 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploration of the medical periodic table: towards new targets†
Nicolas P. E. Barry and Peter J. Sadler*
University of Warwick, Department of Chemistry, Gibbet Hill Road, Warwick, UK. E-mail: N.Barry@warwick.ac.uk; P.J.Sadler@warwick.ac.uk
First published on 1st May 2013
Abstract
Metallodrugs offer potential for unique mechanisms of drug action based on the choice of the metal, its oxidation state, the types and number of coordinated ligands and the coordination geometry. We discuss recent progress in identifying new target sites and elucidating the mechanisms of action of anti-cancer, anti-bacterial, anti-viral, anti-parasitic, anti-inflammatory, and anti-neurodegenerative agents, as well as in the design of metal-based diagnostic agents. Progress in identifying and defining target sites has been accelerated recently by advances in proteomics, genomics and metal speciation analysis. Examples of metal compounds and chelating agents (enzyme inhibitors) currently in clinical use, clinical trials or preclinical development are highlighted.
 Peter Sadler | Peter Sadler obtained his BA, MA and DPhil at the University of Oxford. Subsequently he was a Medical Research Council Research Fellow at the University of Cambridge and National Institute for Medical Research. From 1973–1996 he was Lecturer, Reader and Professor at Birkbeck College, University of London, and from 1996–2007 Crum Brown Chair of Chemistry at the University of Edinburgh. In June 2007 he took up a Chair in Chemistry at the University of Warwick. He is a Fellow of the Royal Society of Edinburgh (FRSE) and the Royal Society of London (FRS), and a European Research Council Advanced Investigator. His research interests are centred on the chemistry of metals in medicine. |
 Nicolas Barry | Nicolas Barry received his MSc degree in Chemistry from the Université de Rennes, France in 2008. From 2008 to 2011 he obtained his PhD in Organometallic Chemistry under the supervision of Professors Süss-Fink and Therrien at the Université de Neuchâtel, Switzerland. Presently, he is a Swiss National Science Foundation postdoctoral fellow in the laboratory of Professor Sadler at the University of Warwick. |
Introduction
A significant number of clinical trials now involve metal compounds, metal-chelating agents (metalloenzyme inhibitors) or other agents which interfere with metabolic pathways for metals, both for therapy and for diagnosis. Some of the areas of current clinical interest are listed in Table 1.| Industry/sponsor | Drug | Use | Stage |
|---|---|---|---|
| Metal-based anticancer therapeutics | |||
| Bioplatin AG | Dicycloplatin (Pt) | Bronchial carcinoma | China Phase I |
| Liver cancer | |||
| Blend Therapeutics | Pt complexes including nanoparticles | Targeted and combination therapy | Pre-clinical |
| Regulon Inc. | Lipoplatin (Pt) (liposomal cisplatin) | Lung cancer | Phase III |
| Lipoxal (Pt) (liposomal oxaliplatin) | Colorectal cancer | Phase II | |
| Sanofi | Oxaliplatin (Eloxatin) (Pt) | Colorectal cancer | Approved |
| Cell Therapeutics Inc. | Dinuclear-platinum complex CT-47463 | Various cancers | Pre-clinical |
| Solasia Pharma K.K. | Darinaparsin (As) (Z10-101/SP-02) | Peripheral T-cell lymphoma | Approved US and some other countries |
| Solid tumours (oral) | Phase I | ||
| Ziopharm/Teva | Trisenox (As) | Acute promyelocytic leukaemia | Approved |
| Lung and others | Phase II | ||
| Cancer Research UK | GSAO (As) | Advanced solid tumours that have not responded to therapy | Phase I |
| Sigea | NAMI-A (Ru) in combination with gemcitabine | Anti-metastatic agent | Phase I/II accomplished |
| NIIKIPHARMA | NKP-1339 (Ru) | Various cancers, e.g. neuroendocrine and NSCLC | Phase I |
| NKP-2235 (Ga) | Cancers with high incidence of bone metastases e.g. NSCLC, breast and prostate cancers, and multiple myeloma | Phase I | |
| Virginia Commonwealth University | Belinostat (PXD101): histone deacetylase inhibitor (chelates Zn) | Relapsed or refractory acute leukaemia or myelodysplastic syndrome | Phase I |
| Patheon Inc. | Vorinostat: HDAC inhibitor (chelates Zn) | Cutaneous T cell lymphoma | Approved |
| Asan Medical Center | Gastric cancer | Phase I | |
| Ohio State University Comprehensive Cancer Center | Advanced staged oropharyngeal squamous cell carcinoma | Phase I | |
| Genentech | Brain cancer | Phase I | |
| Italfarmaco | Givinostat: HDAC inhibitor (chelates Zn) | Chronic myeloproliferative neoplasms | Phase II |
| Boneca Corporation | Boronophenylalanine-based boron neutron capture therapy in combination with cetuximab (B) | Head and neck cancer | Phase I |
| Weill Medical College of Cornell University | Tetrathiomolybdate (Mo) | Breast cancer | Phase II |
| University of Michigan Cancer Center | Esophageal carcinoma | Phase II | |
| Sanofi | Mozobil (plerixafor): (potential chelator-Zn) | Stem cell mobilisation | Approved |
| Viamet Pharmaceuticals, Inc. | VT-464 (lyase selective metalloenzyme inhibitor (Fe in CYP17)) | Prostate cancer | Phase I |
| Mayo Clinic | Auranofin (Au) | Recurrent fallopian tube, ovarian epithelial, peritoneal cavity cancer | Pilot |
| Univ Kansas | Chronic lymphocytic leukaemia (CLL) | Phase II | |
| Small lymphocytic lymphoma | |||
| Prolymphocytic leukaemia | |||
| Other metal-based therapeutics | |||
| Sanofi | Ferroquine (Fe) (SSR97193) | Antimalarial | Phase II |
| VPS-1, Inc. | VT-1161 (lanosterol demethylase metalloenzyme (CYP51) inhibitor) | Antifungal | Phase I |
| Prana Biotechnology | PBT2: hydroxyquinoline derivative – metal chelator | Alzheimer's | Phase II |
| Huntington's | |||
| Shire Pharmaceuticals | Lanthanum carbonate (Fosrenol) (La) | Hyperphosphatemia | Approved |
| Smith & Nephew | Acticoat Absorbant™ Silver Eluting Dressing (Ag) | Prevention of lower extremity revascularization wound complications | Phase IV |
| Ceva (Nature Vet Pty Ltd) | Cu-Algesic (CuII indomethacin) (Cu) | Anti-inflammatory (veterinary-horses, dogs) | Approved |
| Redox Pharmaceutical Corporation | Doxovir (CTC-96) CoIII bis(2-methylimidazole) acacen complex (Co) | Ophthalmic herpetic keratitis and adenoviral conjunctivitis | Phase I completed |
| Herpes labialis | Phase II completed | ||
| The University of Texas Health Science Center | Bismuth subcitrate potassium, metronidazole, tetracycline hydrochloride and omeprazole (Bi) | Helicobacter pylori infection | Phase IV |
| UCSF/UC San Diego | Auranofin (Au) | Treatment of amoebiasis and parasite Giardia intestinalis | Trials planned |
| Photodynamic therapy agents | |||
| Steba Biotech S.A. | Palladium bacteriopherophorbide photosensitizer TOOKAD (Pd) | Localised prostate cancer | Phase III |
| University of Oxford/Steba Biotech S.A. | Predetermined small renal tumour targets | Phase I/II | |
| Diagnostic radiopharmaceuticals | |||
| American College of Radiology Imaging Network | Copper Cu 64-ATSM and PET/CT Scan (Cu) | Cervical cancer | Phase II |
| Lantheus | Cardiolite (99mTc-sestamibi) (Tc) | Folate-receptor-positive tumours | Approved |
| Neurite (99mTc-disicate) (Tc) | |||
| Endocyte Inc. | Etarfolatide (EC20-99mTc) (Tc) | Folate-receptor-positive tumours | Phase III |
| Molecular Insight Pharmaceuticals, Inc. | 99mTc-MIP-1404 (Tc) | Prostate cancer | Phase II |
| Peking Union Medical College Hospital | 68Ga-DOTA-TATE PET/CT Scan (Ga) | Mesenchymal tumours | Phase 0 In development |
| Oncogenic osteomalacia | |||
| Vanderbilt University | Neuroendocrine Cancer | Phase I | |
| Therapeutic radiopharmaceuticals | |||
| Actinium Pharmaceuticals | Actinium-225-Labeled Humanized Anti-CD33 Monoclonal Antibody HuM195 (Ac) | Leukaemia myelodysplastic syndrome | Phase I |
| Lintuzumab-Ac225 (Ac) | Acute myeloid leukaemia | Phase I/II | |
| Lintuzumab-Bi213 (Bi) | Acute myeloid leukaemia | Phase II | |
| Algeta ASA (Bayer) | Alpharadin (223Ra chloride) (Ra) | Bone metastasis from castration-resistant prostate cancer | Phase I/II |
| Radiation therapy oncology group and National Cancer Institute | Samarium-153-lexidronam pentasodium (Sm) | Prostate cancer | Phase II |
| UMC Utrecht | Holmium-166 polylactic microspheres (Ho) | Liver neoplasms | Phase II |
| Centre René Gauducheau | Radiation: IMP-288-lutetium (Lu) | Small cell lung cancer | Phase II |
| Contrast agents and others | |||
| OHSU Knight Cancer Institute | Ferumoxytol and gadolinium magnetic resonance imaging (Fe, Gd) | Malignant brain tumours | Phase II |
| Lantheus Medical Imaging | BMS753951 (Gd) | MRI contrast for coronary arterial wall imaging | Development |
| Pharmacyclics | Motexafin gadolinium (Gd) | Radiation and chemotherapy sensitiser | Development |
| Alexion Pharmaceuticals | Cyclic pyranopterin monophosphate replacement therapy (precursor to Mo cofactor) | Markers for Mo cofactor deficiency | Development |
| Isolated SOX deficiency | |||
These areas have been stimulated by recent successes with platinum anticancer drugs (used as a component of nearly 50% of all cancer treatments), with gadolinium complexes as MRI contrast agents (about 20 million doses administered per year) and 99m-technetium radiopharmaceuticals for γ-ray imaging (used in about 20 million radiodiagnostic procedures each year). However, many other diseases and conditions are of current interest including neurodegeneration, microbial and parasitic (and other neglected tropical diseases) infections and inflammation.1–4 About 24 elements are essential for man, Fig. 1. In principle, we should consider the roles of all essential elements in therapy (control of homeostasis), but this is currently hampered in many cases (e.g. Si, V, Ni, Sn) because their natural biochemistry is poorly understood and we do not know how to diagnose conditions associated with their metabolism. For some elements it is already possible to use genomics to understand medical conditions related to them (e.g. Fe, Cu, Zn), but in other cases it is not clear how, or if, genomes code for these elements. Genomes do not code for the elements themselves, but for particular chemical forms (e.g. vitamin B12 for cobalt). Genomic codes for metals are usually codes for proteins, proteins which are highly, but not totally, selective for particular metal ions. As is evident from the above examples of Pt, Gd and Tc, inorganic medicinal chemistry can make use of non-essential as well as essential elements for the design of drugs and diagnostic agents, Fig. 1.4
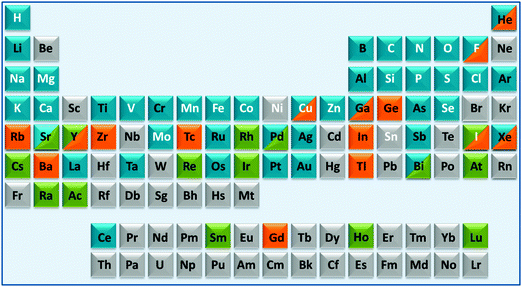 | ||
| Fig. 1 A medical periodic table: essential elements for man (symbols in white font); medical radioisotopes (green fill); elements currently used in therapy (blue fill) or diagnosis (orange fill). The entries (limited to 2 fill colours, illustrative and not comprehensive) are mainly restricted to elements/compounds which are clinically approved or on current clinical trials (e.g. as listed on http://www.clinicaltrials.gov/). Some entries for implants are included (e.g. Ti, Ta). The basis for some of the entries is given in Table S1 (ESI†). | ||
For many centuries the use of metallodrugs has been driven by empiricism. Whilst random screening is still a useful weapon in drug discovery, these days it should be guided largely by rational design. This implies that target sites (usually gene products – proteins) should be identified and verified early in the design process. Immediately this presents challenges for medicinal metal coordination chemistry: either the synthesised complexes need to be inert to redox and ligand substitution reactions, or these reactions need to be controlled under biological conditions en route to the target. In turn, metal- and/or ligand-centred redox reactions provide a unique platform for drug design with concepts quite distinct from those for purely organic drugs.
In this article we highlight some areas of current interest in metallodrugs. We focus especially on their interactions with (proposed) target sites, including DNA and proteins. Despite the fact that successful clinical platinum anticancer drugs have DNA as their major target, DNA is nowadays not considered to be a favoured target site. This is partly because DNA is also likely to be attacked in normal healthy cells as well as in cancer cells. However, downstream processing of platinated DNA can differ in normal and cancer cells so differential cytotoxicity can still be achieved. Moreover, it is becoming evident that anticancer drugs that target a single protein or enzyme are not always successful, since cells readily become resistant to such drugs and utilise alternative metabolic pathways. Metallo-anticancer drugs have the potential advantage of attacking several sites (multi-targeting), which can be an highly effective strategy. It is important to identify these target sites and elucidate the molecular mechanisms of action. As we shall see, a range of new target sites are being proposed for metallodrugs but there is still much progress to be made on target-site validation. Tailoring the design of metallodrugs to treat specific diseases and conditions is likely to be a major part of future personalised medicine, which will include genomic and proteomic profiling of individuals.
1. Metal-based anticancer drugs
1.1. DNA targeting
1.1.1.1. Direct coordinative DNA binding. It is well established that the target site for the anticancer drug cisplatin (1), cis-[PtCl2(NH3)2] (Chart 1, FDA approval 1978), is nuclear DNA.5–15 More than 4000 Pt compounds have now been tested as potential anticancer drugs, with the worldwide approval for clinical use of carboplatin (2, 1989) and oxaliplatin (3, 2002), and of three others – nedaplatin (4), lobaplatin (5) and heptaplatin (6) (Chart 1) for clinical use only in Japan, China and South Korea, respectively. The ability to form DNA adducts is important for the activity of all of these drugs. Table 2 lists the X-ray structures of cis-diam(m)ine Pt–DNA adducts registered in the Protein Data Bank by January 2013. Dicycloplatin (7) is a new complex developed at the University of Beijing by Yang et al. that has passed through Chinese clinical phase I trials. This compound is composed of one molecule of carboplatin and one molecule of cyclobutane-1,1-dicarboxylic acid interacting via hydrogen bonds (see Chart 1).16 The mechanism by which the presence of free ligand affects the activity of this platinum drug is not clear.
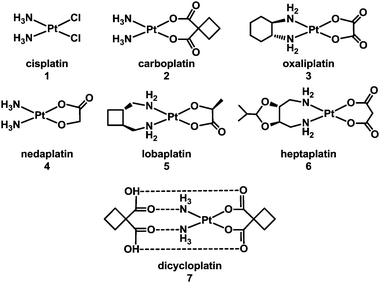 | ||
| Chart 1 Molecular structures of some platinum anticancer drugs that are approved or undergoing clinical trials. | ||
| Platinum complex | Type of Pt–DNA adduct | DNA structural changes | PDB ID and Ref. |
|---|---|---|---|
| Cisplatin | 1,2-cis-{Pt(NH3)2}2+-d(GpG) intrastrand cross-link | Bending (kinking) of duplex; rolling of the adjacent guanines; unwinding of the helix at platination site; widening and flattening of the minor groove opposite the platinum adduct | 3LPV;173M9M, 3M9N, 3M9O;182R8J, 2R8K;192R7Z;201AIO;211A84;221AU523 |
| 1,3-cis-{Pt(NH3)2}2+-d(GpTpG) intrastrand cross-link | 2WTF;241KSB;251DA4, 1DA526 | ||
| 1,3-cis-{Pt(NH3)2}2+-d(GpTpG) intrastrand cross-link in a nucleosome | 3O6227 | ||
| Interstrand cross-link | 1A2E;281DDP29 | ||
| Oxaliplatin | 1,2-{Pt(dach)}2+-d(GpG) intrastrand cross-link | Stronger duplex bending than with cisplatin30 | 2K0T, 2K0U, 2K0V;311PG9, 1PGC;321IHH33 |
| Pyriplatin | Monofunctional platinum–DNA adduct (Pt-N7G binding) | Markedly different transcription inhibition mechanism compared to cisplatin or oxaliplatin | 3CO334 |
| [PtCl(en) (ACRAMTU-S)] (NO3)2 | Dual mode | Platinum-N7G binding in the major groove + ACRAMTU intercalation leading to inhibition of damaged DNA transcription by RNA polymerase II | 1XRW35 |
Compounds based on cis-diam(m)ine PtII structures tend to produce very similar types of adduct on target DNA – as exemplified in Table 2. It is therefore not surprising that they induce similar biological consequences, although protein recognition of the DNA adducts is dependent on the ligands. This consideration led to the hypothesis that development of Pt compounds structurally different from cisplatin which form different types of DNA–Pt adducts, may lead to a different spectrum of biological activity, perhaps complementary to cisplatin.36 In the late 1980s, Farrell began a fruitful investigation of linear multi-platinum complexes, where the platinum centres are separated by aliphatic chains, and the chlorido ligands are cis or trans at the extremities of the complex.37–40 These poly-platinum complexes are generally highly positively-charged. This leads to a stronger initial electrostatic recognition of DNA in comparison with monomeric species. Another critical feature of these multinuclear complexes is flexibility. The potent multi-platinum complex [(trans-PtCl(NH3)2)2{μ-trans-Pt(NH3)2(NH2(CH2)6NH2)2}]4+, BBR3464 (8, Chart 2),41 completed a phase I trial,42 but failed phase II due to instability in blood.43–47
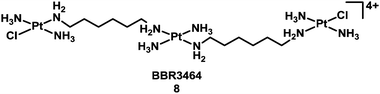 | ||
| Chart 2 Molecular structure of complex 8. | ||
It seems to be the only Pt compound not based on the cisplatin chemotype to have entered human clinical trials.48 Hydrolysis of BBR3464 and DNA adduct formation are well documented.49,50 In general polynuclear platinum compounds bind rapidly to DNA (t1/2ca. 40 min).47 The most relevant feature of the special BBR3464–DNA binding is the lack of severe DNA distortions such as a kink, or significant unwinding of the helix, which are characteristics of DNA adducts of mononuclear platinum complexes. One of the direct consequences of this mild Pt-induced DNA conformational change is that these adducts are poor substrates for recognition by proteins, such as those containing the HMG domain, which binds to rigidly-bent DNA, as induced by cisplatin.51 Overviews of multi-nuclear platinum drugs52,53 and DNA binding to polynuclear platinum complexes54 have been published.
Low-spin octahedral 5d6 PtIV complexes have potential advantages as anticancer drugs because they are more inert to substitution reactions than square-planar PtII complexes and so are expected to undergo fewer side reactions en route to the tumour. Also they often have higher aqueous solubility, a feature exploited long ago by Tobe et al. who synthesised iproplatin (CHIP, JM9, cis,trans,cis-[PtCl2(OH)2(isopropylamine)2]).55 Iproplatin entered phase I and II clinical trials,56,57 and even phase III,58 but ultimately was found to be less active than cisplatin and so not registered for clinical use.59 Tetraplatin (ormaplatin) [PtCl4(D,L-cyclohexane-1,2-diamine)], also showed promise in preclinical studies but caused severe neurotoxicity in treated patients, and trials were subsequently abandoned at phase I.60 The unpredictability of the rate, extent and localisation of in vivo reduction of these PtIV complexes to the active PtII species may be a complication in clinical use.
The need to overcome platinum resistance, to reduce toxic side-effects and broaden the spectrum of activity has also focussed attention on the potential of anticancer drugs containing other metals. Among them, Ru-based-drugs are promising.61 The RuIII anticancer drug imidazolium-trans-dimethylsulfoxide-imidazole-tetrachlororuthenate (NAMI-A) (9) developed in Trieste by Mestroni, Alessio, et al. has reached phase I/II trials in combination with gemcitabine. Another octahedral RuIII complex indazolium-trans-bis(1H-indazole)-tetrachlororuthenate (KP1019) (10), developed by Keppler et al. in Vienna (Chart 3), has completed phase I clinical trials.62,63 Organometallic arene RuII complexes such as [(η6-biphenyl)Ru(en)Cl]+ (en = ethylenediamine)61 (11) and [(η6-p-cymene)Ru(PTA)Cl2]64 (p-cymene = para-cymene and PTA = 1,3,5-triaza-7-phosphaadamantane) (12) (see Chart 3) have been widely studied and several others also show promise.65–76
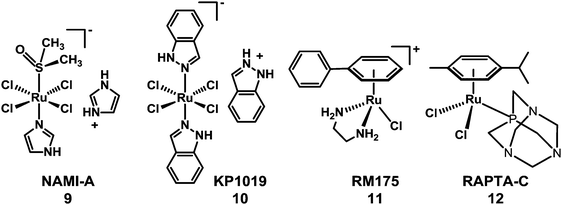 | ||
| Chart 3 Molecular structures of RuIII anticancer complexes 9 and 10, and RuII arene complexes 11 and 12. | ||
The antimetastatic activity of NAMI-A is thought to be due to combined effects on the control of angiogenesis (possibly because it interferes with NO metabolism)77,78 and anti-invasive properties towards tumour cells and blood vessels, and not to interaction with nucleic acids, although it can interact with DNA in vitro.79 DNA has been proposed as one of the biological targets of KP1019, and it also triggers apoptosis.80 However, the cellular mechanism of the activation of apoptosis is not understood.81 These RuIII complexes may undergo activation by reduction to RuIIin vivo.82
Different mechanisms of action have been investigated for explaining the anticancer activity of arene ruthenium complexes. Interactions between complexes containing reactive Ru–Cl and nuclear DNA can occur, through the formation of intermediary aqua complexes able to ruthenate DNA specifically at guanine residues.83,84 If the arene is extended (e.g. biphenyl, dihydroanthracene), DNA binding can involve not only direct coordination to G bases but also arene intercalation (see Section 1.1.1.3.). Ligand oxidation can provide a way to activate thiolato complexes of RuII arenes, which may be of importance when the intracellular thiol glutathione binds to them. Mono- (to give the sulfenate) and bis- (sulfinate) oxygenation appear to be facile, but surprisingly do not weaken the Ru–S bond85 even though this provides a route to nucleobase binding.86 Protonation of the sulfenate oxygen on the other hand does labilise this bond.87
Osmium, the heavier congener of ruthenium and a third row transition metal, commonly exhibits slower kinetics than ruthenium, and is often considered to be relatively inert. However, it is possible to tune the biochemical reactivity of the arene OsII complexes through understanding their aqueous solution chemistry. The rates of hydrolysis of [(η6-arene)Os(XY)Cl]n+ complexes can be controlled by the choice of the chelating ligand XY (faster for XY = O,O, followed by O,N and slowest for N-donor chelating ligands, especially when N,N is a strong π-acceptor such as azopyridine). The pKa values of the aqua adducts also follow this order, being highest for O,O ligands, with coordinated water tending to be more reactive than coordinated hydroxide.88 Ligand substitution rates on arene OsII are often ca. 100× slower than for RuII and coordinated aqua ligands ca. 1.5 pKa units more acidic. Thus, active arene OsII complexes have been designed.89–99 In particular chlorido OsII picolinate complexes can bind to DNA, but interestingly some inert iodido OsII azopyridine complexes exhibit nanomolar potency to a wide range of cancer cells and are also active in vivo.100 The azopyridine complexes appear to have redox mechanisms of action.101 Interestingly injected OsO4 has a long history of use in therapy in Scandinavia for chemical synovectomyin the treatment of chronic synovitis.102
In addition to binding to DNA,103 IrIII Cp* anticancer complexes can also have redox mechanisms of action, readily forming hydride complexes on reaction with coenzyme NADH, and even producing H2 catalytically.104 These findings highlight the existence of new potential targets for such complexes.
1.1.1.2. Non-covalent DNA binders. Metal complexes of an appropriate shape and polarity can intercalate between DNA base pairs.105 Intercalators can be potent mutagens, due to their ability to induce structural and therefore functional changes in duplex DNA, leading to inhibition of transcription, replication and DNA repair processes. Intercalators are potential antibiotics, antibacterials, trypanocides, schistosomicides, and antitumour agents.106,107 Several metallo-intercalator complexes have been reported recently, Table 3.
| Metal | Ligands |
|---|---|
| a dppz = dipyrido[3,2-a:2′,3′-c]-phenazine. | |
| ZnII | Schiff bases;108 trihydroxy-isoflavones109 |
| AuIII | Liriodenines110 |
| AuI | Phosphines111 |
| AgI | bis-Benzimidazole ligands112 |
| CuII | Dipyrido-phenazine and glycinato ligands;113 (phenyl(pyridine-2-yl)methylidene)-benzohydrazides;114 trihydroxy-isoflavones;109 terpyridine ligands115 |
| CuII–PbIV | Phenanthroline derivatives116 |
| PtII | Carbenes;117 ethylenediamine, diaminocyclohexane and phenanthroline derivatives;118 phenylpyridine;119 bipyridyl-thiourea ligands;120 phenanthroline derivatives121 |
| NiII | Trihydroxy-isoflavones;109 (phenyl(pyridine-2-yl)methylidene)-benzohydrazides;114 phenanthroline derivatives122 |
| IrIII | Polypyridine ligands;126,127 cyclopentadienyl ligands123 |
| RhIII | Polypyridine ligands124 |
| CoIII | (Phenyl(pyridine-2-yl)methylidene)-benzohydrazides;114 quinoline derivatives;125 phenanthroline derivatives122 |
| RuII | Dppza ligands;131,132 allopyranoside-grafted ruthenium(II) complex;126 quinone derivatives;127 phenanthroline derivatives118,119,128 |
Besides intercalation and groove binding, highly charged multinuclear complexes can bind strongly to the phosphate backbone of DNA, forming a “phosphate clamp. The trinuclear complex triplatinNC [{trans-Pt(NH3)2(NH2(CH2)6(NH3)}2-μ-{trans-Pt(NH3)2(NH2(CH2)6NH2)2}](NO3)8 (13, see Chart 4) exclusively utilises backbone functional groups to interact with DNA, and can associate with DNA even in the absence of direct coordination to DNA bases.129
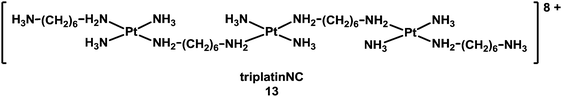 | ||
| Chart 4 Molecular structure of triplatinNC. | ||
1.1.1.3. Coordination plus intercalation: dual mode DNA binders. Metal complexes with σ-bonded aromatic side arms or organometallic complexes with extended π-bonded arenes can act as dual-function complexes, binding to DNA both by direct metal coordination to a DNA base and intercalation between DNA bases through an attached aromatic ligand.105 The utilisation of σ-bonded side arm-containing platinum complexes acting as intercalators was reported in the late 1980s.130,131 For example, Pt complexes incorporating planar aromatic ligands such as acridine orange, 9-aminoacridine, ethidium bromide, and acridinylthiourea (ACRAMTU) bind to DNA by both coordination and intercalation.131–133 The dual mode interaction causes a strong structural modification by increasing the length and unwinding the DNA duplex. On the other hand, helical bending, C3′-endo deoxyribose puckering and rolling are not observed, which distinguishes the dual mode-induced damage from intrastrand cross-link damage.35 The adducts formed by the potent complex [PtCl(en)(ACRAMTU-S)](NO3)2 (14) (Fig. 2) inhibit transcription of the damaged DNA by RNA polymerase II.134
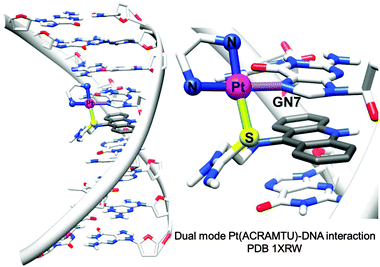 | ||
| Fig. 2 X-ray crystal structure showing the dual mode coordination/intercalation interaction between complex 14 and DNA (PDB ID 1XRW). Adapted from ref. 35. | ||
Additionally, biphenyl RuII complex 11 (Chart 3) not only binds strongly and preferentially to G bases in DNA but also intercalates via the phenyl substituent on the η6-arene.135,136 In monofunctional adducts of {(η6-biphenyl)Ru(en)}2+ with the 14-mer (ATACATGGTACATA)·(TATG*TACCATGTAT) ruthenation occurs at N7 of each guanine residue. At one site (G*), not only was intercalation of the arene between G* and adjacent T observed, but also stacking of a non-intercalated arene on a tilted adjacent thymine on the surface of the major groove.137 Other dual-function coordination complexes have also been investigated,138,139 such as the di-RhII complex cis-[Rh2(dap)(μ-O2CCH3)2-(η1-O2CCH3)(CH3OH)](O2CCH3) (dap = 1,12-diazaperylene).
A series of low-spin 5d6 cyclopentadienyl IrIII organometallic half-sandwich complexes has been shown to form adducts with 9-ethylguanine and/or 9-ethyladenine readily, depending on the chelating ligands. Moreover, in the case of complexes incorporating extended Cp* ligands, the ability to intercalate into DNA appears to contribute to the anticancer potency of these IrIII complexes.123
Combining Pt and other metal ions also provides a strategy for designing complexes able to bind DNA both by coordination and by intercalation. For example, in heterobimetallic complexes containing Pt and Ru centres joined by a linker,140 PtII offers coordinative binding through ligand substitution, and octahedral RuII chelated by terpyridine or extended pyridyl ligands can act as an intercalator as well as possessing useful optical properties.140 Alternatively, the Pt unit is capable of interacting with DNA by π–π stacking or coordination through Cl substitution, and the Ru unit can bind to DNA by electrostatic or surface binding, or partial intercalation.141
1.1.1.4. G-quadruplex binders. Telomeres in human genes contain many repeats of the sequence d(GGTTAG) (G-quartets) and are responsible for maintaining cell division. In these G-rich strands, four-stranded G-quadruplexes can be formed consisting of planar G-quartets stabilised by K+ or Na+. These inhibit the enzyme telomerase. Targeting either telomerase or stabilising G-quartets is an effective strategy for the design of anticancer agents.
The first, and only example to date, of an X-ray crystal structure of human telomeric G-quadruplex DNA bound to a metal complex was reported in 2012.142 NickelII and copperII salphen metal complexes were co-crystallised in the presence of human telomeric DNA and the effective binding of these metal–salphen complexes to human telomeric quadruplexes by direct end-stacking was demonstrated (see Fig. 3). Other methods have also been used to demonstrate the binding of metal complexes to quadruplex DNA. Table 4 lists examples of coordinative and non-coordinative metal-based G-quadruplex DNA binders reported between 2009 and 2012.
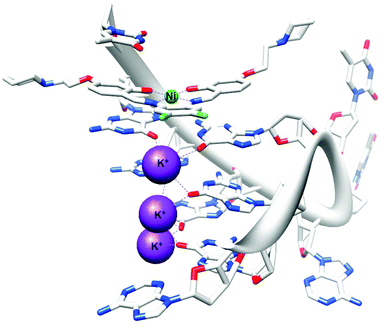 | ||
| Fig. 3 X-ray crystal structure of human telomeric G-quadruplex DNA bound to a salphen–NiII complex (PDB ID 3QSF). Adapted from ref. 142. | ||
| Metal | Ligands/ref. |
|---|---|
| AuIII | Porphyrins;143 pyrazolylpyridine144 |
| CuII | Salphen derivatives;142 terpyridine-based ligands145 |
| PtII | Carboxamide phenanthrolines and bis-carboxamide pyridines;146 terpyridines;145,147,148 Schiff bases;149 phenanthroimidazoles;150 triarylpyridines;151 perylene derivatives152 |
| Tetranuclear PtII | Quinoxalines153 |
| PtII–ZnII | Porphyrazine derivatives154 |
| Dimetallic CuII, PtII and ZnII | Terpyridine-based ligands155 |
| PdII | Terpyridine-based ligands;145 carboxamide phenanthrolines and bis-carboxamide pyridines146 |
| NiII | Salphen derivatives142 |
| IrIII | 2,2-Biquinolines156 |
| CoIII, MnIII, Octanuclear RuII | Porphyrins157,158 |
| RuII | Polypyridyl ligands;159 phenanthroline derivatives;160,161 Dppz ligands162 |
| RuII | Phenanthroline derivatives |
| Dinuclear RuII | Bipyridines and tetrapyrido-phenazines;163 tetraazaphenanthrenes and tetrapyridoacridines164 |
1.1.2.1. Metallodrugs with sequence specificity. New metal drug candidates should (ideally) interact with a specific biological target. For DNA, incorporation of ligands possessing sequence specificity can provide a strategy to enhance the selectivity of binding. For example, mononuclear and dinuclear PtII complexes incorporating linear and hairpin polyamide ligands (imidazole, pyrrole and β-alanine subunits, see Chart 5) are capable of recognising sequences up to seven base-pairs in length, providing a foundation for the synthesis of hairpin polyamides with multiple platinum groups leading to high DNA-sequence-selectivity, water-solubility and potentially highly active complexes.165,166
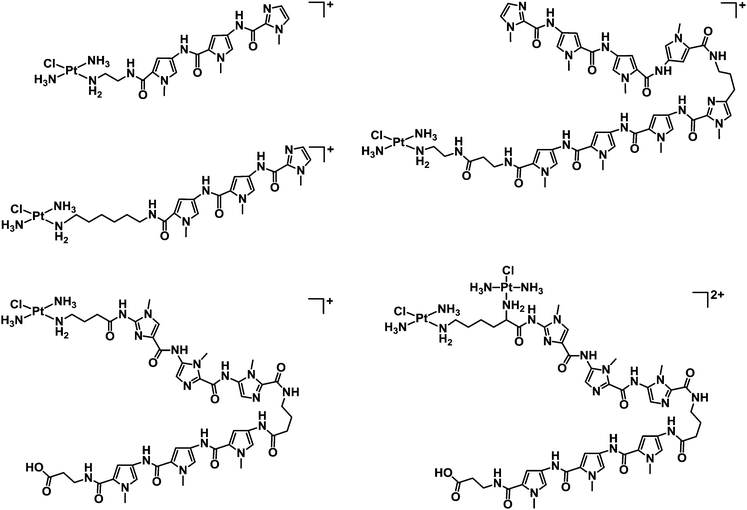 | ||
| Chart 5 Molecular structures of some mononuclear and dinuclear PtII complexes incorporating linear and hairpin polyamide ligands. | ||
Specific recognition of DNA sequences can be also used to overcome side-effects and resistance to platinum drugs. Similarly incorporation of an oligonucleotide into a Pt complex allows sequence recognition within double stranded DNA by the formation of triple helices.167 Various pathways to metal-containing oligonucleotides have been designed, such as conjugation of an end-functionalised oligonucleotide, chelation of an oligonucleotide to the metal or incorporation of metal-containing phosphoramidites or hydrogenophosphonates. Oligodeoxynucleotide-tethered bifunctional cis-dichlorido Pt complexes168 can retain cross-linking ability.
1.1.2.2. Protein-mediated DNA recognition. The design of metallodrugs incorporating groups capable of interacting specifically with DNA sequences is a challenge. An elegant strategy is to use a presenter protein and a small molecule that can bind to both protein and target.169 The formation of such ternary complexes has been described for organic drugs, such as the immunosuppressive antibiotic rapamycin,170 and has been illustrated as a concept for metallodrugs by an arene-Ru-biotin derivative171 embedded in the tetrameric streptavidin protein (Fig. 4).172 The supramolecular structure binds G-quadruplex DNA with selectivity towards DNA telomeres, even in the presence of competing targets (such as glutathione). However, because of a poor cellular uptake, the in vivo activity of the system was not investigated.
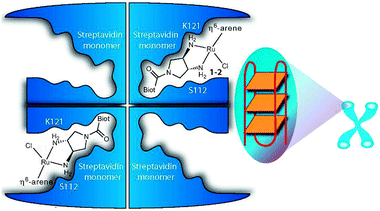 | ||
Fig. 4 Presenter protein strategy for targeting telomeric DNA with Ru metallodrugs. The Ru drug embedded into tetrameric streptavidin in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio forms a supramolecular complex that may allow extensive interactions with DNA (here depicted as G-quadruplex telomeric DNA; G4A). Reprinted from ref. 172 with permission. Copyright (2010) John Wiley & Sons. :1 molar ratio forms a supramolecular complex that may allow extensive interactions with DNA (here depicted as G-quadruplex telomeric DNA; G4A). Reprinted from ref. 172 with permission. Copyright (2010) John Wiley & Sons. | ||
1.2. Protein targeting
A number of proteins (e.g. kinases, bacterial Zn enzymes) have been proposed as potential biological targets for metal complexes or chelating agents in the case of metalloprotein targets.173–187 Modern bioanalytical techniques with high sensitivity and selectivity have facilitated the discovery of potential protein targets for metal complexes.188 However, as for DNA, the ability of a metallodrug to interact with amino acids, peptides or proteins in vitro does not necessarily imply a direct involvement in the cellular mechanism of action of this metallodrug. The involvement of particular targets has to be validated (e.g. by knockdown experiments, silencing gene expression by RNAi techniques). This involves blocking production of the protein in the cell and observing the effects on the activity of the drug candidate. So far, this has been rarely done for metallodrugs. Correlations of activity with protein binding (including inverse) can be useful for structure–activity relationships.189We now illustrate recent work on interactions of metallodrugs with potential protein targets. The examples are illustrative and not comprehensive.
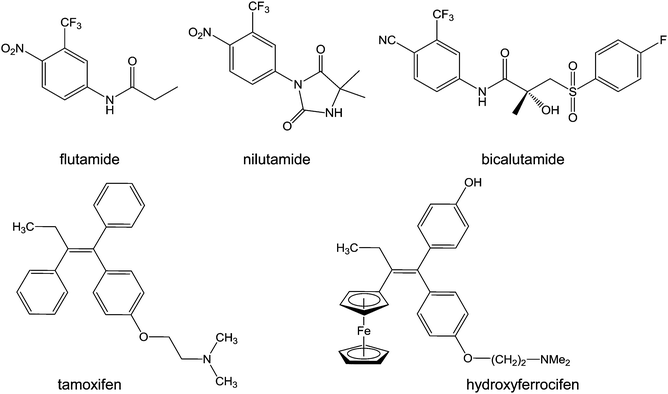 | ||
| Chart 6 Molecular structures of flutamide, nilutamide, bicalutamide, tamoxifen, and hydroxyferrocifen. | ||
Ferrocenyl organometallic complexes incorporating steroidal androgens testosterone and dihydrotestosterone (DHT) exhibit a strong antiproliferative effect on the hormone-independent prostate cancer cells PC-3 with IC50 values in the low micromolar range.195
Tamoxifen ((Z)-2-[4-(1,2-diphenyl-1-butenyl)phenoxy]-N,N-dimethylethanamine, Chart 6) is a bioavailable selective estrogen receptor modulator commonly prescribed for preventing and treating hormone-dependent breast cancers. Its metabolite, hydroxytamoxifen, is the active form which competitively binds to estrogen receptors. Despite this clinical success, tamoxifen suffers from critical limitations: ineffectiveness towards ER-tumours (a third of hormone-dependent tumours), development of resistance mechanisms (another third), and increases of uterine and endometrial cancer risks.196
Ferrocenyl tamoxifen derivatives, obtained by substitution of the β-phenyl ring of hydroxytamoxifen by a ferrocenyl fragment (Chart 6), show promise for the treatment of both hormone-dependent and independent breast cancer cells.197 The replacement of the phenyl group by ferrocene reduces receptor affinity by about 40%, whilst the increase in length of the dimethylaminoalkyl chain has an adverse effect on receptor binding. In addition to tamoxifen-like binding to the estrogen receptors, ferrocifens are likely to be activated by oxidation to quinone methides in cells, suggesting a dual biological mode of action. The mechanism of formation of these quinone methides from ferrocenyl phenols is a two-step pathway involving two successive single-electron oxidations, accompanied by deprotonation which stabilises the quinone radical, resulting in the quinone methide structure.88 The proposed quinone methides have been characterised, and indeed appear to be the active metabolites of ferrocifens.198
Ruthenocenyl tamoxifen analogues behave essentially as antiestrogens.199 Electrochemical studies of such complexes showed that the oxidation of the ruthenocenyl fragment is irreversible and leads to rapid decomposition of the organometallic entity.197 Similarly, cyclopentadienyl Re analogues do not exhibit antiproliferative activity, ({CpRe(CO)3} acting as a spectator group).200 Therefore, the redox activity of the ferrocenyl group is of central importance, along with the antiestrogenic properties of the hydroxytamoxifen derivative, for providing a unique dual mechanism of action of these ferrocenyl tamoxifen derivatives.
The anticancer activity of titanocene dichloride has been known for decades, but its efficacy in Phase II clinical trials in patients with metastatic renal cell carcinoma201 or metastatic breast cancer202 was too low to be pursued. The titanocenyl-derivative of tamoxifen203 has estrogenic effects on hormone-dependent breast cancer cells in the nanomolar range. Unfortunately, hydrolysis of the titanocene fragment leads to the generation of TiIV species that behave in a similar way to estradiol towards the estrogen receptor. This explains the estrogenic effect observed for Cp2TiCl2 and precludes the use of Cp2TiCl2 derivatives of tamoxifen as drug candidates for the treatment of breast cancer. It also poses the problem of the possible role of titanium salts as endocrine disruptors.197 Nonetheless, the recent development of highly active water-soluble ring-substituted cationic titanocene dichloride derivatives has reactivated interest in such organometallic complexes.204–206
Another promising way of targeting hormonal receptors is utilisation of carboranes as estrogen receptor agonists and antagonists. Estrogen receptors are over-expressed in estrogen-receptor-positive tumours, such as in some breast cancers.207 Carboranes are versatile pharmacophores and possess unique properties that make them useful in inorganic chemistry.208,209 Compounds incorporating carborane cages structurally close to estradiol,210 can act as efficient estrogen receptor agonists.211Closo carborane tamoxifen is more stable than tamoxifen itself, and the Z carborane tamoxifen isomer exhibits similar inhibition properties as tamoxifen.
Mitochondrial proteins are potential targets for arsenic compounds which can bind strongly to vicinal (Cys) thiol groups, increase ROS production and induce apoptotic signalling pathways. Several studies have highlighted the potential of arsenic metallodrugs in cancer therapy. Arsenic(III) trioxide (As2O3) has been used as a therapeutic agent for over 2000 years.212–214 More recently Ehrlich screened hundreds of organoarsenic compounds for biological activity and in 1910 introduced Salvarsan (arsphenamine) for the treatment of syphilis. Currently, As2O3, (ATO, ‘Trisenox’, Table 1) is the most effective single agent for the treatment of acute promyelocytic leukaemia.215 In aqueous solution ATO exists as the trihydroxide As(OH)3 and is taken up into cells via the aquaporins (especially aquaglyceroporins216)-membrane transport proteins.
GSAO (4-(N-(S-glutathionylacetyl)amino) phenylarsonous acid) is a promising new compound, known to inhibit adenine nucleotide translocase (ANT) in the inner membrane of mitochondria. A phase I clinical study is in progress in patients with solid tumours refractory to standard therapy (Table 1).217 Metabolism of GSAO is required for biological activity. This metabolism involves a two-step mechanism, first cleavage by γ-glutamyltranspeptidase at the cell surface to give GCAO (4-(N-(S-cysteinylglycylacetyl)amino) phenylarsonous acid). Then GCAO enters cells via an organic ion transporter and is metabolised by dipeptidases to CAO (4-(N-(S-cysteinylacetyl)amino) phenylarsonous acid) in the cytosol (Fig. 5). Finally, CAO enters the mitochondrial matrix and AsIII cross-links cysteine residues 57 and 257 of human ANT1,218 so inhibiting this enzyme.
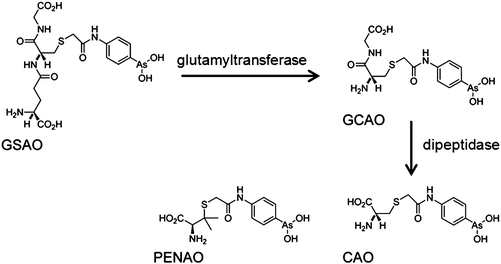 | ||
| Fig. 5 Metabolism of GSAO and molecular structure of PENAO. Adapted from ref. 218. | ||
The analogue of CAO, PENAO (4-(N-(S-penicillaminylacetyl)amino) phenylarsonous acid) (Fig. 5), is accumulated in cells ca. 85-fold faster than GSAO, has a 44× higher antiproliferative activity and 20× higher antitumour efficiency in mice. In 2012, patients with solid tumours refractory to standard therapy were recruited for Phase I/IIa dose escalation studies.218
In general, many positively-charged lipophilic complexes are taken up by mitochondria, for example cartionic gold(I) phosphines and carbenes.219,220 Another example is inert polypyridyl RuII complexes which target mitochondrial function and induce apoptosis. A strategy for combinatorial parallel coordination chemistry has been recently used, giving access to a library of more than 500 monocationic polypyridyl ruthenium complexes.221 These complexes were screened for cytotoxicity towards cancer cells, and structure–activity relationships led to the discovery of a lead complex [Ru(tBu2bpy)2(phox)]PF6 (tBu2bpy = 4,4′-di-tert-buty-2,2′-bipyridine and Hphox = 2-(2′-hydroxyphenyl)oxazoline) (15, Chart 7), which is active at submicromolar concentrations in clinically relevant Burkitt-like lymphoma cells. This complex strongly reduces the mitochondrial membrane potential, suggesting involvement of the intrinsic pathway of programmed cell death. These complexes are chiral, but all compounds in this study were formed as mixtures of enantiomers or diastereoisomers. It would be interesting to test the separated enantiomers for activity.
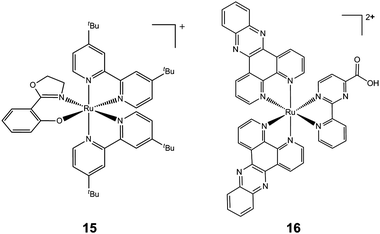 | ||
| Chart 7 Molecular structures of complexes 15 and 16. Only one enantiomer is shown. | ||
Another example of mitochondrial targeting is the complex [Ru(dppz)2(CppH)]2+ (CppH = 2-(2′-pyridyl)pyrimidine-4-carboxylic acid; dppz = dipyrido[3,2-a:2′,3′-c]phenazine, 16, Chart 7), which impairs the mitochondrial membrane potential in HeLa cells as early as 2 h after treatment and induces apoptosis.222
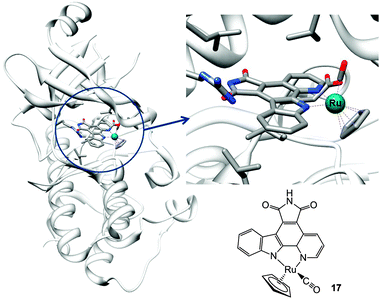 | ||
| Fig. 6 Molecular structure of complex 17 and X-ray crystal structure showing its interactions in the ATP pocket of PIM2 kinase (PDB ID 2IWI). Adapted from ref. 233. | ||
Aberrant activity of tumour necrosis factor-α (TNF-α, a pro-inflammatory cytokine involved in the regulation of many key biological processes, e.g. haematopoiesis, immunity, and inflammation)234 is associated with a number of diseases, such diabetes, tumourigenesis, and autoinflammatory diseases. Clinical trials in ovarian cancer suggest that synthetic therapeutic antibodies (e.g. infliximab), which bind directly to TNF-α, may be effective in blocking its interaction with the tumour necrosis factor receptor (TNFR).235–237 Despite this success, synthetic antibodies suffer from limitations, such as development of anti-antibody response, which has led to the search of alternative small-molecule-based therapies as inhibitors of TNF-α. Based on the approach developed for kinase inhibition, inert octahedral metal complexes, such as the cyclometalated biquinoline iridium(III) complex 18 (see Chart 8), hold potential as direct TNF-α inhibitors.238
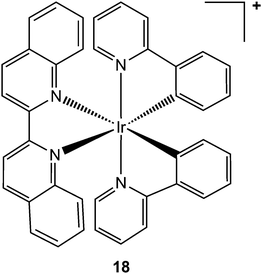 | ||
| Chart 8 Molecular structure of complex 18. One enantiomer is shown. | ||
Interest in targeting the thioredoxin system – including thioredoxin (Trx), thioredoxin reductase (TrxR) and NADPH – led in 2006, to the submission of a New Drug Application to the FDA for Motexafin gadolinium, an inhibitor of thioredoxin reductase and ribonucleotide reductase, for the treatment of lung cancers with brain metastases. This compound is currently in clinical development as a radiation and chemotherapy sensitiser (Table 1). Thioredoxin reductase remains an attractive target for metallodrugs (Table 5), particularly for gold complexes,239–243 although there is a need to validate this as a target for these metal complexes in vivo. Thioredoxin reductase (TrxR) contains FAD and NADPH binding domains and a redox-active disulphide (Cys–Cys) bond in its active site. It transfers electrons to thioredoxin which, in turn, reduces disulphide bonds and other substrates. Mammalian TrxRs contain a second redox-active site, a C-terminal –Cys-SeCys– (where SeCys is selenocysteine).244
| Metal | Compound/ligands |
|---|---|
| Se | Organic selenium-containing amino acids245 |
| As | Arsenic trioxide246 |
| Hg | Methylmercury247 |
| AuI | Carbene derivatives;240,242,248,249 Auranofin;250 thiosemicarbazones;242 Benzimidazol-2-ylidene;251N,N′-disubstituted cyclic thiourea252 |
| AuI and AuIII | Glyoxaldehyde-bis(thiosemicarbazones)253 |
| AgI | Carbene derivatives;249 silver-nanoparticles254 |
| PtII | Nedaplatin255 |
| PtIV | cis-[Pt(NH3)2Cl4] and trans-[PtCl2(CN)4]2−;256 |
| Pd | K2PdCl4, and K2PdCl6;257 |
| RuII | Carbene derivatives258 |
| CrVI | Chromates259 |
1.3. Metallodrug delivery and activation
Platinum(IV) complexes of the type [Pt(OH)2(N3)2(amine1)(amine2)] have strong azide-to-PtIV charge-transfer bands, are stable in the dark, and towards the intracellular reducing agent glutathione.263 After short treatment and short irradiation times (e.g. 1 h), they react rapidly with DNA bases such as guanine and are potently cytotoxic. Interestingly, the trans diam(m)ine diazido complexes appear to be more effective as photoactivatable anticancer agents than the cis isomers (Fig. 7).264 These complexes are also more effective than cisplatin when used under conditions appropriate for clinical phototherapeutic drugs (short treatment times, short irradiation times).
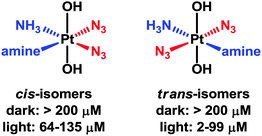 | ||
| Fig. 7 Photocytotoxicity of trans-diam(m)ine PtIV diazido complexes versus their cis-isomers, with associated IC50 values. | ||
The DNA lesions are unusual and include interstrand crosslinks. Trans,trans,trans-[Pt(OH)2(N3)2(NH3)(py)] is active in vivo on activation with blue light in an oesophageal cancer model.265Trans,trans,trans-[Pt(OH)2(N3)2(py)2] (py = pyridine) undergoes photoreduction when irradiated by UVA, blue or green light,266 and also produces azidyl radicals which can be quenched by L-Trp.267 Therefore these diazido complexes may have a dual mechanism of action involving the production of reactive PtII and radicals from the PtIV prodrug. Unlike conventional photosensitizers (which convert 3O2 to excited 1O2: photodynamic therapy), these complexes would not rely of O2 for activity, a potential advantage since tumours are often relatively hypoxic.
Other metal complexes are potent light activated anticancer agents. For instance, strained octahedral tris-bipyridyl ruthenium(II) complexes have been shown to be inert until triggered by visible light.268 Under irradiation, a light-activated ligand release mechanism occurs, leading to DNA binding, and to an increase in cytotoxicity of 2 orders of magnitude in cancer cells (with potencies superior to cisplatin against 3D tumour spheroids). The use of intramolecular strain is a promising strategy for developing light-activated Ru complexes for PDT applications.
CO is also a natural signalling molecule which can be released from a metal centre by light activation,272 and metal carbonyls have been extensively investigated as potential CO-donating pharmaceuticals.273,274In vivo, CO appears to have a role as a messenger, has anti-inflammatory properties and an ability to suppress organ graft rejection. The design of metal complexes that can release CO at a predicable rate is therefore valuable as a relatively non-toxic source of CO. Cell viability studies of HT29 colon cancer cells treated with the CO-releasing compound [Mn(CO)3(tpm)]PF6 (tpm = tris(pyrazolyl)methane) have revealed a significant photoinduced cytotoxicity, comparable to that of the established anticancer agent 5-fluorouracil.275
A large number of carrier systems have been developed in recent years, spanning functionalised carbon nanotubes, nanorods, metal–organic frameworks, metalla-cages, nanoparticles, liposomes, nanogels, proteins, and polymers. This wide diversity of systems offers an interesting pool of carriers, and each system possesses unique advantages.260 The delivery system can also influence the activity of metallodrugs. For example, a PtIV complex has been recently tethered via amide linkages to gold nanoparticles (AuNPs) functionalized with thiolated oligonucleotides. The resulting systems exhibit 12-fold higher activity than free cisplatin towards A549 lung cancer cells.278 The delivery of a lethal dose of cisplatin to prostate cancer cells by the encapsulation of PtIV prodrug in prostate-specific membrane nanoparticles (NPs) has also been recently reported.279 The utilisation of prostate-specific membrane antigen to target aptamers overexpressed in tumour cells, allows an specific delivery of the active drug into cancer cells. The release of the PtIV complex from the nanoparticles is followed by reduction to PtII (cisplatin), which can subsequently form 1,2-d(GpG) intrastrand cross-links on nuclear DNA.
Ruthenium metalla-cages also show promise as drug delivery systems. Supramolecular metalla-prisms based on arene ruthenium complexes can encapsulate square-planar acetylacetonato PdII and PtII complexes in their cavities and deliver them into cells, (e.g. [19]6+, see Chart 9).280,281 The activity of these carceplexes towards human ovarian cancer cell lines is more than an order of magnitude higher than the empty metalla-cage.281 Metalla-prisms can be synthesised with larger portal sizes (e.g. [20]6+, [21]6+, and [22]6+, see Chart 9).282,283 The guest can be released from such metalla-cages without rupture of the cage,284–288 and the extent of drug release correlates with the portal size of the cage.289 The host–guest capability of these systems has been used for encapsulating hydrophobic pyrenyl-cycloplatinate complexes,290 pyrenyl-containing dendrimers of different generations, for targeting cancer cells via the EPR effect,291–293 and for incorporating guests having affinity for G-quadruplex DNA.294 They can also be used as vehicles for intracellular delivery of photosensitisers, with possible use in photodynamic therapy.295 These systems being robust, highly water-soluble, and versatile are promising for drug delivery.
![Molecular structures of metalla-cages [19]6+, [20]6+, [21]6+, and [22]6+.](/image/article/2013/CC/c3cc41143e/c3cc41143e-c9.gif) | ||
| Chart 9 Molecular structures of metalla-cages [19]6+, [20]6+, [21]6+, and [22]6+. | ||
2. Anti-viral, anti-microbial and anti-diabetic metallodrugs
2.1. Anti-viral metallodrugs
000 people die from hepatitis.296 This disease can be treated by combination therapy involving pegylated recombinant interferon and ribavirin. However, interferon is not always well tolerated, some HCV genotypes respond better to interferon than others, and both interferon and ribavirin are not selective for HCV or viral disease in general.297 Moreover, the absence of an effective vaccine is stimulating the search for new drugs. A possible strategy for such development is to target specifically the critical RNA sequences present in hepatitis C virus but rare in the genome of infected host cells, For instance, the synthesis of a catalytic metallodrug that targets stem-loop IIb of the internal ribosomal entry site (IRES) RNA of hepatitis C virus has been recently reported.298 This metal complex incorporates an amino-terminal copper and nickel binding motif (ATCUN) that is found naturally at the N-terminus of many albumins299 as well as natural peptides such as histatin 5300 and neuromedin,301 and which binds copper and nickel with very high affinity.302 The ATCUN ligand provides a stabilisation of the redox states Cu(III)/Cu(II) and prevents the formation of the labile Cu(I) state. A C-terminal tetrapeptide (YrFK-amide) targeting domain is also coordinated to the metal centre in order to provide a selective recognition of the HCV IRES stem-loop IIb domain (Chart 10). Interestingly, neither the targeting peptide alone YrFK-amide, lacking the metal binding ATCUN motif, nor the metal binding ATCUN domain alone show any cellular efficacy. However, the complex reacts in vitro with the HCV IRES stem-loop IIb domain and inactivates catalytically and irreversibly the replication of hepatitis C virus with a turnover number of about 32.298 Despite this low turnover number, this preliminary study represents a promising new approach to the design of anti-hepatitis drugs.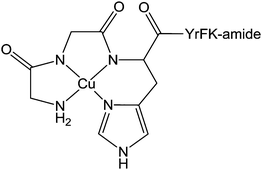 | ||
| Chart 10 Molecular structure of Cu(II)-YrFK-amide complex. | ||
The enzyme integrase (IN) catalyses the integration of viral DNA into the host cell DNA and is a particularly interesting target for HIV inhibitors. The active centre of the enzyme possesses two MgII ions held in place by a triad of protein carboxylate side chains, as well as an HHCC zinc finger site. Ligands which can chelate to the MgII ions might be effective enzyme inhibitors and drugs. Such a drug is raltegravir, approved by the FDA in 2007 (see Fig. 8). Despite its clinical success it suffers from an overall dose burden, and a lack of potency. To help to overcome rising raltegravir resistance, Cohen et al. have synthesised a series of raltegravir-chelator derivatives (RCD) as HIV integrase inhibitors in order to assess the role of the metal-binding group.304 The binding mode for this series of RCD molecules was elucidated by docking simulations, based on a crystal structure of prototype foamy virus (PFV) integrase in complex with raltegravir (PDB ID 3OYA). The docking of raltegravir and of RCD-1 (structure in Fig. 8) into PFV IN gives identical binding modes (Fig. 8), showing that the O,O,O donor atom triad of both raltegravir and RCD can bind to the MgII ions, forming 5- and 6-membered chelate rings.
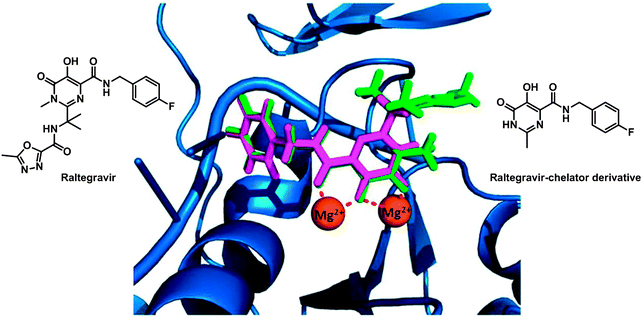 | ||
| Fig. 8 Molecular structures of raltegravir and RCD-1. Comparison of the computational docking of RCD-1 (magenta) in the PFV IN versus the reported crystal structure of raltegravir (green) bound to PFV IN (PDB ID 3OYA). Adapted from ref. 304. | ||
Carbamoyl pyridine scaffolds can also chelate two MgII ions in the active site of HIV-1 IN with nanomolar affinity.305 Carcelli et al. have shown that incorporation of RuII arene substituents can increase the potency of quinolone and hydroxypyrimidine-carboxamide HIV-1 IN inhibitors.306
An effective anti-HIV strategy is to target the membrane protein CXCR-4, a seven-helix transmembrane G-protein-coupled receptor and one of the several chemokine receptors that HIV uses to infect CD4+ T cells. The bis-macrocycle xylyl-bicyclam has potent anti-HIV activity but its use in the treatment of HIV was hindered by its lack of oral availability and cardiac disturbances.307 During clinical trials, its effectiveness in mobilising stem cells from the bone marrow was discovered, which led to clinical approval by the FDA in 2008 for this purpose (e.g. in transplant therapy) as the drug Mozobil.308
Metal ions such as ZnII and CuII bind to cyclam strongly309 and relatively rapidly,310 and it seems likely that metal complexation by xylyl-bicyclam is involved in the mechanism of action of the drug in vivo. The affinity of xylyl-bicyclam for the CXCR4 receptor is enhanced by factors of 7, 36, and 50 by incorporation of CuII, ZnII, or NiII, respectively, into the cyclam rings. Dizinc xylybicyclam tetraacetate forms the unusual folded cis-V configuration with an acetate carboxylate bound to ZnII on one side of the cyclam ring and acetate forming a double H-bond to cyclam NH groups on the other side of the ring.311 In a model of the Zn drug docked onto human CXCR4, a similar coordination can be achieved involving the carboxylates of Asp171, Asp162 and Glu288. Hydrophobic interactions involving Trp side chains and the periphery of the cyclam rings together with metal–carboxylate binding and H-bonding are observed in the X-ray crystal structure of CuII–cyclam and CuII2–bicyclam adducts of the protein lysozyme (Fig. 9).312
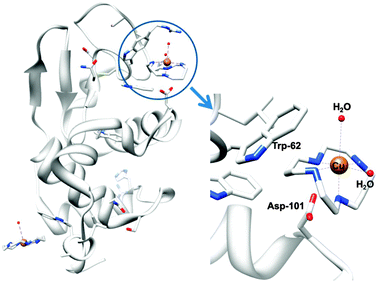 | ||
| Fig. 9 X-ray crystal structure of a CuII–cyclam adduct of lysozyme (PDB ID 1YIK). A different viewing point is adopted in the enlargement on the right side, for clarity. Adapted from ref. 312. | ||
Configurationally constrained metal–cyclam complexes have potential as even more potent antiHIV agents. For example, ZnII hexyl-dimethyl-cyclam complexes are active, but more active are Zn2((5-5′-[1,4-phenylenebis(methylene)]-bis(1,5,8,12-tetraazabicyclo[10.2.2]hexadecane))(OAc)4,313 and Ni2(1-[4-aminomethylbenzyl]-1,4,8,11-tetraazabicyclo-[10.2.2]hexadecane)(NO3)2.314 These studies illustrate that direct metal coordination, H-bonding and hydrophobic interactions with the ligands can all play important roles in metallodrug-protein target recognition.
2.2. Anti-microbial agents
000 among HIV-negative people and 430000 HIV-associated TB deaths).315 Moreover, 27000 cases of extremely drug-resistant TB in both developed and underdeveloped countries were reported in 2005; The most recently developed anti-tuberculosis drug is more than 40 years old. New anti-tuberculosis drugs with different biological mechanisms are needed, and although metal complexes might provide efficient alternatives, very few examples of such complexes have been reported, and in most cases, their mechanisms of action have rarely been thoroughly studied.Nonetheless, some recent studies have demonstrated the potential of metal-based drugs for the development of anti-tuberculosis agents. Among them, the redox activation of isoniazid iron(II) complexes is of interest.316 Indeed, isoniazid has been used as a front-line drug in the treatment of TB, although resistant TB strains have limited its use. Isoniazid is a prodrug that needs to be activated by an electron transfer reaction. This reaction is catalysed by the catalase-peroxidase KatG, and leads to the formation of an intermediate isonicotinic acyl radical that promptly reacts with NADH, generating a NAD–isoniazid adduct. This adduct is an efficient inhibitor of the enoyl reductase enzyme InhA, a major target for anti-Mycobacterium tuberculosis agents.317
About 50% of isolated isoniazid-resistant strains have either a deletion or mutations in the katG gene and this KatG enzyme disruption (blocking electron transfer reactions) has been shown to be the major cause of isoniazid resistance.318 For overcoming such resistance, the coordination of isoniazid to metal complexes (e.g. cyanoferrates) might allow a rapid oxidation of the metal centre, triggering isoniazid activation intramolecularly, independently of the activation by KatG enzymes (see Fig. 10 for a possible mechanism of action). This hypothetical mechanism still needs to be supported by experimental evidence, but preliminary studies on an isoniazid-containing pentacyanoferrate complex show that an inner-sphere electron transfer reaction occurs between the metal and isoniazid, thereby activating this prodrug and overcoming the necessity for KatG. This metal complex inhibits both wild-type InhA and its isoniazid-resistant mutant InhA I21V, even in the absence of KatG and NADH, bypassing the enzymatic activation. Interestingly, the ruthenium analogue (isoniazid)pentacyanoruthenate(II) complex does not inhibit InhA, probably because its very high electrochemical potential is outside the biological range.
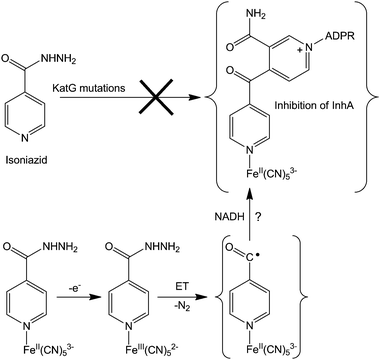 | ||
| Fig. 10 Possible mechanism for overcoming isoniazid resistance using an FeII complex. Based on ref. 316. | ||
A wide range of inorganic compounds from organometallic complexes to metal organic frameworks, nanoparticles or metallic surfaces have also been recently investigated for killing or inhibiting microbial growth, including mercury,322 silver,323 gold wires324 and nanoparticles,325 copper,326 cobalt,327 platinum,326 palladium,328 ruthenium,329 iron (chelation therapy),330,331 manganese complexes,332 titanium dioxide nanoparticles,333 noble metal nanoparticles,334,335 and nanostructures as antibacterial drug delivery systems.336
2.3. Anti-diabetic metallodrugs
Diabetes mellitus (DM) is a group of chronic metabolic diseases characterised by persistent hyperglycaemia associated with absolute or relative deficiency in insulin secretion from the beta cells of pancreas. The dysfunction of insulin receptors might also be associated with diabetes. The current (285 million patients worldwide) and dramatically-increasing incidence of this disease is stimulating the search for new anti-diabetic agents.337 Potential insulin mimetic metallodrugs have received much interest in the last two decades. The aim of developing new oral insulin mimetic drugs that act independently of insulin, for both insulin-dependent and insulin-independent diabetes, is to overcome side effects such as hyperinsulinemic hypoglycaemia due to current insulin preparations and synthetic drugs used in clinic.Zinc and vanadium complexes have been widely studied as potential metallopharmaceutics for treating diabetes mellitus. Zinc plays an essential role in the physiology, structure and function of insulin,338 and vanadium is also an essential element. The complex bis(maltolato)oxovanadium(IV) (BMOV, Chart 11), underwent phase II clinical trials but failed due to effects on the kidneys of patients. To reduce toxicity, Wei et al. have synthesised a vanadium complex which incorporates a modified pyranone derivative (BBOV, Chart 11).339 BBOV has low in vivo toxicity and oral administration of this complex to STZ-induced diabetic rats leads to a dramatic reduction of hyperglycaemia, along with an increase of the impaired glucose tolerance activity.
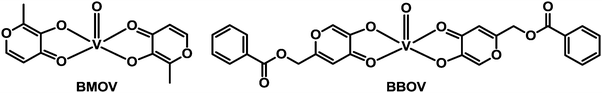 | ||
| Chart 11 Molecular structures of vanadyl anti-diabetic drug candidates BMOV and BBOV. | ||
A third metal of interest for development of anti-diabetic drugs is chromium, an element for which there is now no good evidence that it is essential.340,341Table 6 summarises recent examples of anti-diabetic CrIII complexes and their activity.
| CrIII ligands | Activity |
|---|---|
| Nicotinates, phenylalanines, histidinates | Decrease of blood glucose level342–345 |
| Polysaccharides | No positive effect346 |
| Picolinates | Improvement of serum lipid metabolism |
| Improvement of glucose metabolism347,348 | |
| Propionates | Amelioration of insulin resistance symptoms349 |
| Rutin, folate and stachyose ligands | Control of blood glucose350 |
| Malate | Control of blood glucose level, liver glycogen level, and of the activities of aspartate transaminase, alanine transaminase, and alkaline phosphatase351 |
Tetrahedral vanadate, molybdate, and tungstate complexes can compete with phosphate substrates for binding to phosphatases,352 and are potent inhibitors of muscle glycogen phosphorylases by competing with glucose-1-phosphate.353 Sodium tungstate Na2WO4 has been particularly investigated, due to the restoration of hepatic glucose metabolism by the stable oxoanion [WO4]2−.354 Moreover, [WO4]2− appears to mimic most of the metabolic effects of insulin and stimulate insulin output.355 The Keggin anion [PW12O40]3− has been also investigated for its ability to mimic insulin.356–358 Sodium molybdate Na2MoO4 is similarly effective for preventing or treating of diabetic mellitus in the early stages of the disease.359 However none of these compounds has yet received clinical approval as a new antidiabetic drug.
There is also increasing interest in the potential use of vanadium compounds for treating leishmaniasis, Chagas' disease and amoebiasis, and viral infections.360
3. Anti-parasitic, anti-inflammatory and anti-neurodegenerative metallodrugs
3.1. Anti-parasitic metallodrugs
Plasmodium falciparum is one of the five species of Plasmodium parasite that causes the most lethal form of malaria. Plasmodium falciparum is particularly sensitive to oxidative stress,361 and targeting of thioredoxin reductase is an approach that is being widely studied.362 Auranofin (Fig. 11) and a few related gold complexes strongly inhibit Plasmodium falciparum growth, probably due to a direct inhibition of Plasmodium falciparum thioredoxin reductase. In 2012, based on studies demonstrating that auranofin is a pro-drug giving rise to the active fragment {Au(PEt3)}+ (while the tetraacetylthioglucose ligand is excreted in vivo),363 molecular docking experiments364 were carried out and suggest that {Au(PEt3)}+ can bind to a N atom of an histidine in the active site of Plasmodium falciparum thioredoxin reductase, Fig. 11. This mode of binding to a protein His sidechain (despite the presence of Cys thiol sulfurs) was observed previously for {Au(PEt3)}+ in the X-ray structure of cyclophilin-3.365 Gold(I) also has a high affinity for selenocysteine which provides selectivity for inhibition of thioredoxin reductase over glutathione reductase.251
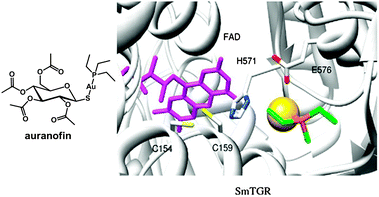 | ||
| Fig. 11 Molecular structure of auranofin. Docked conformation of {Au(PEt3)}+ in truncated SmTGR. FAD is in violet sticks. The gold atom is a yellow sphere. Residues interacting with the gold atom are in sticks. Adapted from ref. 364. | ||
Another approach for designing metallodrugs with potency against malaria is to coordinate biologically-active quinolone derivatives to metal centres. The ferrocene–quinoline conjugate ferroquine has been on clinical trials (Chart 12, Table 1).366
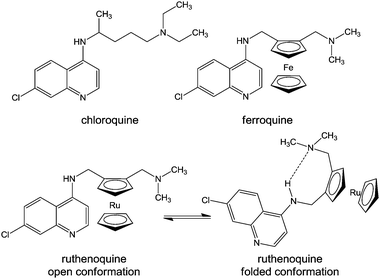 | ||
| Chart 12 Molecular structures of chloroquine, ferroquine, and ruthenoquine. The intramolecular hydrogen bond facilitating membrane permeability of ruthenoquine is shown. | ||
Chloroquine is thought to interfere with the digestion of haemoglobin in the blood stages of the malaria life cycle. Even though some similarities between the biological mode of action of chloroquine and ferroquine have been observed,367 ferroquine exhibits additional mechanisms.368 In particular, ferrocene can undergo a one-electron oxidation, yielding the ferrocenium cation, which may generate hydroxyl radicals under physiological solutions,369,370 leading to potential DNA371 and cell membrane damage.372 Moreover, the weaker base properties and higher lipophilicity at physiological pH of ferroquine compared to chloroquine, as well as intramolecular H-bonding with the lateral side chain of ferroquine (in non polar conditions) leads to the improved ability of ferroquine to cross membranes and a higher accumulation in the digestive vacuole.373
Ferroquine and ruthenoquine analogues possess slightly different mechanisms of action, Ruthenoquine does not produce reactive oxygen species (ROS) under the oxidative conditions of the parasitic digestive vacuole of Plasmodium falciparum.374 However, both compounds are active against drug-susceptible and drug-resistant strains of Plasmodium falciparum.375 Ruthenocenic derivatives of chloroquine with and without an intramolecular hydrogen bond have been used to study the localisation and quantification of ruthenoquine in Plasmodium falciparum-infected erythrocytes.374 This study suggested that the presence of intramolecular H-bonding (Chart 12) substantially improves the membrane permeability and transport of the drug to its target.
000 deaths a year.376 The World Health Organisation estimates that amoebiasis is the fourth leading cause of death due to protozoan infections (after malaria, Chagas disease, and trichomoniasis). Current medications for the treatment of amoebiasis are based on nitroimidazole derivatives, such metronidazole, and tinidazole. However, the appearance of resistant E. histolytica strains377 and side effects associated to these drugs, are strong limitations for this family of compounds. New drugs are therefore needed with new mechanisms of action.Metal complexes of active antiamoebiasis drugs have been investigated including AuI, RuII and CuII complexes of metronidazole ([1-(2-hydroxyethyl)-2-methyl-5-nitro-1H-imidazole]) which have higher activity than uncomplexed metronidazole.378 Cyclooctadiene RuII complexes of thiosemicarbazone derivatives are also more active than metronidazole.379 Recently, an automated, high-throughput screen has been developed to facilitate drug screening for Entamoeba histolytica.380 Interestingly, this screening identified that auranofin is 10× more potent against E. histolytica than metronidazole. The capability of this drug to inhibit E. histolytica thioredoxin reductase prevents the reduction of thioredoxin and enhances the sensitivity of trophozoites to reactive oxygen-mediated killing. This new use of auranofin represents a promising therapy for amoebiasis, and the drug has been granted orphan-drug status by the FDA (Table 1).
3.2. Anti-inflammatory metallodrugs
In the blood, the FeIII binding site of transferrin is also a strong site for BiIII.391 The recent 2.4 Å X-ray crystal structure of human transferrin with Bi in the N-lobe and Fe in the C-lobe shows Bi bound in a partially-opened cleft via only one of the two binding cleft Tyr side-chains together with nitrilotriacetate, carbonate and water.392
3.3. Neurodegenerative diseases
Neurodegenerative diseases (NDs), the progressive loss of structure or function of neurons – are attracting much attention due to the global increase of life expectancy in modern societies; 35 million people currently live with dementia; Alzheimer's disease (AD), Parkinson's disease (PD), and prion diseases (PrDs) are the most prevalent NDs.393 However the lack of knowledge of the origins, mechanisms, and development factors for these diseases results in poorly efficient drugs, which treat symptoms at best (for ADs and PDs, but no medication available for PrDs),394 and do not reverse or slow down disease progression.Senile plaques, neurofibrillary tangles, neutrophil threads, amyloid-β peptide (Aβ) deposition, selective loss of neurons and decreased synaptic density in post-mortem brains are the pathognomonic indicators of Alzheimer's disease. Aberrant metal biochemistry in the pathogenesis of AD has been demonstrated.395,396 Related to the synaptic activity, Cu and Zn are of importance, since millimolar concentrations of Zn are released upon neuronal activation, whilst Cu is involved in the regulation of synaptic functions. Amyloid-β peptide (Aβ) is released upon neuronal activation, and Aβ deposition and oxidative stress may be attributed to interactions between Aβ and metal ions. Bush et al. have recently reported the post-hoc analysis of a Phase IIa double-blind, randomised, placebo-controlled clinical trial for a copper/zinc ionophore, PBT2,397 an hydroxyquinoline derivative that facilitates the clearance of Aβ aggregates in the cortex by targeting the zinc and copper ions that mediate the assembly of these aggregates in amyloid and diffuse deposits, effectively detoxifying the Aβ.398 The output of this study shows clear improvement for treated patients compared to placebo group.397
In 2012, the bioinorganic chemistry of Alzheimer's disease was surveyed by Kepp,399 and the role that metal-Aβ association species play in AD was comprehensively reviewed by Pithadia and Lim.400 Inhibition of the interactions between Aβ and metal ions is a promising therapeutic approach for developing new and effective anti-AD agents. This inhibition may be achieved by utilisation of competitive agents (for occupying the metal binding site on Aβ), or by using chelating agents. Chelation therapy is a powerful tool for metal depletion and excretion and has been extensively studied in recent years for the treatment of Wilson's disease and neurodegenerative diseases.401,402
Metal chelators, especially inhibitors of histone deacetylases (HDACs), are also of much interest in anticancer therapy, as exemplified by suberoylanilide hydroxamic acid (SAHA, vorinostat, Zolinza) which inhibits Zn(II)-dependent class I and class II histone deacetylases (HDACs). Histone acetylation plays a key role in controlling the affinity of histones for DNA and gene expression. Deacetylation of Lys on histones produces a positive charge on its side chain and increases DNA affinity.
In 2013, Telpoukhovskaia and Orvig, surveyed how coordination chemistry might play a role in anti-neurodegenerative drug development, by understanding the binding preferences of metal ions for key proteins involved in the propagation of NDs, and by using the tools of inorganic chemistry for investigating the competition of synthetic ligands with proteins for metal ions.403
4. Diagnostic and therapeutic radiopharmaceuticals
There is an increasing interest in the development of both diagnostic and therapeutic radiopharmaceuticals (Fig. 1), as illustrated by the large number of ongoing clinical trials worldwide (Table 1). The development of radiopharmaceuticals is aided by their rapid passage from the laboratory into the clinic, since very small doses are usually administered, posing a negligible toxicity hazard.Therapeutic radiopharmaceuticals are useful for delivering locally cytotoxic doses of ionising radiation. The radionuclides used emit β−-particles (electrons) or α-particles (used in targeted α-therapy (TAT)). Most radiotherapeutic nuclides in the clinic are β− emitters, such 32P, 47Sc, 64Cu, 67Cu, 89Sr, 90Y, 105Rh, 111Ag, 117mSn, 131I, 149Pm, 153Sm, 166Ho, 177Lu, 186Re, 188Re.404 Recent advances in this field include the first clinical trial using an α-particle emitting 225Ac complex, labelled with a humanised antibody, lintuzumab, which targets the CD33 antigen expressed on the blast cells of most cases of acute myeloid leukaemia. The phase I clinical trial was initiated by the Scheinberg group at Memorial Sloan-Kettering Cancer Center with a primary goal to define both safety and the maximum tolerated dose of 225Ac TAT in patients with advanced myeloid leukaemia (AML) through a dose escalation series.405 Promising results have led to the ongoing phase I/II clinical trial, sponsored by Actinium Pharmaceuticals (Table 1). This complex is also undergoing a phase I clinical trial for the treatment of leukaemia myelodysplastic syndrome. The Scheinberg group reported the first proof-of-concept 213Bi TAT clinical trial, again targeting CD33 with antibody lintuzumab to treat 18 patients with advanced myeloid leukaemia in a phase I trial.406 This compound is now undergoing a phase II clinical trials (Table 1). Jurcic et al. also conducted a follow up study with 213Bi TAT, wherein 13 newly diagnosed patients and 18 patients with relapsed/refractory acute myeloid leukaemia were first treated with continuous cytarabine (cytosine arabinoside) infusion for 5 days.407 They observed marrow blast reductions at all dose levels.
Two types of radioimaging are used in clinic: single-photon emission computed tomography (SPECT), and positron emission tomography (PET). SPECT is based on the utilisation of pharmaceuticals labeled with a γ-emitting radionuclide, while PET requires a radiopharmaceutical labeled with a positron β+-emitting radionuclide. Useful γ-emitting nuclides include 99mTc, 67Ga, 111In, and 201Tl; useful β+-emitting nuclides include 55Co, 64Cu, 66Ga, 68Ga, 82Rb, 86Y.404 Recent clinical developments of such radioimaging agents include for instance 68Ga-DOTA-TATE408 and 64Cu-ATSM for PET/CT scans409 (Table 1, and Chart 13 for the molecular structure of 68Ga-DOTA-TATE).
 | ||
| Chart 13 Molecular structure of 68Ga-DOTA-TATE. | ||
Multimodality imaging (positron emission tomography-computed tomography (PET-CT)) has been used in clinic for more than a decade, and is nowadays one of the main cancer imaging techniques.410 Following this clinical success, combined PET/magnetic resonance (MR) systems for clinical use have been recently developed. Ga-DOTATOC PET/MR imaging is feasible in patients, with a good diagnostic image quality (average visual rating PET/CT, 2.83; PET/MR, 2.08). Moreover, detectability of focal PET lesions is equivalent to PET/CT on a patient basis and organ-system basis.411 The clinical value of Ga-DOTATOC PET/MR with additional diagnostic MR protocols needs to be evaluated against PET/CT with multiphase contrast-enhanced CT protocols in future studies.
Conclusions
The exploration of the medical periodic table presents exciting challenges. Inorganic compounds, and metal complexes in particular, offer mechanisms of drug action that can be quite distinct from those of organic drugs. About 13 metal ions are essential for mammalian life, which can all be used in therapy. However, not only can essential metals be used, but also non-essential metals, a strategy that might be particularly important for fighting bacteria which are becoming increasingly resistant to organic drugs. Also radionuclides with suitable ligands offer targeted agents both for diagnosis and therapy. The ligands can play critical roles in the activity of all metallodrugs and diagnostic agents. It is important to identify the parts of metallodrugs which are critical for activity (the pharmacophores).We have tried to focus here particularly on the discovery of new targets for metallodrugs. These days an understanding of targets and mechanisms of action is essential if a drug is to receive approval for clinical use. Such understanding will eventually become important when patients can be screened on a personal-medicine basis for the optimum drug to treat their particular conditions. There are signs that rapid progress is currently being made, aided by advances in metal analysis and especially speciation techniques, and by the methods of modern molecular biology (proteomics and genomics), which can be fruitfully applied to the identification of target sites and understanding of metabolic pathways.
Although many new targets are currently being proposed for metallodrugs, and provide promising new leads for development, few have been validated in vivo. Advances in controlling metallodrug activation are also needed so that metallodrugs reach target sites and do not undergo unwanted side-reactions. From a general point of view, the field will benefit greatly from the application of the drug design principles used for the development of organic drugs. In this sense, we have tried to highlight some recent and promising strategies. Specific G-quadruplex binders, metallodrugs with DNA sequence specificity, inert polypyridyl metal complexes specifically able to target kinases, catalytic metallodrugs, and multi-labeled nanoparticle drug delivery systems are striking examples of the new strategies that can be used for designing metal-based anticancer drug candidates. The potential of inorganic and organometallic complexes as antibiotics, and for the treatment of other diseases, such as viral and parasitic diseases is also apparent.
We also have highlighted some organic drugs which are targeted to metal ions, often to the metal in a metalloenzyme. Organic drugs are often designed so they contain H-bond acceptors (lone pair donors). These acceptor atoms are also potential metal binding sites but perhaps not always recognised as such.
There is an urgent need to find new and effective therapies for a wide range of diseases and conditions. As highlighted in Table 1, there is current clinical and industrial interest in such advances.
Acknowledgements
We thank the Swiss National Science Foundation (Grant No. PA00P2-145308 to NPEB), the ERC (Grant No. 247450 to PJS), EPSRC (Grant No. EP/F034210/1) and EC COST Action CM1105 for support. We also thank Roger Alberto, Enzo Alessio, Martin Brechbiel, Peter Caravan, Seth Cohen, Nicholas Farrell, Stephen Lippard, Neil Moore, Thomas O'Halloran, Christopher Orvig, Jonathan Sessler, and Holden Thorp for supplying information on current commercial and clinical developments, Thomas Ward for providing Fig. 4, and Eduardo Sousa for discussion of Fig. 10.References
- A. Mukherjee and P. J. Sadler, in Metals in Medicine: Therapeutic Agents in Wiley Encyclopedia of Chemical Biology, ed. T. P. Begley, John Wiley & Sons, Hoboken, 2009, vol. 3, pp. 80–126 Search PubMed.
- P. Caravan, in Metals in Medicine: Imaging Agents in Wiley Encyclopedia of Chemical Biology, ed. T. P. Begley, John Wiley & Sons, Hoboken, 2009, vol. 3, pp. 66–79 Search PubMed.
- P. Faller and C. Hureau, Chem.–Eur. J., 2012, 18, 15910–15920 CrossRef CAS.
- N. J. Farrer and P. J. Sadler, in Bioinorganic Medicinal Chemistry, ed. E. Alessio, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 1–47 Search PubMed.
- H. C. Harder and B. Rosenberg, Int. J. Cancer, 1970, 6, 207–216 CrossRef CAS.
- J. A. Howle and G. R. Gale, Biochem. Pharmacol., 1970, 19, 2757–2762 CrossRef CAS.
- J. M. Pascoe and J. J. Roberts, Biochem. Pharmacol., 1974, 23, 1345–1357 CrossRef CAS.
- R. J. Knox, F. Friedlos, D. A. Lydall and J. J. Roberts, Cancer Res., 1986, 46, 1972–1979 CAS.
- E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467–2498 CrossRef CAS.
- A. Eastman, Biochemistry, 1983, 22, 3927–3933 CrossRef CAS.
- A. L. Pinto and S. J. Lippard, Biochim. Biophys. Acta, Rev. Cancer, 1985, 780, 167–180 CrossRef CAS.
- A. Eastman, Biochemistry, 1986, 25, 3912–3915 CrossRef CAS.
- D. B. Zamble and S. J. Lippard, in Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug, ed. B. Lippert, VHCA, Verlag Helvetica Chimica Acta and Wiley-VCH, Zürich and Weinheim, 1999, pp. 71–110 Search PubMed.
- U.-M. Ohndorf and S. J. Lippard, in Structural aspects of Pt–DNA adduct recognition by proteins in DNA Damage Recognition, Taylor and Francis, New York, 2006, pp. 239–261 Search PubMed.
- A. Eastman, in Cisplatin, ed. B. Lippert, 1999, pp. 111–134 Search PubMed.
- S. Li, H. Huang, H. Liao, J. Zhan, Y. Guo, B.-Y. Zou, W.-Q. Jiang, Z.-Z. Guan and X.-Q. Yang, Int. J. Clin. Pharmacol. Ther., 2013, 51, 96–105 CAS.
- R. C. Todd and S. J. Lippard, J. Inorg. Biochem., 2010, 104, 902–908 CrossRef CAS.
- J. H. Y. Wong, J. A. Brown, Z. Suo, P. Blum, T. Nohmi and H. Ling, EMBO J., 2010, 29, 2059–2069 CrossRef CAS.
- A. Alt, K. Lammens, C. Chiocchini, A. Lammens, J. C. Pieck, D. Kuch, K.-P. Hopfner and T. Carell, Science, 2007, 318, 967–970 CrossRef CAS.
- G. E. Damsma, A. Alt, F. Brueckner, T. Carell and P. Cramer, Nat. Struct. Mol. Biol., 2007, 14, 1127–1133 CAS.
- P. M. Takahara, A. C. Rosenzweig, C. A. Frederick and S. J. Lippard, Nature, 1995, 377, 649–652 CrossRef CAS.
- A. Gelasco and S. J. Lippard, Biochemistry, 1998, 37, 9230–9239 CrossRef CAS.
- D. Yang, S. S. G. E. van Boom, J. Reedijk, J. H. van Boom and A. H. J. Wang, Biochemistry, 1995, 34, 12912–12920 CrossRef CAS.
- T. Reissner, S. Schneider, S. Schorr and T. Carell, Angew. Chem., Int. Ed., 2010, 49, 3077–3080 CrossRef CAS.
- L. G. Marzilli, J. S. Saad, Z. Kuklenyik, K. A. Keating and Y. Xu, J. Am. Chem. Soc., 2001, 123, 2764–2770 CrossRef CAS.
- C. J. van Garderen and L. P. A. van Houte, Eur. J. Biochem., 1994, 225, 1169–1179 CrossRef CAS.
- R. C. Todd and S. J. Lippard, Chem. Biol., 2010, 17, 1334–1343 CrossRef CAS.
- F. Coste, J.-M. Malinge, L. Serre, W. Shepard, M. Roth, M. Leng and C. Zelwer, Nucleic Acids Res., 1999, 27, 1837–1846 CrossRef CAS.
- H. Huang, L. Zhu, B. R. Reid, G. P. Drobny and P. B. Hopkins, Science, 1995, 270, 1842–1845 CAS.
- Y. Wu, D. Bhattacharyya, C. L. King, I. Baskerville-Abraham, S.-H. Huh, G. Boysen, J. A. Swenberg, B. Temple, S. L. Campbell and S. G. Chaney, Biochemistry, 2007, 46, 6477–6487 CrossRef CAS.
- D. Bhattacharyya, S. Ramachandran, S. Sharma, W. Pathmasiri, C. L. King, I. Baskerville-Abraham, G. Boysen, J. A. Swenberg, S. L. Campbell, N. V. Dokholyan and S. G. Chaney, PLoS One, 2011, 6, e23582 CAS.
- Y. Wu, P. Pradhan, J. Havener, G. Boysen, J. A. Swenberg, S. L. Campbell and S. G. Chaney, J. Mol. Biol., 2004, 341, 1251–1269 CrossRef CAS.
- B. Spingler, D. A. Whittington and S. J. Lippard, Inorg. Chem., 2001, 40, 5596–5602 CrossRef CAS.
- K. S. Lovejoy, R. C. Todd, S. Zhang, M. S. McCormick, J. A. D'Aquino, J. T. Reardon, A. Sancar, K. M. Giacomini and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8902–8907 CrossRef CAS.
- H. Baruah, M. W. Wright and U. Bierbach, Biochemistry, 2005, 44, 6059–6070 CrossRef CAS.
- V. Brabec and J. Kasparkova, Drug Resist. Updates, 2005, 8, 131–146 CrossRef CAS.
- J. W. Cox, S. J. Berners-Price, M. S. Davies, Y. Qu and N. Farrell, J. Am. Chem. Soc., 2001, 123, 1316–1326 CrossRef CAS.
- N. Farrell, T. G. Appleton, Y. Qu, J. D. Roberts, A. P. S. Fontes, K. A. Skov, P. Wu and Y. Zou, Biochemistry, 1995, 34, 15480–15486 CrossRef CAS.
- P. K. Wu, Y. Qu, B. Van Houten and N. Farrell, J. Inorg. Biochem., 1994, 54, 207–220 CrossRef CAS.
- S. J. Berners-Price, M. S. Davies, J. W. Cox, D. S. Thomas and N. Farrell, Chem.–Eur. J., 2003, 9, 713–725 CrossRef CAS.
- N. P. Farrell, S. G. De Almeida and K. A. Skov, J. Am. Chem. Soc., 1988, 110, 5018–5019 CrossRef CAS.
- C. Sessa, G. Capri, L. Gianni, F. Peccatori, G. Grasselli, J. Bauer, M. Zucchetti, L. Vigano, A. Gatti, C. Minoia, P. Liati, S. Van den Bosch, A. Bernareggi, G. Camboni and S. Marsoni, Ann. Oncol., 2000, 11, 977–983 CrossRef CAS.
- J. D. Roberts, J. Peroutka and N. Farrell, J. Inorg. Biochem., 1999, 77, 51–57 CrossRef CAS.
- C. Manzotti, G. Pratesi, E. Menta, R. D. Domenico, E. Cavalletti, H. H. Fiebig, L. R. Kelland, N. Farrell, D. Polizzi, R. Supino, G. Pezzoni and F. Zunino, Clin. Cancer Res., 2000, 6, 2626–2634 CAS.
- G. Pratesi, P. Perego, D. Polizzi, S. C. Righetti, R. Supino, C. Caserini, C. Manzotti, F. C. Giuliani, G. Pezzoni, S. Tognella, S. Spinelli, N. Farrell and F. Zunino, Br. J. Cancer, 1999, 80, 1912–1919 CrossRef CAS.
- P. Perego, C. Caserini, L. Gatti, N. Carenini, S. Romanelli, R. Supino, D. Colangelo, I. Viano, R. Leone, S. Spinelli, G. Pezzoni, C. Manzotti, N. Farrell and F. Zunino, Mol. Pharmacol., 1999, 55, 528–534 CAS.
- V. Brabec, J. Kasparkova, O. Vrana, O. Novakova, J. W. Cox, Y. Qu and N. Farrell, Biochemistry, 1999, 38, 6781–6790 CrossRef CAS.
- N. Farrell, Met. Ions Biol. Syst., 2004, 42, 251–296 CAS.
- A. Hegmans, S. J. Berners-Price, M. S. Davies, D. S. Thomas, A. S. Humphreys and N. Farrell, J. Am. Chem. Soc., 2004, 126, 2166–2180 CrossRef CAS.
- M. S. Davies, D. S. Thomas, A. Hegmans, S. J. Berners-Price and N. Farrell, Inorg. Chem., 2002, 41, 1101–1109 CrossRef CAS.
- U.-M. Ohndorf, M. A. Rould, Q. He, C. O. Pabo and S. J. Lippard, Nature, 1999, 399, 708–712 CrossRef CAS.
- N. J. Wheate and J. G. Collins, Coord. Chem. Rev., 2003, 241, 133–145 CrossRef CAS.
- N. J. Wheate and J. G. Collins, Curr. Med. Chem.: Anti-Cancer Agents, 2005, 5, 267–279 CrossRef CAS.
- J. B. Mangrum and N. P. Farrell, Chem. Commun., 2010, 46, 6640–6650 RSC.
- P. D. Braddock, T. A. Connors, M. Jones, A. R. Khokhar, D. H. Melzack and M. L. Tobe, Chem.–Biol. Interact., 1975, 11, 145–161 CrossRef CAS.
- V. H. Bramwell, D. Crowther, S. O'Malley, R. Swindell, R. Johnson, E. H. Cooper, N. Thatcher and A. Howell, Cancer Treat. Rep., 1985, 69, 409–416 CAS.
- P. J. Creaven, L. Pendyala and S. Madajewicz, Drugs Exp. Clin. Res., 1986, 12, 287–292 CAS.
- H. Anderson, J. Wagstaff, D. Crowther, R. Swindell, M. J. Lind, J. McGregor, M. S. Timms, D. Brown and P. Palmer, Eur. J. Cancer Clin. Oncol., 1988, 24, 1471–1479 CrossRef CAS.
- P. D. Bonomi, D. M. Finkelstein, J. C. Ruckdeschel, R. H. Blum, M. D. Green, B. Mason, R. Hahn, D. C. Tormey, J. Harris and R. Comis, J. Clin. Oncol., 1989, 7, 1602–1613 CAS.
- R. J. Schilder, F. P. LaCreta, R. P. Perez, S. W. Johnson, J. M. Brennan, A. Rogatko, S. Nash, C. McAleer, T. C. Hamilton and D. Roby, Cancer Res., 1994, 54, 709–717 CAS.
- F. Wang, H. Chen, S. Parsons, I. D. H. Oswald, J. E. Davidson and P. J. Sadler, Chem.–Eur. J., 2003, 9, 5810–5820 CrossRef CAS.
- A. Bergamo and G. Sava, Dalton Trans., 2007, 1267–1272 RSC.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CAS.
- P. J. Dyson and G. Sava, Dalton Trans., 2006, 1929–1933 RSC.
- C. S. Allardyce, P. J. Dyson, D. J. Ellis and S. L. Heath, Chem. Commun., 2001, 1396–1397 RSC.
- R. E. Morris, R. E. Aird, P. d. S. Murdoch, H. Chen, J. Cummings, N. D. Hughes, S. Parsons, A. Parkin, G. Boyd, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2001, 44, 3616–3621 CrossRef CAS.
- B. Wu, M. S. Ong, M. Groessl, Z. Adhireksan, C. G. Hartinger, P. J. Dyson and C. A. Davey, Chem.–Eur. J., 2011, 17, 3562–3566 CrossRef CAS.
- P. Nowak-Sliwinska, J. R. v. Beijnum, A. Casini, A. A. Nazarov, G. Wagnières, H. v. d. Bergh, P. J. Dyson and A. W. Griffioen, J. Med. Chem., 2011, 54, 3895–3902 CrossRef CAS.
- P. Govender, A. K. Renfrew, C. M. Clavel, P. J. Dyson, B. Therrien and G. S. Smith, Dalton Trans., 2011, 40, 1158–1167 RSC.
- G. S. Smith and B. Therrien, Dalton Trans., 2011, 40, 10793–10800 RSC.
- T. Bugarcic, A. Habtemariam, R. J. Deeth, F. P. A. Fabbiani, S. Parsons and P. J. Sadler, Inorg. Chem., 2009, 48, 9444–9453 CrossRef CAS.
- F. Barragan, P. Lopez-Senin, L. Salassa, S. Betanzos-Lara, A. Habtemariam, V. Moreno, P. J. Sadler and V. Marchan, J. Am. Chem. Soc., 2011, 133, 14098–14108 CrossRef CAS.
- A. Kisova, L. Zerzankova, A. Habtemariam, P. J. Sadler, V. Brabec and J. Kasparkova, Mol. Pharmaceutics, 2011, 8, 949–957 CrossRef CAS.
- M. Pernot, T. Bastogne, N. P. E. Barry, B. Therrien, G. Koellensperger, S. Hann, V. Reshetov and M. Barberi-Heyob, J. Photochem. Photobiol., B, 2012, 117, 80–89 CrossRef CAS.
- I. Romero-Canelón, L. Salassa and P. J. Sadler, J. Med. Chem., 2013, 56, 1291–1300 CrossRef.
- I. Romero-Canelon, A. M. Pizarro, A. Habtemariam and P. J. Sadler, Metallomics, 2012, 4, 1271–1279 RSC.
- A. Vacca, M. Bruno, A. Boccarelli, M. Coluccia, D. Ribatti, A. Bergamo, S. Garbisa, L. Sartor and G. Sava, Br. J. Cancer, 2002, 86, 993–998 CrossRef CAS.
- L. Morbidelli, S. Donnini, S. Filippi, L. Messori, F. Piccioli, P. Orioli, G. Sava and M. Ziche, Br. J. Cancer, 2003, 88, 1484–1491 CrossRef CAS.
- G. Sava, E. Alessio, A. Bergamo and G. Mestroni, in Topics in Biological Inorganic Chemistry, ed. M. J. Clarke and P. J. Sadler, Springer, 1999, vol. 1, pp. 143–169 Search PubMed.
- S. Kapitza, M. Pongratz, M. A. Jakupec, P. Heffeter, W. Berger, L. Lackinger, B. K. Keppler and B. Marian, J. Cancer Res. Clin. Oncol., 2005, 131, 101–110 CrossRef CAS.
- M. Groessl, Y. O. Tsybin, C. G. Hartinger, B. K. Keppler and P. J. Dyson, JBIC, J. Biol. Inorg. Chem., 2010, 15, 677–688 CrossRef CAS.
- G. Süss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- H. Chen, J. A. Parkinson, R. E. Morris and P. J. Sadler, J. Am. Chem. Soc., 2002, 125, 173–186 CrossRef.
- H. Chen, J. A. Parkinson, S. Parsons, R. A. Coxall, R. O. Gould and P. J. Sadler, J. Am. Chem. Soc., 2002, 124, 3064–3082 CrossRef CAS.
- T. Sriskandakumar, H. Petzold, P. C. A. Bruijnincx, A. Habtemariam, P. J. Sadler and P. Kennepohl, J. Am. Chem. Soc., 2009, 131, 13355–13361 CrossRef CAS.
- F. Wang, J. Xu, A. Habtemariam, J. Bella and P. J. Sadler, J. Am. Chem. Soc., 2005, 127, 17734–17743 CrossRef CAS.
- H. Petzold, J. Xu and P. J. Sadler, Angew. Chem., Int. Ed., 2008, 47, 3008–3011 CrossRef CAS.
- A. L. Noffke, A. Habtemariam, A. M. Pizarro and P. J. Sadler, Chem. Commun., 2012, 48, 5219–5246 RSC.
- A. F. A. Peacock, A. Habtemariam, R. Fernandez, V. Walland, F. P. A. Fabbiani, S. Parsons, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Am. Chem. Soc., 2006, 128, 1739–1748 CrossRef CAS.
- A. F. A. Peacock, M. Melchart, R. J. Deeth, A. Habtemariam, S. Parsons and P. J. Sadler, Chem.–Eur. J., 2007, 13, 2601–2613 CrossRef CAS.
- Y. Fu, A. Habtemariam, A. M. B. H. Basri, D. Braddick, G. J. Clarkson and P. J. Sadler, Dalton Trans., 2011, 40, 10553–10562 RSC.
- S. H. v. Rijt, H. Kostrhunova, V. Brabec and P. J. Sadler, Bioconjugate Chem., 2011, 22, 218–226 CrossRef.
- Y. Fu, A. Habtemariam, A. M. Pizarro, S. H. v. Rijt, D. J. Healey, P. A. Cooper, S. D. Shnyder, G. J. Clarkson and P. J. Sadler, J. Med. Chem., 2010, 53, 8192–8196 CrossRef CAS.
- S. H. v. Rijt, A. Mukherjee, A. M. Pizarro and P. J. Sadler, J. Med. Chem., 2010, 53, 840–849 CrossRef.
- M. Hanif, A. A. Nazarov, C. G. Hartinger, W. Kandioller, M. A. Jakupec, V. B. Arion, P. J. Dyson and B. K. Keppler, Dalton Trans., 2010, 39, 7345–7352 RSC.
- N. P. E. Barry, F. Edafe, P. J. Dyson and B. Therrien, Dalton Trans., 2010, 39, 2816–2820 RSC.
- N. P. E. Barry, O. Zava, P. J. Dyson and B. Therrien, J. Organomet. Chem., 2012, 705, 1–6 CrossRef CAS.
- A. F. A. Peacock, S. Parsons and P. J. Sadler, J. Am. Chem. Soc., 2007, 129, 3348–3357 CrossRef CAS.
- S. H. van Rijt, A. F. A. Peacock, R. D. L. Johnstone, S. Parsons and P. J. Sadler, Inorg. Chem., 2009, 48, 1753–1762 CrossRef CAS.
- S. D. Shnyder, Y. Fu, A. Habtemariam, S. H. van Rijt, P. A. Cooper, P. M. Loadman and P. J. Sadler, MedChemComm, 2011, 2, 666–668 RSC.
- Y. Fu, M. J. Romero, A. Habtemariam, M. E. Snowden, L. Song, G. J. Clarkson, B. Qamar, A. M. Pizarro, P. R. Unwin and P. J. Sadler, Chem. Sci., 2012, 3, 2485–2494 RSC.
- R. Bessant, A. Steuer, S. Rigby and M. Gumpel, Rheumatology, 2003, 42, 1036–1043 CrossRef CAS.
- Z. Liu, A. Habtemariam, A. M. Pizarro, S. Fletcher, A. Kisova, O. Vrana, L. Salassa, P. Bruijnincx, G. Clarkson, V. Brabec and P. J. Sadler, J. Med. Chem., 2011, 54, 3011–3026 CrossRef CAS.
- S. Betanzos-Lara, Z. Liu, A. M. Pizarro, B. Qamar, A. Habtemariam and P. J. Sadler, Angew. Chem., Int. Ed., 2012, 51, 3897–3900 CrossRef CAS.
- H.-K. Liu and P. J. Sadler, Acc. Chem. Res., 2011, 44, 349–359 CrossRef CAS.
- R. Martinez and L. Chacon-Garcia, Curr. Med. Chem., 2005, 12, 127–151 CrossRef CAS.
- L. Salassa, Eur. J. Inorg. Chem., 2011, 4931–4947 CrossRef CAS.
- T. Mukherjee, J. C. Pessoa, A. Kumar and A. R. Sarkar, Dalton Trans., 2012, 41, 5260–5271 RSC.
- L.-J. Tang, X. Chen, Y.-N. Sun, J. Ye, J. Lu, Y. Han, X. Jiang, C.-C. Cheng, C.-C. He, P.-H. Qiu and X.-K. Li, J. Inorg. Biochem., 2011, 105, 1623–1629 CrossRef CAS.
- Z.-F. Chen, Y.-C. Liu, Y. Peng, X. Hong, H.-H. Wang, M.-M. Zhang and H. Liang, JBIC, J. Biol. Inorg. Chem., 2012, 17, 247–261 CrossRef CAS.
- J. C. Lima and L. Rodriguez, Anti-Cancer Agents Med. Chem., 2011, 11, 921–928 CrossRef CAS.
- H. Wu, J. Yuan, Y. Bai, G. Pan, H. Wang, J. Kong, X. Fan and H. Liu, Dalton Trans., 2012, 41, 8829–8838 RSC.
- A. Terenzi, L. Tomasello, A. Spinello, G. Bruno, C. Giordano and G. Barone, J. Inorg. Biochem., 2012, 117, 103–110 CrossRef CAS.
- P. Krishnamoorthy, P. Sathyadevi, A. H. Cowley, R. R. Butorac and N. Dharmaraj, Eur. J. Med. Chem., 2011, 46, 3376–3387 CrossRef CAS.
- V. M. Manikandamathavan, V. Rajapandian, A. J. Freddy, T. Weyhermüller, V. Subramanian and B. U. Nair, Eur. J. Med. Chem., 2012, 57, 449–458 CrossRef CAS.
- F. Arjmand, S. Parveen, M. Afzal, L. Toupet and T. Ben Hadda, Eur. J. Med. Chem., 2012, 49, 141–150 CrossRef CAS.
- R. Wai-Yin Sun, A. Lok-Fung Chow, X.-H. Li, J. J. Yan, S. Sin-Yin Chui and C.-M. Che, Chem. Sci., 2011, 2, 728–736 RSC.
- J. Moretto, B. Chauffert, F. Ghiringhelli, J. R. Aldrich-Wright and F. Bouyer, Invest. New Drugs, 2011, 29, 1164–1176 CrossRef CAS.
- J. Liu, C.-H. Leung, A. L.-F. Chow, R. W.-Y. Sun, S.-C. Yan and C.-M. Che, Chem. Commun., 2011, 47, 719–721 RSC.
- G. Marverti, A. Ligabue, M. Montanari, D. Guerrieri, M. Cusumano, M. L. DiPietro, L. Troiano, E. DiVono, S. Iotti, G. Farruggia, F. Wolf, M. G. Monti and C. Frassineti, Invest. New Drugs, 2011, 29, 73–86 CrossRef CAS.
- N. Shahabadi and L. Nemati, DNA Cell Biol., 2012, 31, 883–890 CrossRef CAS.
- S. Ramakrishnan, E. Suresh, A. Riyasdeen, M. A. Akbarsha and M. Palaniandavar, Dalton Trans., 2011, 40, 3245–3256 RSC.
- Z. Liu, A. Habtemariam, A. M. Pizarro, S. A. Fletcher, A. Kisova, O. Vrana, L. Salassa, P. C. A. Bruijnincx, G. J. Clarkson, V. Brabec and P. J. Sadler, J. Med. Chem., 2011, 54, 3011–3026 CrossRef CAS.
- A. V. Vargiu and A. Magistrato, Inorg. Chem., 2012, 51, 2046–2057 CrossRef CAS.
- C. N. Sudhamania, H. S. B. Naika and D. Girija, Nucleosides, Nucleotides Nucleic Acids, 2012, 31, 130–146 Search PubMed.
- X.-L. Zhao, Y.-Z. Ma and K.-Z. Wang, J. Inorg. Biochem., 2012, 113, 66–76 CrossRef CAS.
- S. P. Foxon, C. Green, M. G. Walker, A. Wragg, H. Adams, J. A. Weinstein, S. C. Parker, A. J. H. M. Meijer and J. A. Thomas, Inorg. Chem., 2011, 51, 463–471 CrossRef.
- X.-L. Zhao, M.-J. Han, A.-G. Zhang and K.-Z. Wang, J. Inorg. Biochem., 2012, 107, 104–110 CrossRef CAS.
- S. Komeda, T. Moulaei, K. K. Woods, M. Chikuma, N. P. Farrell and L. D. Williams, J. Am. Chem. Soc., 2006, 128, 16092–16103 CrossRef CAS.
- K. R. Barnes and S. J. Lippard, in Metal Complexes in Tumor Diagnosis and as Anticancer Agents, ed. A. Sigel and H. Sigel, New York, 2004, vol. 42, pp. 143–177 Search PubMed.
- H. Baruah, C. G. Barry and U. Bierbach, Curr. Top. Med. Chem., 2004, 4, 1537–1549 CrossRef CAS.
- M. V. Keck and S. J. Lippard, J. Am. Chem. Soc., 1992, 114, 3386–3390 CrossRef CAS.
- W. J. Sundquist, D. P. Bancroft, L. Chassot and S. J. Lippard, J. Am. Chem. Soc., 1988, 110, 8559–8560 CrossRef CAS.
- H. Kostrhunova, J. Malina, A. J. Pickard, J. Stepankova, M. Vojtiskova, J. Kasparkova, T. Muchova, M. L. Rohlfing, U. Bierbach and V. Brabec, Mol. Pharmaceutics, 2011, 8, 1941–1954 CrossRef CAS.
- H. Chen, J. A. Parkinson, S. Parsons, R. A. Coxall, R. O. Gould and P. J. Sadler, J. Am. Chem. Soc., 2002, 124, 3064–3082 CrossRef CAS.
- O. Novakova, H. Chen, O. Vrana, A. Rodger, P. J. Sadler and V. Brabec, Biochemistry, 2003, 42, 11544–11554 CrossRef CAS.
- H.-K. Liu, S. J. Berners-Price, F. Wang, J. A. Parkinson, J. Xu, J. Bella and P. J. Sadler, Angew. Chem., Int. Ed., 2006, 45, 8153–8156 CrossRef CAS.
- M. Kang, A. Chouai, H. T. Chifotides and K. R. Dunbar, Angew. Chem., Int. Ed., 2006, 45, 6148–6151 CrossRef CAS.
- A. Frodl, D. Herebian and W. S. Sheldrick, J. Chem. Soc., Dalton Trans., 2002, 3664–3673 RSC.
- R. L. Williams, H. N. Toft, B. Winkel and K. J. Brewer, Inorg. Chem., 2003, 42, 4394–4400 CrossRef CAS.
- K. van der Schilden, F. Garcia, H. Kooijman, A. L. Spek, J. G. Haasnoot and J. Reedijk, Angew. Chem., Int. Ed., 2004, 43, 5668–5670 CrossRef CAS.
- N. H. Campbell, N. H. A. Karim, G. N. Parkinson, M. Gunaratnam, V. Petrucci, A. K. Todd, R. Vilar and S. Neidle, J. Med. Chem., 2011, 55, 209–222 CrossRef.
- R. W.-Y. Sun, C. K.-L. Li, D.-L. Ma, J. J. Yan, C.-N. Lok, C.-H. Leung, N. Zhu and C.-M. Che, Chem.–Eur. J., 2010, 16, 3097–3113 CrossRef CAS.
- K. Suntharalingam, D. Gupta, P. J. S. Miguel, B. Lippert and R. Vilar, Chem.–Eur. J., 2010, 16, 3613–3616 CrossRef CAS.
- E. Largy, F. Hamon, F. Rosu, V. Gabelica, E. De Pauw, A. Guédin, J.-L. Mergny and M.-P. Teulade-Fichou, Chem.–Eur. J., 2011, 17, 13274–13283 CrossRef CAS.
- J. E. Reed, A. J. P. White, S. Neidle and R. Vilar, Dalton Trans., 2009, 2558–2568 RSC.
- K. Suntharalingam, A. J. P. White and R. Vilar, Inorg. Chem., 2009, 48, 9427–9435 CrossRef CAS.
- K. Suntharalingam, A. J. P. White and R. Vilar, Inorg. Chem., 2009, 48, 9427–9435 CrossRef CAS.
- P. Wu, D.-L. Ma, C.-H. Leung, S.-C. Yan, N. Zhu, R. Abagyan and C.-M. Che, Chem.–Eur. J., 2009, 15, 13008–13021 CrossRef CAS.
- K. J. Castor, J. Mancini, J. Fakhoury, N. Weill, R. Kieltyka, P. Englebienne, N. Avakyan, A. Mittermaier, C. Autexier, N. Moitessier and H. F. Sleiman, ChemMedChem, 2012, 7, 85–94 CrossRef CAS.
- J.-T. Wang, Y. Li, J.-H. Tan, L.-N. Ji and Z.-W. Mao, Dalton Trans., 2011, 40, 564–566 RSC.
- L. Rao, J. D. Dworkin, W. E. Nell and U. Bierbach, J. Phys. Chem. B, 2011, 115, 13701–13712 CrossRef CAS.
- X.-H. Zheng, H.-Y. Chen, M.-L. Tong, L.-N. Ji and Z.-W. Mao, Chem. Commun., 2012, 48, 7607–7609 RSC.
- I. Manet, F. Manoli, M. P. Donzello, C. Ercolani, D. Vittori, L. Cellai, A. Masi and S. Monti, Inorg. Chem., 2011, 50, 7403–7411 CrossRef CAS.
- K. Suntharalingam, A. J. P. White and R. Vilar, Inorg. Chem., 2010, 49, 8371–8380 CrossRef CAS.
- H. Yang, V. P. Ma, D. S. Chan, H. Z. He, C. H. Leung and D. L. Ma, Curr. Med. Chem., 2013, 20, 576–582 CAS.
- C. Romera, O. Bombarde, R. Bonnet, D. Gomez, P. Dumy, P. Calsou, J.-F. Gwan, J.-H. Lin, E. Defrancq and G. Pratviel, Biochimie, 2011, 93, 1310–1317 CrossRef CAS.
- N. P. E. Barry, N. H. Abd Karim, R. Vilar and B. Therrien, Dalton Trans., 2009, 10717–10719 RSC.
- G. L. Liao, X. Chen, L. N. Ji and H. Chao, Chem. Commun., 2012, 48, 10781–10783 RSC.
- D. Sun, Y. Liu, D. Liu, R. Zhang, X. Yang and J. Liu, Chem.–Eur. J., 2012, 18, 4285–4295 CrossRef CAS.
- D. Sun, R. Zhang, F. Yuan, D. Liu, Y. Zhou and J. Liu, Dalton Trans., 2012, 41, 1734–1741 RSC.
- J. Sun, Y. An, L. Zhang, H.-Y. Chen, Y. Han, Y.-J. Wang, Z.-W. Mao and L.-N. Ji, J. Inorg. Biochem., 2011, 105, 149–154 CrossRef CAS.
- T. Wilson, M. P. Williamson and J. A. Thomas, Org. Biomol. Chem., 2010, 8, 2617–2621 CAS.
- S. Rickling, L. Ghisdavu, F. Pierard, P. Gerbaux, M. Surin, P. Murat, E. Defrancq, C. Moucheron and A. Kirsch-De Mesmaeker, Chem.–Eur. J., 2010, 16, 3951–3961 CrossRef CAS.
- R. I. Taleb, D. Jaramillo, N. J. Wheate and J. R. Aldrich-Wright, Chem.–Eur. J., 2007, 13, 3177–3186 CrossRef CAS.
- N. J. Wheate, R. I. Taleb, A. M. Krause-Heuer, R. L. Cook, S. Wang, V. J. Higgins and J. R. Aldrich-Wright, Dalton Trans., 2007, 5055–5064 RSC.
- I. Dieter-Wurm, M. Sabat and B. Lippert, J. Am. Chem. Soc., 1992, 114, 357–359 CrossRef CAS.
- S. K. Sharma and L. W. McLaughlin, J. Inorg. Biochem., 2004, 98, 1570–1577 CrossRef CAS.
- J. Clardy, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1826–1827 CrossRef CAS.
- L. A. Banaszynski, C. W. Liu and T. J. Wandless, J. Am. Chem. Soc., 2005, 127, 4715–4721 CrossRef CAS.
- M. Creus, A. Pordea, T. Rossel, A. Sardo, C. Letondor, A. Ivanova, I. LeTrong, R. E. Stenkamp and T. R. Ward, Angew. Chem., Int. Ed., 2008, 47, 1400–1404 CrossRef CAS.
- J. M. Zimbron, A. Sardo, T. Heinisch, T. Wohlschlager, J. Gradinaru, C. Massa, T. Schirmer, M. Creus and T. R. Ward, Chem.–Eur. J., 2010, 16, 12883–12889 CrossRef CAS.
- G. Gasser and N. Metzler-Nolte, in Bioinorganic Medicinal Chemistry, ed. E. Alessio, Wiley-VCH Verlag GmbH & Co. KGaA, 2011, pp. 351–382 Search PubMed.
- A. R. Timerbaev, C. G. Hartinger, S. S. Aleksenko and B. K. Keppler, Chem. Rev., 2006, 106, 2224–2248 CrossRef CAS.
- G. Sava, G. Jaouen, E. A. Hillard and A. Bergamo, Dalton Trans., 2012, 41, 8226–8234 RSC.
- A. Casini, A. Guerri, C. Gabbiani and L. Messori, J. Inorg. Biochem., 2008, 102, 995–1006 CrossRef CAS.
- A. Casini and L. Messori, Curr. Top. Med. Chem., 2011, 11, 2647–2660 CrossRef CAS.
- S. Rau and S. Zheng, Curr. Top. Med. Chem., 2012, 12, 197–209 CrossRef CAS.
- M. Groessl and P. J. Dyson, Curr. Top. Med. Chem., 2011, 11, 2632–2646 CrossRef CAS.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS.
- U. Jungwirth, C. R. Kowol, B. K. Keppler, C. G. Hartinger, W. Berger and P. Heffeter, Antioxid. Redox Signaling., 2011, 15, 1085–1127 CrossRef CAS.
- A. Casini, C. Hartinger, A. Nazarov and P. Dyson, in Medicinal Organometallic Chemistry, ed. G. Jaouen and N. Metzler-Nolte, Springer Berlin Heidelberg, 2010, vol. 32, pp. 57–80 Search PubMed.
- P. K. Sasmal, C. N. Streu and E. Meggers, Chem. Commun., 2013, 49, 1581–1587 RSC.
- A. L. Merkel, E. Meggers and M. Ocker, Expert Opin. Invest. Drugs, 2012, 21, 425–436 CrossRef CAS.
- E. Meggers, Angew. Chem., Int. Ed., 2011, 50, 2442–2448 CrossRef CAS.
- E. Meggers, Chem. Commun., 2009, 1001–1010 RSC.
- K. J. Kilpin and P. J. Dyson, Chem. Sci., 2013, 4, 1410–1419 RSC.
- M. Groessl and C. Hartinger, Anal. Bioanal. Chem., 2012, 1–18 Search PubMed.
- S. M. Meier, M. Hanif, W. Kandioller, B. K. Keppler and C. G. Hartinger, J. Inorg. Biochem., 2012, 108, 91–95 CrossRef CAS.
- A. Jemal, R. Siegel, E. Ward, T. Murray, J. Xu and M. J. Thun, Ca– Cancer J. Clin., 2007, 57, 43 CrossRef.
- R. Neri, E. Peets and A. Watnick, Biochem. Soc. Trans., 1979, 7, 565 CAS.
- A. U. Decensi, F. Boccardo, D. Guarneri, N. Positano, M. C. Paoletti, M. Costantini, G. Martorana and L. Giuliani, J. Urol., 1991, 146, 377 CAS.
- H. Tucker, J. W. Crook and G. J. Chesterson, J. Med. Chem., 1988, 31, 954 CrossRef CAS.
- T. Hara, J. Miyazaki, H. Araki, M. Yamaoka, N. Kanzaki, M. Kusaka and M. Miyamoto, Cancer Res., 2003, 63, 149 CAS.
- S. Top, C. l. Thibaudeau, A. Vessières, E. Brulé, F. Le Bideau, J.-M. Joerger, M.-A. Plamont, S. Samreth, A. Edgar, J. r. m. Marrot, P. Herson and G. r. Jaouen, Organometallics, 2009, 28, 1414–1424 CrossRef CAS.
- Y. L. K. Tan, P. Pigeon, S. Top, E. Labbe, O. Buriez, E. A. Hillard, A. Vessieres, C. Amatore, W. K. Leong and G. Jaouen, Dalton Trans., 2012, 41, 7537–7549 RSC.
- G. Jaouen, S. Top and A. Vessieres, in Bioorganometallics, ed. G. Jaouen, Wiley-VCH, Weinheim, 2006, pp. 65–95 Search PubMed.
- D. Hamels, P. M. Dansette, E. A. Hillard, S. Top, A. Vessières, P. Herson, G. Jaouen and D. Mansuy, Angew. Chem., Int. Ed., 2009, 48, 9124–9126 CrossRef CAS.
- P. Pigeon, S. Top, A. Vessières, M. Huché, E. A. Hillard, E. Salomon and G. Jaouen, J. Med. Chem., 2005, 48, 2814–2821 CrossRef CAS.
- G. Jaouen, S. Top, A. Vessieres, G. Leclercq and M. J. McGlinchey, Curr. Med. Chem., 2004, 11, 2505–2517 CrossRef CAS.
- G. Lummen, H. Sperling, H. Luboldt, T. Otto and H. Rubben, Cancer Chemother. Pharmacol., 1998, 42, 415 CrossRef CAS.
- N. Kröger, U. R. Kleeberg, K. Mross, L. Edler and D. K. Hossfeld, Onkologie, 2000, 23, 60 CrossRef.
- S. Top, E. B. Kaloun, A. Vessieres, I. Laios, G. Leclercq and G. Jaouen, J. Organomet. Chem., 2002, 565, 29–35 Search PubMed.
- O. R. Allen, L. Croll, A. L. Gott, R. J. Knox and P. C. McGowan, Organometallics, 2004, 23, 288 CrossRef CAS.
- L. Kater, J. Claffey, M. Hogan, P. Jesse, B. Kater, S. Strauß, M. Tacke and A. Prokop, Toxicol. In Vitro, 2012, 26, 119–124 CrossRef CAS.
- C. M. Dowling, S. Cuffe, M. Tacke, A. Deally, M. Hogan, J. M. Fitzpatrick and R. W. G. Watson, Lett. Drug Des. Discovery, 2012, 9, 226–233 CrossRef CAS.
- A. F. Armstrong and J. F. Valliant, Dalton Trans., 2007, 4240–4251 RSC.
- N. P. E. Barry and P. J. Sadler, Chem. Soc. Rev., 2012, 41, 3264–3279 RSC.
- N. P. E. Barry, R. J. Deeth, G. J. Clarkson, I. Prokes and P. J. Sadler, Dalton Trans., 2013, 42, 2580–2587 RSC.
- Y. Endo, T. Iijima, Y. Yamakoshi, M. Yamaguchi, H. Fukasawa and K. Shudo, J. Med. Chem., 1999, 42, 1501–1504 CrossRef CAS.
- M. Calvaresi and F. Zerbetto, J. Chem. Inf. Model., 2011, 51, 1882–1896 CrossRef CAS.
- K. H. Antman, Oncologist, 2001, 6, 1–2 CrossRef CAS.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2010, 54, 3–25 CrossRef.
- R. Sun, P. Board and A. Blackburn, Mol. Cancer, 2011, 10, 142 CrossRef CAS.
- S. J. Ralph, Met. Based Drugs, 2008, 2008, 260146 CrossRef.
- B. P. Rosen and Z. Liu, Environ. Int., 2009, 35, 512–515 CrossRef CAS.
- M. A. Elliott, S. J. Ford, E. Prasad, L. J. Dick, H. Farmer, P. J. Hogg and G. W. Halbert, Int. J. Pharm., 2012, 426, 67–75 CrossRef CAS.
- D. Park, J. Chiu, G. Perrone, P. Dilda and P. Hogg, Cancer Cell Int., 2012, 12, 11 CrossRef CAS.
- J. L. Hickey, R. A. Ruhayel, P. J. Barnard, M. V. Baker, S. J. Berners-Price and A. Filipovska, J. Am. Chem. Soc., 2008, 130, 12570–12571 CrossRef CAS.
- L. E. Wedlock, M. R. Kilburn, J. B. Cliff, L. Filgueira, M. Saunders and S. J. Berners-Price, Metallomics, 2011, 3, 917–925 RSC.
- S. P. Mulcahy, K. Grundler, C. Frias, L. Wagner, A. Prokop and E. Meggers, Dalton Trans., 2010, 39, 8177–8182 RSC.
- V. Pierroz, T. Joshi, A. Leonidova, C. Mari, J. Schur, I. Ott, L. Spiccia, S. Ferrari and G. Gasser, J. Am. Chem. Soc., 2012, 134, 20376–20387 CrossRef CAS.
- G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, Science, 2002, 298, 1912–1934 CrossRef CAS.
- E. Meggers, Curr. Opin. Chem. Biol., 2007, 11, 287–292 CrossRef CAS.
- E. Meggers, G. E. Atilla-Gokcumen, H. Bregman, J. Maksimoska, S. P. Mulcahy, N. Pagano and D. S. Williams, Synlett, 2007, 1177–1189 CrossRef CAS.
- J. Maksimoska, L. Feng, K. Harms, C. Yi, J. Kissil, R. Marmorstein and E. Meggers, J. Am. Chem. Soc., 2008, 130, 15764–15765 CrossRef CAS.
- J. Maksimoska, D. S. Williams, G. E. Atilla-Gokcumen, K. S. M. Smalley, P. J. Carroll, R. D. Webster, P. Filippakopoulos, S. Knapp, M. Herlyn and E. Meggers, Chem.–Eur. J., 2008, 14, 4816–4822 CrossRef.
- C.-H. Leung, H. Yang, V. P.-Y. Ma, D. S.-H. Chan, H.-J. Zhong, Y.-W. Li, W.-F. Fong and D.-L. Ma, MedChemComm, 2012, 3, 696–698 RSC.
- A. Wilbuer, D. H. Vlecken, D. J. Schmitz, K. Kräling, K. Harms, C. P. Bagowski and E. Meggers, Angew. Chem., Int. Ed., 2010, 49, 3839–3842 CrossRef CAS.
- E. Meggers, Curr. Opin. Chem. Biol., 2007, 11, 287–292 CrossRef CAS.
- A. Kastl, A. Wilbuer, A. L. Merkel, L. Feng, P. Di Fazio, M. Ocker and E. Meggers, Chem. Commun., 2012, 48, 1863–1865 RSC.
- F. Barragán, P. López-Senín, L. Salassa, S. Betanzos-Lara, A. Habtemariam, V. Moreno, P. J. Sadler and V. Marchán, J. Am. Chem. Soc., 2011, 133, 14098–14108 CrossRef.
- A. N. Bullock, S. Russo, A. Amos, N. Pagano, H. Bregman, J. É. Debreczeni, W. H. Lee, F. v. Delft, E. Meggers and S. Knapp, PLoS One, 2009, 4, e7112 Search PubMed.
- H. Wajant, K. Pfizenmaier and P. Scheurich, Cell Death Differ., 2003, 10, 45–65 CrossRef CAS.
- K. Chatzantoni and A. Mouzaki, Curr. Top. Med. Chem., 2006, 6, 1707–1714 CrossRef CAS.
- S. Madhusudan, S. R. Muthuramalingam, J. P. Braybrooke, S. Wilner, K. Kaur, C. Han, S. Hoare, F. Balkwill and T. S. Ganesan, J. Clin. Oncol., 2005, 23, 5950–5959 CrossRef CAS.
- M. L. Harrison, E. Obermueller, N. R. Maisey, S. Hoare, K. Edmonds, N. F. Li, D. Chao, K. Hall, C. Lee, E. Timotheadou, K. Charles, R. Ahern, D. M. King, T. Eisen, R. Corringham, M. DeWitte, F. Balkwill and M. Gore, J. Clin. Oncol., 2007, 25, 4542–4549 CrossRef CAS.
- C.-H. Leung, H.-J. Zhong, H. Yang, Z. Cheng, D. S.-H. Chan, V. P.-Y. Ma, R. Abagyan, C.-Y. Wong and D.-L. Ma, Angew. Chem., Int. Ed., 2012, 51, 9010–9014 CrossRef CAS.
- A. Meyer, A. Gutiérrez, I. Ott and L. Rodríguez, Inorg. Chim. Acta, 2013, 398, 72–76 CrossRef CAS.
- E. Schuh, C. Pflüger, A. Citta, A. Folda, M. P. Rigobello, A. Bindoli, A. Casini and F. Mohr, J. Med. Chem., 2012, 55, 5518–5528 CrossRef CAS.
- A. Casini and L. Messori, Curr. Top. Med. Chem., 2011, 11, 2647–2660 CrossRef CAS.
- R. Rubbiani, S. Can, I. Kitanovic, H. Alborzinia, M. Stefanopoulou, M. Kokoschka, S. Mönchgesang, W. S. Sheldrick, S. Wölfl and I. Ott, J. Med. Chem., 2011, 54, 8646–8657 CrossRef CAS.
- G. Boscutti, L. Feltrin, D. Lorenzon, S. Sitran, D. Aldinucci, L. Ronconi and D. Fregona, Inorg. Chim. Acta, 2012, 393, 304–317 CrossRef CAS.
- D. Mustacich and G. Powis, Biochem J., 2000, 346, 1–8 CrossRef CAS.
- A. S. Rahmanto and M. J. Davies, IUBMB Life, 2012, 64, 863–871 CrossRef CAS.
- E. M. Beauchamp and A. Üren, Vitam. Horm., 2012, 88, 333–354 CrossRef CAS.
- V. Branco, J. Canário, J. Lu, A. Holmgren and C. Carvalho, Free Radical Biol. Med., 2012, 52, 781–793 CrossRef CAS.
- W. Liu, K. Bensdorf, M. Proetto, U. Abram, A. Hagenbach and R. Gust, J. Med. Chem., 2011, 54, 8605–8615 CrossRef CAS.
- M. Pellei, V. Gandin, M. Marinelli, C. Marzano, M. Yousufuddin, H. V. R. Dias and C. Santini, Inorg. Chem., 2012, 51, 9873–9882 CrossRef CAS.
- F. Saccoccia, F. Angelucci, G. Boumis, M. Brunori, A. E. Miele, D. L. Williams and A. Bellelli, J. Inorg. Biochem., 2012, 108, 105–111 CrossRef CAS.
- R. Rubbiani, I. Kitanovic, H. Alborzinia, S. Can, A. Kitanovic, L. A. Onambele, M. Stefanopoulou, Y. Geldmacher, W. S. Sheldrick, G. Wolber, A. Prokop, S. Wölfl and I. Ott, J. Med. Chem., 2010, 53, 8608–8618 CrossRef CAS.
- K. Yan, C.-N. Lok, K. Bierla and C.-M. Che, Chem. Commun., 2010, 46, 7691–7693 RSC.
- J. Lessa, K. O. Ferraz, J. Guerra, L. Miranda, C. D. Romeiro, E. Souza-Fagundes, P. Barbeira and H. Beraldo, BioMetals, 2012, 25, 587–598 CrossRef CAS.
- M. Srivastava, S. Singh and W. T. Self, Environ. Health Perspect., 2012, 120, 56–61 CrossRef CAS.
- Y. Wang, H. Lu, D. Wang, S. Li, K. Sun, X. Wan, E. W. Taylor and J. Zhang, Toxicol. Appl. Pharmacol., 2012, 265, 342–350 CrossRef CAS.
- S. Huo, S. Shen, D. Liu and T. Shi, J. Phys. Chem. B, 2012, 116, 6522–6528 CrossRef CAS.
- S. Prast-Nielsen, M. Cebula, I. Pader and E. S. J. Arnér, Free Radical Biol. Med., 2010, 49, 1765–1778 CrossRef CAS.
- L. Oehninger, M. Stefanopoulou, H. Alborzinia, J. Schur, S. Ludewig, K. Namikawa, A. Munoz-Castro, R. W. Koster, K. Baumann, S. Wolfl, W. S. Sheldrick and I. Ott, Dalton Trans., 2013, 42, 1657–1666 RSC.
- J. M. Myers, W. E. Antholine and C. R. Myers, Toxicology, 2011, 281, 37–47 CrossRef CAS.
- J. S. Butler and P. J. Sadler, Cur. Opin. Chem. Biol, 2013, 17, 175–188 CrossRef CAS.
- L. Salassa, H. I. A. Phillips and P. J. Sadler, Phys. Chem. Chem. Phys., 2009, 11, 10311–10316 RSC.
- N. A. Smith and P. J. Sadler, Philos. Trans. R. Soc., A, 2013 DOI:10.1098/rsta.2013.0125.
- P. J. Bednarski, F. S. Mackay and P. J. Sadler, Anti-Cancer Agents Med. Chem., 2007, 7, 75–93 CrossRef CAS.
- N. J. Farrer, J. A. Woods, V. P. Munk, F. S. Mackay and P. J. Sadler, Chem. Res. Toxicol., 2009, 23, 413–421 CrossRef.
- A. F. Westendorf, J. A. Woods, K. Korpis, N. J. Farrer, L. Salassa, K. Robinson, V. Appleyard, K. Murray, R. Gruenert, A. M. Thompson, P. J. Sadler and P. J. Bednarski, Mol. Cancer Ther., 2012, 11, 1894–1904 CrossRef CAS.
- N. J. Farrer, J. A. Woods, L. Salassa, Y. Zhao, K. S. Robinson, G. Clarkson, F. S. Mackay and P. J. Sadler, Angew. Chem., Int. Ed., 2010, 49, 8905–8908 CrossRef CAS.
- J. S. Butler, J. A. Woods, N. J. Farrer, M. E. Newton and P. J. Sadler, J. Am. Chem. Soc., 2012, 134, 16508–16511 CrossRef CAS.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS.
- K. Bian and F. Murad, Front. Biosci., 2003, 8, D264–D278 CrossRef CAS.
- S. P. Fricker, Met. Ions Biol. Syst., 2004, 41, 421–480 CAS.
- F. Marquele-Oliveira, D. C. de Almeida Santana, S. F. Taveira, D. M. Vermeulen, A. R. Moraes de Oliveira, R. S. da Silva and R. F. V. Lopez, J. Pharm. Biol. Anal., 2010, 53, 843–851 CrossRef CAS.
- U. Schatzschneider, Inorg. Chim. Acta, 2011, 374, 19–23 CrossRef CAS.
- T. R. Johnson, B. E. Mann, J. E. Clark, R. Foresti, C. J. Green and R. Motterlini, Angew. Chem., Int. Ed., 2003, 42, 3722–3729 CrossRef CAS.
- R. Motterlini, B. E. Mann and R. Foresti, Expert Opin. Invest. Drugs., 2005, 14, 1305–1318 CrossRef CAS.
- J. Niesel, A. Pinto, H. W. Peindy-N'Dongo, K. Merz, I. Ott, R. Gust and U. Schatzschneider, Chem. Commun., 2008, 1798–1800 RSC.
- Y. Matsumura and H. Maeda, Cancer. Res., 1986, 46, 6387–6392 CAS.
- H. Maeda, Adv. Enzyme Regul., 2001, 41, 189–207 CrossRef CAS.
- S. Dhar, W. L. Daniel, D. A. Giljohann, C. A. Mirkin and S. J. Lippard, J. Am. Chem. Soc., 2009, 131, 14652–14653 CrossRef CAS.
- S. Dhar, F. X. Gu, R. Langer, O. C. Farokhzad and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17356–17361 CrossRef CAS.
- B. Therrien, G. Süss-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, Angew. Chem., Int. Ed., 2008, 47, 3773–3776 CrossRef CAS.
- O. Zava, J. Mattsson, B. Therrien and P. J. Dyson, Chem.–Eur. J., 2010, 16, 1428–1431 CrossRef CAS.
- N. P. E. Barry and B. Therrien, Eur. J. Inorg. Chem., 2009, 4695–4700 CrossRef CAS.
- J. Freudenreich, N. P. E. Barry, G. Süss-Fink and B. Therrien, Eur. J. Inorg. Chem., 2010, 2400–2405 CrossRef CAS.
- N. P. E. Barry, O. Zava, P. J. Dyson and B. Therrien, Aust. J. Chem., 2010, 63, 1529–1537 CrossRef CAS.
- N. P. E. Barry, O. Zava, J. Furrer, P. J. Dyson and B. Therrien, Dalton Trans., 2010, 39, 5272–5277 RSC.
- N. P. E. Barry, F. Edafe and B. Therrien, Dalton Trans., 2011, 40, 7172–7180 RSC.
- L. E. H. Paul, B. Therrien and J. Furrer, Inorg. Chem., 2011, 51, 1057–1067 CrossRef.
- L. H. Paul, B. Therrien and J. Furrer, JBIC, J. Biol. Inorg. Chem., 2012, 17, 1053–1062 CrossRef CAS.
- N. P. E. Barry, O. Zava, P. J. Dyson and B. Therrien, Chem.–Eur. J., 2011, 17, 9669–9677 CrossRef CAS.
- N. P. E. Barry, O. Zava, W. Wu, J. Zhao and B. Therrien, Inorg. Chem. Commun., 2012, 18, 25–28 CrossRef CAS.
- A. Pitto-Barry, N. P. E. Barry, O. Zava, R. Deschenaux, P. J. Dyson and B. Therrien, Chem.–Eur. J., 2011, 17, 1966–1971 CrossRef CAS.
- A. Pitto-Barry, N. P. E. Barry, O. Zava, R. Deschenaux and B. Therrien, Chem.–Asian J., 2011, 6, 1595–1603 CrossRef CAS.
- A. Pitto-Barry, O. Zava, P. J. Dyson, R. Deschenaux and B. Therrien, Inorg. Chem., 2012, 51, 7119–7124 CrossRef CAS.
- K. Suntharalingam, A. Łęczkowska, M. A. Furrer, Y. Wu, M. K. Kuimova, B. Therrien, A. J. P. White and R. Vilar, Chem.–Eur. J., 2012, 18, 16277–16282 CrossRef CAS.
- F. Schmitt, J. Freudenreich, N. P. E. Barry, L. Juillerat-Jeanneret, G. Süss-Fink and B. Therrien, J. Am. Chem. Soc., 2012, 134, 754–757 CrossRef CAS.
- World Health Organization, 2012, Fact_sheet_N164.
- T. Shimakami, R. E. Lanford and S. M. Lemon, Curr. Opin. Pharmacol., 2009, 9, 537–544 CrossRef CAS.
- S. Bradford and J. A. Cowan, Chem. Commun., 2012, 48, 3118–3120 RSC.
- B. Sarkar and Y. Wigfield, Biochem. Cell Biol., 1968, 46, 601–607 CrossRef CAS.
- J. Grogan, C. J. McKnight, R. F. Troxler and F. G. Oppenheim, FEBS Lett., 2001, 491, 76–80 CrossRef CAS.
- C. Harford and B. Sarkar, Biochem. Biophys. Res. Commun., 1995, 209, 877–882 CrossRef CAS.
- S.-J. Lau, T. P. A. Kruck and B. Sarkar, J. Biol. Chem., 1974, 249, 5878–5884 CAS.
- E. D. Clercq, Med. Res. Rev., 2002, 22, 531–565 CrossRef.
- A. Agrawal, J. DeSoto, J. L. Fullagar, K. Maddali, S. Rostami, D. D. Richman, Y. Pommier and S. M. Cohen, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2251–2256 CrossRef CAS.
- T. Kawasuji, B. A. Johns, H. Yoshida, T. Taishi, Y. Taoda, H. Murai, R. Kiyama, M. Fuji, T. Yoshinaga, T. Seki, M. Kobayashi, A. Sato and T. Fujiwara, J. Med. Chem., 2012, 55, 8735–8744 CrossRef CAS.
- M. Carcelli, A. Bacchi, P. Pelagatti, G. Rispoli, D. Rogolino, T. W. Sanchez, M. Sechi and N. Neamati, J. Inorg. Biochem., 2013, 118, 74–82 CrossRef CAS.
- D. Schols, J. A. Esté, G. Henson and E. D. Clercq, Antiviral Res., 1997, 35, 147–156 CrossRef CAS.
- E. De Clercq, Pharmacol. Ther., 2010, 128, 509–518 CrossRef CAS.
- R. M. Izatt, K. Pawlak, J. S. Bradshaw and R. L. Bruening, Chem. Rev., 1991, 91, 1721–1785 CrossRef CAS.
- S. J. Paisey and P. J. Sadler, Chem. Commun., 2004, 306–307 RSC.
- X. Liang, J. A. Parkinson, M. Weishäupl, R. O. Gould, S. J. Paisey, H.-s. Park, T. M. Hunter, C. A. Blindauer, S. Parsons and P. J. Sadler, J. Am. Chem. Soc., 2002, 124, 9105–9112 CrossRef CAS.
- T. M. Hunter, I. W. McNae, X. Liang, J. Bella, S. Parsons, M. D. Walkinshaw and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2288–2292 CrossRef CAS.
- G. C. Valks, G. McRobbie, E. A. Lewis, T. J. Hubin, T. M. Hunter, P. J. Sadler, C. Pannecouque, E. De Clercq and S. J. Archibald, J. Med. Chem., 2006, 49, 6162–6165 CrossRef CAS.
- R. Smith, D. Huskens, D. Daelemans, R. E. Mewis, C. D. Garcia, A. N. Cain, T. N. C. Freeman, C. Pannecouque, E. D. Clercq, D. Schols, T. J. Hubin and S. J. Archibald, Dalton Trans., 2012, 41, 11369–11377 RSC.
- World-Health-Organization, Global Tuberculosis Report, 2012.
- E. Sousa, L. Basso, D. Santos, I. Diógenes, E. Longhinotti, L. França Lopes and Í. Sousa Moreira, JBIC, J. Biol. Inorg. Chem., 2012, 17, 275–283 CrossRef CAS.
- R. Rawat, A. Whitty and P. J. Tonge, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13881–13886 CrossRef CAS.
- S. W. Yu, S. Girotto, C. Lee and R. S. Magliozzo, J. Biol. Chem., 2003, 278, 14769–14775 CrossRef CAS.
- N. C. Lloyd, H. W. Morgan, B. K. Nicholson and R. S. Ronimus, Angew. Chem., Int. Ed., 2005, 44, 941–944 CrossRef CAS.
- M. Patra, G. Gasser and N. Metzler-Nolte, Dalton Trans., 2012, 41, 6350–6358 RSC.
- J. E. Bandow and N. Metzler-Nolte, ChemBioChem, 2009, 10, 2847–2850 CrossRef CAS.
- N. K. Kaushik, A. Mishra, A. Ali, J. S. Adhikari, A. K. Verma and R. Gupta, JBIC, J. Biol. Inorg. Chem., 2012, 17, 1217–1230 CrossRef CAS.
- I. Chevrier, J. L. Sague, P. S. Brunetto, N. Khanna, Z. Rajacic and K. M. Fromm, Dalton Trans., 2013, 42, 217–231 RSC.
- M. Jongsma, F. H. Pelser, H. Mei, J. Atema-Smit, B. Belt-Gritter, H. Busscher and Y. Ren, Clin. Oral Investig., 2012, 1–10 Search PubMed.
- W. Wan and J. T. Yeow, J. Nanosci. Nanotechnol., 2012, 12, 4601–4606 CrossRef CAS.
- N. S. Ng, P. Leverett, D. E. Hibbs, Q. Yang, J. C. Bulanadi, M. J. Wu and J. R. Aldrich-Wright, Dalton Trans., 2013, 42, 3196–3209 RSC.
- A. Choudhary, R. Sharma and M. Nagar, Int. Res. J. Pharm. Pharmacol., 2011, 1, 172–187 Search PubMed.
- H. Khan, A. Badshah, G. Murtaz, M. Said, Z.-u. Rehman, C. Neuhausen, M. Todorova, B. J. Jean-Claude and I. S. Butler, Eur. J. Med. Chem., 2011, 46, 4071–4077 CrossRef CAS.
- A. I. Ramos, T. M. Braga and S. S. Braga, Mini–Rev. Med. Chem., 2012, 12, 227–235 CrossRef CAS.
- T. Zhou, Y. Ma, X. Kong and R. C. Hider, Dalton Trans., 2012, 41, 6371–6389 RSC.
- H. J. Vogel, Biochem. Cell Biol., 2012, 90, 233–244 CrossRef CAS.
- A. A. Osowole, I. Ott and O. M. Ogunlana, Int. J. Inorg. Chem., 2010 DOI:10.1155/2012/206417.
- L. Clément, C. Hurel and N. Marmier, Chemosphere, 2013, 90, 1083–1090 CrossRef.
- R. R. Arvizo, S. Bhattacharyya, R. A. Kudgus, K. Giri, R. Bhattacharya and P. Mukherjee, Chem. Soc. Rev., 2012, 41, 2943–2970 RSC.
- M. K. Rai, S. D. Deshmukh, A. P. Ingle and A. K. Gade, J. Appl. Microbiol., 2012, 112, 841–852 CrossRef CAS.
- A. Brandelli, Mini-Rev. Med. Chem., 2012, 12, 731–741 CrossRef CAS.
- American Diabetes Association, 2010, 33, S62–S69.
- M. C. Foster, R. D. Leapman, M. X. Li and I. Atwater, Biophys. J., 1993, 64, 525–532 CrossRef CAS.
- Y.-B. Wei and X.-D. Yang, Biometals, 2012, 25, 1261–1268 CrossRef CAS.
- K. Bona, S. Love, N. Rhodes, D. McAdory, S. Sinha, N. Kern, J. Kent, J. Strickland, A. Wilson, J. Beaird, J. Ramage, J. Rasco and J. Vincent, JBIC, J. Biol. Inorg. Chem., 2011, 16, 381–390 CrossRef.
- J. B. Vincent, Dalton Trans., 2010, 39, 3787–3794 RSC.
- D. S. Jennings, P. B. Brevard, J. A. Flohr and J. W. Gloeckner, J. Am. Diet. Assoc., 1997, 97, A65 CrossRef.
- X. Yang, K. Palanichamy, A. C. Ontko, M. N. A. Rao, C. X. Fang, J. Ren and N. Sreejayan, FEBS Lett., 2005, 579, 1458–1464 CrossRef CAS.
- X.-P. Yang, S.-Y. Li, F. Dong, J. Ren and N. Sreejayan, J. Inorg. Biochem., 2006, 100, 1187–1193 CrossRef CAS.
- A. Dogukan, M. Tuzcu, V. Juturu, G. Cikim, I. Ozercan, J. Komorowski and K. Sahin, J. Renal Nutr., 2010, 20, 112–120 CrossRef CAS.
- L. Zhang, Y. Cao, L.-L. Pen, Z.-J. Wen and Y.-S. Yang, J. Phys. Sci., 2002, 24, 69–71 Search PubMed.
- D.-S. Kim, T.-W. Kim, I.-K. Park, J.-S. Kang and A.-S. Om, Metabolism, 2002, 51, 589–594 CrossRef CAS.
- L. K. Trent and D. Tiedingcancel, J. Sports Med. Phys. Fitness, 1995, 35, 273–280 CAS.
- E. Król and Z. Krejpcio, Food Chem. Toxicol., 2010, 48, 2791–2796 CrossRef.
- F. Li, X. Wub, Y. Zou, T. Zhao, M. Zhang, W. Feng and L. Yang, Food Chem. Toxicol., 2012, 50, 1623–1631 CrossRef CAS.
- X.-Y. Wu, F. Li, T. Zhao, G.-H. Mao, J. Li, H.-Y. Qu, Y.-N. Ren and L.-Q. Yang, Biol. Trace Elem. Res., 2012, 148, 91–101 CrossRef CAS.
- D. Rehder, Coord. Chem. Rev., 1999, 182, 297–322 CrossRef.
- P. J. Stankiewicz and M. J. Gresser, Biochemistry, 1988, 27, 206–212 CrossRef CAS.
- A. Barbera, J. E. Rodriguez-Gil and J. J. Guinovart, J. Biol. Chem., 1994, 269, 20047–20053 CAS.
- S. Piquer, S. Barceló-Batllori, M. Julià, N. Marzo, B. Nadal, J. J. Guinovart and R. Gomis, Biochem. Biophys. Res. Commun., 2007, 358, 385–391 CrossRef CAS.
- S. Uskokovic-Markovic, M. Milenkovic, A. Topic, J. Kotur-Stevuljevic, A. Stefanovic and J. Antic-Stankovic, J. Pharm. Pharm. Sci., 2007, 10, 340–349 CAS.
- I. Holclajtner-Antunovic, D. Bajuk-Bogdanovic, M. Todorovic, U. Mioc, J. Zakrzewska and S. Uskokovic-Markovic, Can. J. Chem., 2008, 86, 996–1004 CrossRef CAS.
- A. Topic, M. Milenkovic, S. Uskokovic-Markovic and D. Vucicevic, Biol. Trace Elem. Res., 2010, 134, 296–306 CrossRef CAS.
- S. R. Panneerselvam and S. Govindasamy, Clin. Chim. Acta, 2004, 345, 93–98 CrossRef CAS.
- D. Rehder, Future Med. Chem., 2012, 4, 1823–1837 CrossRef CAS.
- A. R. Sannella, A. Casini, C. Gabbiani, L. Messori, A. R. Bilia, F. F. Vincieri, G. Majori and C. Severini, FEBS Lett., 2008, 582, 844–847 CrossRef CAS.
- T. Jaeger and L. Flohe, Biofactors, 2006, 27, 109–120 CrossRef.
- K. P. Bhabak, B. J. Bhuyan and G. Mugesh, Dalton Trans., 2011, 40, 2099–2111 RSC.
- A. Caroli, S. Simeoni, R. Lepore, A. Tramontano and A. Via, Biochem. Biophys. Res. Commun., 2012, 417, 576–581 CrossRef CAS.
- J. Zou, P. Taylor, J. Dornan, S. P. Robinson, M. D. Walkinshaw and P. J. Sadler, Angew. Chem., Int. Ed., 2000, 39, 2931–2934 CrossRef CAS.
- F. Dubar, J. Khalife, J. Brocard, D. Dive and C. Biot, Molecules, 2008, 13, 2900–2907 CrossRef CAS.
- C. Biot, D. Taramelli, I. Forfar-Bares, L. A. Maciejewski, M. Boyce, G. Nowogrocki, J. S. Brocard, N. Basilico, P. Olliaro and T. J. Egan, Mol. Pharmaceutics, 2005, 2, 185–193 CrossRef CAS.
- C. Biot, F. Nosten, L. Fraisse, D. Ter-Minassian, J. Khalife and D. Dive, Parasite, 2011, 18, 207–214 CrossRef CAS.
- N. Chavain, V. Vezin, D. Dive, N. Touati, J.-F. Paul, E. Buisine and C. Biot, Mol. Pharmaceutics, 2008, 5, 510–516 Search PubMed.
- D. Osella, M. Ferrali, P. Zanello, F. Laschi, M. Fontani, C. Nervi and G. Cavigiolio, Inorg. Chim. Acta, 2000, 306, 42–48 CrossRef CAS.
- H. Tamura and M. Miwa, Chem. Lett., 1997, 1177–1178 CrossRef CAS.
- M. Salmain and N. Metzler-Nolte, in Ferrocenes, ed. P. Stepnicka, John Wiley & Sons, Chichester, UK, 2008, pp. 499–639 Search PubMed.
- C. Biot, N. Chavain, F. Dubar, B. Pradines, X. Trivelli, J. Brocard, I. Forfar and D. Dive, J. Organomet. Chem., 2009, 694, 845–854 CrossRef CAS.
- C. Biot, F. Dubar, J. Khalife and C. Slomianny, Metallomics, 2012, 4, 780–783 RSC.
- F. Dubar, T. J. Egan, B. Pradines, D. Kuter, K. K. Ncokazi, D. Forge, J.-F. o. Paul, C. Pierrot, H. Kalamou, J. Khalife, E. Buisine, C. Rogier, H. Vezin, I. Forfar, C. Slomianny, X. Trivelli, S. Kapishnikov, L. Leiserowitz, D. Dive and C. Biot, ACS Chem. Biol., 2010, 6, 275–287 CrossRef.
- J. C. Garcia-Ramos, Y. Toledano-Magana, L. G. Talavera-Contreras, M. Flores-Alamo, V. Ramirez-Delgado, E. Morales-Leon, L. Ortiz-Frade, A. G. Gutierrez, A. Vazquez-Aguirre, C. Mejia, J. C. Carrero, J. P. Laclette, R. Moreno-Esparza and L. Ruiz-Azuara, Dalton Trans., 2012, 41, 10164–10174 RSC.
- P. L. Johanson, Parasitol. Today, 1993, 9, 183 CrossRef.
- F. Athar, K. Husain, M. Abid, S. M. Agarwal, S. J. Coles, M. B. Hursthouse, M. R. Maurya and A. Azam, Chem. Biodiversity, 2005, 2, 1320–1330 CAS.
- S. Singh, F. Athar, M. R. Maurya and A. Azam, Eur. J. Med. Chem., 2006, 41, 592–598 CrossRef CAS.
- A. Debnath, D. Parsonage, R. M. Andrade, C. He, E. R. Cobo, K. Hirata, S. Chen, G. Garcia-Rivera, E. Orozco, M. B. Martinez, S. S. Gunatilleke, A. M. Barrios, M. R. Arkin, L. B. Poole, J. H. McKerrow and S. L. Reed, Nat. Med., 2012, 18, 956–960 CrossRef CAS.
- J. E. Pope, O. Hong and B. E. Koehler, J. Rheumatol., 2002, 29, 255–260 Search PubMed.
- Z. Trávníček, P. Štarha, J. Vančo, T. Šilha, J. Hošek, P. Suchý and G. Pražanová, J. Med. Chem., 2012, 55, 4568–4579 CrossRef.
- S. Gunatilleke, C. Oliveira, J. A. McCammon and A. Barrios, JBIC, J. Biol. Inorg. Chem., 2008, 13, 555–561 CrossRef CAS.
- N. Yang and H. Sun, in Biological Chemistry of Arsenic, Antimony and Bismuth, ed. H. Sun, John Wiley & Sons, Chichester, 2011, pp. 53–81 Search PubMed.
- P. C. Andrews, M. Busse, G. B. Deacon, R. L. Ferrero, P. C. Junk, J. G. MacLellan and A. Vom, Dalton Trans., 2012, 41, 11798–11806 RSC.
- P. C. Andrews, R. L. Ferrero, P. C. Junk, J. G. Maclellan and R. M. Peiris, Aust. J. Chem., 2012, 65, 883–891 CrossRef CAS.
- N. Yang and H. Sun, Coord. Chem. Rev., 2007, 251, 2354–2366 CrossRef CAS.
- R. Urgesi, R. Cianci and M. E. Riccioni, Clin. Exp. Gastroenterol., 2012, 5, 151–157 CrossRef CAS.
- J. Liang, J. Li, Y. Han, J. Xia, Y. Yang, W. Li, S. Zhang, Y. Wu, Y. Yuan, Z. Li, Y. Du, M. Chen, B. Chen, B. Jiang, Y. Bai, Q. Wen, K. Wu and D. Fan, Helicobacter, 2012, 17, 458–465 CrossRef CAS.
- C.-N. Tsang, K.-S. Ho, H. Sun and W.-T. Chan, J. Am. Chem. Soc., 2011, 133, 7355–7357 CrossRef CAS.
- H. Sun, H. Li, A. B. Mason, R. C. Woodworth and P. J. Sadler, J. Biol. Chem., 2001, 276, 8829–8835 CrossRef CAS.
- N. Yang, H. Zhang, M. Wang, Q. Hao and H. Sun, Sci. Rep., 2012, 2, 999 Search PubMed.
- World-Health-Organization, Dementia—a public health priority, UK, 2012.
- A. Samii, J. G. Nutt and B. R. Ransom, Lancet, 2004, 363, 1783–1793 CrossRef CAS.
- V. B. Kenche and K. J. Barnham, Br. J. Pharmacol., 2011, 163, 211–219 CrossRef CAS.
- P. J. Crouch and K. J. Barnham, Acc. Chem. Res., 2012, 45, 1604–1611 CrossRef CAS.
- N. G. Faux, C. W. Ritchie, A. Gunn, A. Rembach, A. Tsatsanis, J. Bedo, J. Harrison, L. Lannfelt, K. Blennow, H. Zetterberg, M. Ingelsson, C. L. Masters, R. E. Tanzi, J. L. Cummings, C. M. Herd and A. I. Bush, J. Alzheimer’s Dis., 2010, 20, 509–516 CAS.
- A. I. Bush, J. Alzheimer’s Dis., 2008, 15, 223–240 CAS.
- K. P. Kepp, Chem. Rev., 2012, 112, 5193–5239 CrossRef CAS.
- A. S. Pithadia and M. H. Lim, Curr. Opin. Chem. Biol., 2012, 16, 67–73 CrossRef CAS.
- P. Delangle and E. Mintz, Dalton Trans., 2012, 41, 6359–6370 RSC.
- A. Budimir, Acta Pharm., 2011, 61, 1–14 CrossRef CAS.
- M. A. Telpoukhovskaia and C. Orvig, Chem. Soc. Rev., 2013, 42, 1836–1846 RSC.
- P. J. Sadler, C. Muncie and M. A. Shipman, in Biological Inorganic Chemistry: Structure and Reactivity, ed. I. Bertini, J. Stieffel, H. B. Gray and J. S. Valentine, University Science Books, Sausalito, 2007, pp. 95–136 Search PubMed.
- M. Miederer, M. R. McDevitt, G. Sgouros, K. Kramer, N.-K. V. Cheung and D. A. Scheinberg, J. Nucl. Med., 2004, 45, 129–137 CAS.
- T. L. Rosenblat, M. R. McDevitt, D. A. Mulford, N. Pandit-Taskar, C. R. Divgi, K. S. Panageas, M. L. Heaney, S. Chanel, A. Morgenstern, G. Sgouros, S. M. Larson, D. A. Scheinberg and J. G. Jurcic, Clin. Cancer Res., 2010, 16, 5303–5311 CrossRef CAS.
- J. G. Jurcic, S. M. Larson, G. Sgouros, M. R. McDevitt, R. D. Finn, C. R. Divgi, Å. M. Ballangrud, K. A. Hamacher, D. Ma, J. L. Humm, M. W. Brechbiel, R. Molinet and D. A. Scheinberg, Blood, 2002, 100, 1233–1239 CAS.
- D. Wild, J. B. Bomanji, P. Benkert, H. Maecke, P. J. Ell, J. C. Reubi and M. E. Caplin, J. Nucl. Med., 2013, 54, 364–372 CrossRef CAS.
- M. Bourgeois, H. Rajerison, F. Guerard, M. Mougin-Degraef, J. Barbet, N. Michel, M. Cherel and A. Faivre-Chauvet, Nucl. Med. Rev. Cent. East. Eur., 2011, 14, 90–95 CrossRef.
- F. C. Gaertner, S. Fürst and M. Schwaiger, Cancer Imaging, 2013, 13, 36–52 CrossRef.
- F. C. Gaertner, A. J. Beer, M. Souvatzoglou, M. Eiber, S. Fürst, S. I. Ziegler, F. Brohl, M. Schwaiger and K. Scheidhauer, Invest. Radiol., 2013, 48, 263–272 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3cc41143e |
| This journal is © The Royal Society of Chemistry 2013 |