.
- (a) T. F. Corbin, R. C. Hahn and H. Shecter, Org. Synth., 1973, 5, 328 Search PubMed; (b) A. Kalir, Org. Synth., 1973, 5, 825 Search PubMed.
- (a) M. Yamaura, T. Suzuki, H. Hashimoto, J. Yoshimura and C. Shin, Bull. Chem. Soc. Jpn., 1985, 58, 2812–2920 CrossRef CAS; (b) Z. Lidert and S. Gronowitz, Synthesis, 1980, 322–324 CrossRef CAS.
- R. H. Mitchell, Y.-H. Lai and R. V. Williams, J. Org. Chem., 1979, 44, 4733–4735 CrossRef CAS.
- (a) H. M. Gilow and D. E. Burton, J. Org. Chem., 1981, 46, 2221–2225 CrossRef CAS; (b) S. Martina, V. Enkelmann, G. Wegner and A.-D. Schlüter, Synthesis, 1991, 613–615 CrossRef CAS; (c) R. M. Kellogg, A. P. Schaap, E. T. Harper and H. Wynberg, J. Org. Chem., 1968, 33, 2902–2909 CrossRef CAS; (d) Y. Goldberg and H. Alper, J. Org. Chem., 1993, 58, 3072–3075 CrossRef CAS.
- (a) G. A. Olah and J. M. Bollinger, J. Am. Chem. Soc., 1967, 89, 4744–4752 CrossRef CAS; (b) G. A. Olah and J. M. Bollinger, J. Am. Chem. Soc., 1968, 90, 947–953 CrossRef CAS; (c) G. A. Olah, J. M. Bollinger and J. Brinich, J. Am. Chem. Soc., 1968, 90, 2587–2594 CrossRef CAS.
- F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry Part A: Structure and Mechanisms, Springer, New York, 5th edn, 2007, pp. 473–497 Search PubMed.
- (a) A. Castellanos and S. P. Fletcher, Chem.–Eur. J., 2011, 17, 5766–5776 CrossRef CAS; (b) S. A. Snyder, D. S. Treitler and A. P. Brucks, Aldrichimica Acta, 2011, 44, 27–40 CAS; (c) C. K. Tan, Z. Ling and Y.-Y. Yeung, Synlett, 2011, 1335–1339 CAS; (d) U. Hennecke, Chem.–Asian J., 2012, 7, 456–465 CrossRef CAS; (e) S. E. Denmark, W. E. Kuester and M. T. Burk, Angew. Chem., Int. Ed., 2012, 51, 10938–10953 CrossRef CAS; (f) K. Murai and H. Fujioka, Heterocycles, 2013, 87, 763–805 CrossRef CAS.
- (a) R. S. Brown, R. W. Nagorski, A. J. Bennet, R. E. D. McClung, G. H. M. Aarts, M. Klobukowski, R. McDonald and B. D. Santarsiero, J. Am. Chem. Soc., 1994, 116, 2448–2456 CrossRef CAS; (b) A. A. Neverov and R. S. Brown, J. Org. Chem., 1996, 61, 962–968 CrossRef CAS; (c) R. S. Brown, Acc. Chem. Res., 1997, 30, 131–137 CrossRef CAS.
- S. E. Denmark, M. T. Burk and A. J. Hoover, J. Am. Chem. Soc., 2010, 132, 1232–1233 CrossRef CAS.
- (a) D. C. Whitehead, R. Yousefi, A. Jaganathan and B. Borhan, J. Am. Chem. Soc., 2010, 132, 3298–3300 CrossRef CAS; (b) R. Yousefi, D. C. Whitehead, J. M. Mueller, R. J. Staples and B. Borhan, Org. Lett., 2011, 113, 608–611 CrossRef; (c) A. Jaganathan, A. Garzan, D. C. Whitehead, R. J. Staples and B. Borhan, Angew. Chem., Int. Ed., 2011, 50, 2593–2596 CrossRef CAS; (d) K. C. Nicolaou, N. L. Simmons, Y. Ying, P. M. Heretsch and J. S. Chen, J. Am. Chem. Soc., 2011, 133, 8134–8137 CrossRef CAS.
- (a) G. E. Veitch and E. N. Jacobsen, Angew. Chem., Int. Ed., 2010, 49, 7332–7335 CrossRef CAS; (b) M. C. Dobish and J. N. Johnston, J. Am. Chem. Soc., 2012, 134, 6068–6071 CrossRef CAS; (c) C. S. Brindle, C. S. Yeung and E. N. Jacobsen, Chem. Sci., 2013, 4, 2100–2104 RSC.
- (a) W. Zhang, S. Zheng, N. Liu, J. B. Werness, I. A. Guzei and W. Tang, J. Am. Chem. Soc., 2010, 132, 3664–3665 CrossRef CAS; (b) W. Zhang, N. Liu, C. M. Schienebec, K. Decloux, S. Zheng, J. B. Werness and W. Tang, Chem.–Eur. J., 2012, 18, 7296–7305 CAS.
- W. Zhang, H. Xu, H. Xu and W. Tang, J. Am. Chem. Soc., 2009, 131, 3832–3833 CrossRef CAS.
- (a) K. Murai, T. Matsushita, A. Nakamura, S. Fukushima, M. Shimura and H. Fujioka, Angew. Chem., Int. Ed., 2010, 49, 9174–9177 CrossRef CAS; (b) K. Murai, A. Nakamura, T. Matsushita, M. Shimura and H. Fujioka, Chem.–Eur. J., 2012, 18, 8448–8453 CrossRef CAS.
- L. Zhou, C. K. Tan, X. Jiang, F. Chen and Y.-Y. Yeung, J. Am. Chem. Soc., 2010, 132, 15474–15476 CrossRef CAS.
- C. K. Tan, L. Zhou and Y.-Y. Yeung, Org. Lett., 2011, 13, 2738–2741 CrossRef CAS.
- C. K. Tan, C. Le and Y.-Y. Yeung, Chem. Commun., 2012, 48, 5793–5795 RSC.
- J. Chen, L. Zhou, C. K. Tan and Y.-Y. Yeung, J. Org. Chem., 2012, 77, 999–1009 CrossRef CAS.
- X. Jiang, C. K. Tan, L. Zhou and Y.-Y. Yeung, Angew. Chem., Int. Ed., 2012, 51, 7771–7775 CrossRef CAS.
- (a) D. A. Mulholland, K. McFarland and R. Randrianarivelojoisa, Biochem. Syst. Ecol., 2006, 34, 365–369 CrossRef CAS; (b) I. Čorić, S. Müller and B. List, J. Am. Chem. Soc., 2010, 132, 17370–17373 CrossRef.
- (a) L. Zhou, J. Chen, C. K. Tan and Y.-Y. Yeung, J. Am. Chem. Soc., 2011, 133, 9164–9167 CrossRef CAS; (b) J. Chen, L. Zhou and Y.-Y. Yeung, Org. Biomol. Chem., 2012, 10, 3808–3811 RSC; (c) L. Zhou, D. W. Tay, J. Chen, G. Y. C. Leung and Y.-Y. Yeung, Chem. Commun., 2013, 49, 4412–4414, 10.1039/c2cc36578b.
- (a) V. Gutmann, Coord. Chem. Rev., 1975, 15, 207–237 CrossRef CAS; (b) V. Gutmann, The Donor-Acceptor Approach to Molecular Interactions, Plenum, New York, 1978 Search PubMed; (c) S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560–1638 CrossRef CAS.
- (a) P. D. Boyle and S. M. Godfrey, Coord. Chem. Rev., 2001, 233, 265–299 CrossRef; (b) J. L. Dutton, G. J. Farrar, M. J. Sgro, T. L. Battista and P. J. Ragogna, Chem.–Eur. J., 2009, 15, 10263–10271 CrossRef CAS.
- S. E. Denmark and M. T. Burk, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20655–20660 CrossRef CAS.
- C. K. Tan, F. Chen and Y.-Y. Yeung, Tetrahedron Lett., 2011, 52, 4892–4895 CrossRef CAS.
- F. Chen, C. K. Tan and Y.-Y. Yeung, J. Am. Chem. Soc., 2013, 135, 1232–1235 CrossRef CAS.
- U. Hennecke, C. H. Müller and R. Fröhlich, Org. Lett., 2011, 13, 860–863 CrossRef CAS.
- S. E. Denmark and M. T. Burk, Org. Lett., 2012, 14, 256–259 CrossRef CAS.
- D. Huang, H. Wang, F. Xue, H. Guan, L. Li, X. Peng and Y. Shi, Org. Lett., 2011, 13, 6350–6353 CrossRef CAS.
- Y.-M. Wang, J. Wu, C. Hoong, V. Rauniyar and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 12928–12931 CrossRef CAS.
- Z.-M. Chen, Q.-W. Zhang, Z.-H. Chen, H. Li, Y.-Q. Tu, F.-M. Zhang and J.-M. Tian, J. Am. Chem. Soc., 2011, 133, 8818–8821 CrossRef CAS.
- G.-X. Li, Q.-Q. Fu, X.-M. Zhang, J. Jiang and Z. Tang, Tetrahedron: Asymmetry, 2012, 23, 245–251 CrossRef CAS.
- W. Zhang, N. Liu, C. M. Schienebeck, X. Zhou, I. I. Izhar, I. A. Guzei and W. Tang, Chem. Sci., 2013, 4, 2652–2656, 10.1039/c3sc50446h.
- Y. Cai, X. Liu, Y. Hui, J. Jiang, W. Wang, W. Chen, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2010, 49, 6160–6164 CrossRef CAS.
- Y. Cai, X. Liu, J. Jiang, W. Chen, L. Lin and X. Feng, J. Am. Chem. Soc., 2011, 133, 5636–5639 CrossRef CAS.
- Y. Cai, X. Liu, J. Li, W. Chen, W. Wang, L. Lin and X. Feng, Chem.–Eur. J., 2011, 17, 14916–14921 CrossRef CAS.
- Y.-Y. Yeung, S. Hong and E. J. Corey, J. Am. Chem. Soc., 2006, 128, 6310–6311 CrossRef CAS.
- Y.-Y. Yeung, X. Gao and E. J. Corey, J. Am. Chem. Soc., 2006, 128, 9644–9645 CrossRef CAS.
- S. Hajra, D. Sinha and M. Bhowmick, J. Org. Chem., 2007, 72, 1852–1855 CrossRef CAS.
- S. Hajra, M. Bhowmick and D. Sinha, J. Org. Chem., 2006, 71, 9237–9240 CrossRef CAS.
- S. Hajra, B. Majit and S. Bar, Org. Lett., 2007, 9, 2783–2786 CrossRef CAS.
- S. Hajra, S. Bar, D. Sinha and B. Majit, J. Org. Chem., 2008, 73, 4320–4322 CrossRef CAS.
- C. D. Braddock, D. S. Millan, Y. Pérez-Fuertes, R. H. Pouwer, R. N. Sheppard, S. Solanki and A. J. P. White, J. Org. Chem., 2009, 74, 1835–1841 CrossRef.
- J. Tanuwidjaja, S.-S. Ng and T. F. Jamison, J. Am. Chem. Soc., 2009, 131, 12084–12085 CrossRef CAS.
- (a) S. G. Davies, M. E. C. Polywka and S. E. Thomas, Tetrahedron Lett., 1985, 26, 1461–1464 CrossRef CAS; (b) S. G. Davies, M. E. C. Polywka and S. E. Thomas, J. Chem. Soc., Perkin Trans. 1, 1986, 1277–1282 RSC; (c) F. Bravo, F. E. McDonald, W. A. Neiwert and K. I. Hardcastle, Org. Lett., 2004, 6, 4487–4489 CrossRef CAS.
- K. J. Bonney, C. D. Braddock, A. J. P. White and M. Yaqoob, J. Org. Chem., 2011, 76, 97–104 CrossRef CAS.
- (a) S. A. Snyder, D. S. Treitler, A. P. Brucks and W. Sattler, J. Am. Chem. Soc., 2011, 133, 15898–15901 CrossRef CAS; (b) S. A. Snyder, A. P. Brucks, D. S. Treitler and I. Moga, J. Am. Chem. Soc., 2012, 134, 17714–17721 CrossRef CAS.
- (a) Y. A. Serguchev, M. V. Ponomarenko, L. F. Lourie and A. N. Chernega, J. Fluorine Chem., 2003, 123, 207–215 CrossRef CAS; (b) V. Nair, R. S. Menon, P. B. Beneesh, V. Sreekumar and S. Bindu, Org. Lett., 2004, 6, 767–769 CrossRef CAS; (c) V. Nair, P. B. Beneesh, V. Sreekumar, S. Bindu, R. S. Menon and A. Deepthi, Tetrahedron Lett., 2005, 46, 201–203 CrossRef CAS; (d) T. L. Church, C. M. Byrne, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2007, 129, 8156–8162 CrossRef CAS; (e) T. Abe, H. Takeda, Y. Miwa, K. Yamada, R. Yanada and M. Ishikura, Helv. Chim. Acta, 2010, 93, 233–241 CrossRef CAS.
- L. Zhou, C. K. Tan, J. Zhou and Y.-Y. Yeung, J. Am. Chem. Soc., 2010, 132, 10245–10247 CrossRef CAS.
- R. B. Rothman and M. H. Baumann, Curr. Top. Med. Chem., 2006, 6, 1846 CrossRef.
- L. Zhou, J. Zhou, C. K. Tan, J. Chen and Y.-Y. Yeung, Org. Lett., 2011, 13, 2448–2451 CrossRef CAS.
- L. Zhou, J. Chen, J. Zhou and Y.-Y. Yeung, Org. Lett., 2011, 13, 5804–5807 CrossRef CAS.
- J. Chen, S. Chng, L. Zhou and Y.-Y. Yeung, Org. Lett., 2011, 13, 6456–6459 CrossRef CAS.
- J. Zhou, L. Zhou and Y.-Y. Yeung, Org. Lett., 2012, 14, 5250–5253 CrossRef CAS.
- Z. Ke and Y.-Y. Yeung, Org. Lett., 2013, 15, 1906–1909 CrossRef CAS.
- T. Murase, S. Sato and M. Fujita, Angew. Chem., Int. Ed., 2007, 46, 1083–1085 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |