 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organometallic anticancer complexes of lapachol: metal centre-dependent formation of reactive oxygen species and correlation with cytotoxicity†
Wolfgang
Kandioller
*ab,
Evelyn
Balsano
a,
Samuel M.
Meier
ab,
Ute
Jungwirth
bc,
Simone
Göschl
a,
Alexander
Roller
a,
Michael A.
Jakupec
ab,
Walter
Berger
bc,
Bernhard K.
Keppler
ab and
Christian G.
Hartinger
*d
aUniversity of Vienna, Institute of Inorganic Chemistry, Währinger Str. 42, 1090 Vienna, Austria. E-mail: wolfgang.kandioller@univie.ac.at; Fax: +43 1 4277 9526; Tel: +43 1 4277 52609
bUniversity of Vienna, Research Platform “Translational Cancer Therapy Research”, Währinger Str. 42, A-1090 Vienna, Austria
cMedical University Vienna, Institute of Cancer Research and Comprehensive Cancer Center, Department of Medicine I, A-1090, Borschkegasse 8a, Vienna, Austria
dThe University of Auckland, School of Chemical Sciences, Private Bag 92019, Auckland 1142, New Zealand. E-mail: c.hartinger@auckland.ac.nz; Tel: +64 9 373 7955 ext. 83220
First published on 6th March 2013
Abstract
Organometallic RuII, OsII and RhIII complexes of lapachol induce apoptosis in human tumour cell lines in the low μM range by a mode of action involving oxidative stress, especially in the case of the ruthenium compound.
Multi-targeted drugs are molecules whose different components impact multiple separate biotargets.1 In cancer chemotherapy this approach provides a means of overcoming major disadvantages of currently applied drugs by influencing pharmacological properties, metabolism and resistance development, enabling tuneable antitumour activity, “intramolecular combination therapy”, and introduction of more selective targeted properties.1 One approach to multi-targeted compounds is the combination of anticancer metal complexes with bioactive ligands, as reported for ethacrynic acid, flavonol derivatives and other compound classes.2–4 Lapachol (2-hydroxy-3-(3-methylbut-2-en-1-yl)naphthalene-1,4-dione) and flavonols share with hydroxypyr(id)ones used in medicinal chemistry the same 5-membered ring coordination motif of O,O-bidentate anionic ligands.5 Furthermore, lapachol has antibiotic and anticancer properties and was investigated in clinical trials as an anticancer agent.6,7 Its mode of action is supposed to be related to the generation of reactive oxygen species (ROS), which harm DNA and subsequently induce apoptosis, and biologically active Bi and Sb complexes were reported recently.8,9 Active organometallic RuII(arene) complexes have clearly emerged as highly promising candidates to overcome the disadvantages of clinically-used platinum drugs.10 The RAPTA family and ethylenediamine complexes are the most prominent representatives of this compound class and are at an advanced preclinical development stage.11 Therefore, the combination of bioactive lapachol with an organometallic moiety is a promising strategy, with the metal centre altering the chemical and biological properties of the ligand.
The organometallic complexes 1a–c were synthesised by deprotonating commercially available lapachol L with NaOMe followed by conversion with the respective dimer [MCl2(arene)]2 (M = RuII1a, OsII1b, RhIII1c; arene = η6-p-cymene for RuII, OsII and η5-pentamethylcyclopentadiene for RhIII) to the corresponding organometallics 1a–c in good to excellent yields (75–96%). The complexes were characterised by NMR, ESI-MS and elemental analysis, confirming the proposed structure of the compounds. In addition, single crystals of 1a and 1b suitable for X-ray diffraction analysis (Table S1, ESI†) were obtained from dichloromethane/n-hexane by using the slow diffusion method. Complexes 1a and 1b crystallise in the monoclinic space group P21/n and adopt the pseudo-octahedral “piano-stool” configuration (Fig. 1,‡), which is typical for this class of organometallic compounds. Lapachol acts as an anionic bidentate O,O-chelating ligand leading to the formation of a five-membered, non-planar ring with envelope conformation. The M–O2 distances were slightly shorter (2.0764(10) Å for 1a, 2.077(2) Å for 1b) than the corresponding M–O1 bond lengths (2.1072(11) Å for 1a, 2.128(2) Å for 1b), which is in good agreement with data obtained for related organometallic complexes.
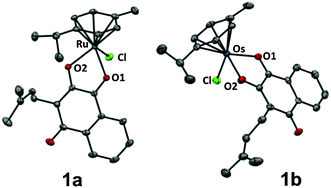 | ||
| Fig. 1 Molecular structure of the MII(η6-p-cymene) complexes 1a (M = Ru) and 1b (M = Os) drawn at 50% probability level. The hydrogen atoms were omitted for clarity. | ||
The coordination of the keto group to the metal centre induces an elongation of the C–O bond length of the coordinated carbonyl group (1.259(4) Å for 1b) as compared to the uncoordinated carbonyl in para-position (1.240(4) Å for 1b), which confirms the [1,4]-dioxo form of the attached ligand.
The aqueous chemistry of the lapachol-containing organometallics 1a–c was studied by ESI-IT-MS (Scheme 1). In all cases, aquation gave the respective aqua complexes (2a–c; Scheme 1), accompanied by ligand release and resulting in the formation of chlorido-bridged (2c) or hydroxido-bridged (2a, 2b) dimers. The extent of ligand release followed the order 2c < 2a < 2b, i.e. compound 2c is largely stable for at least 24 h, while ligand release was most pronounced for 2b as determined by analysis of relative abundances of ions in mass spectra (see Fig. S1, ESI†).
![Synthesis of the organometallic lapachol (L) complexes 1a–c and their subsequent aquation to the aqua complexes 2a–c: (i) NaOMe, [MCl2(arene)]2 (a M = RuII, b M = OsII, arene = η6-p-cymene; c M = RhIII; arene = η5-pentamethylcyclopentadiene).](/image/article/2013/CC/c3cc40432c/c3cc40432c-s1.gif) | ||
| Scheme 1 Synthesis of the organometallic lapachol (L) complexes 1a–c and their subsequent aquation to the aqua complexes 2a–c: (i) NaOMe, [MCl2(arene)]2 (a M = RuII, b M = OsII, arene = η6-p-cymene; c M = RhIII; arene = η5-pentamethylcyclopentadiene). | ||
The reactivity of metal complexes towards biomolecules is a crucial parameter for their biological activity. Therefore, 1a–c were exposed to a mixture containing the DNA model 9-ethylguanine (EtG) and the amino acids glycine (Gly), L-cysteine (Cys), L-histidine (His) and L-methionine (Met). EtG adducts were only transiently formed during the first hour and were only observed for 1a and 1b, while Gly adducts were not detected. Compounds 1a–c formed structurally similar products, i.e., His and mainly Met adducts detected as [M(aa) − H]+ ions (M = (Cp*)Rh, (cym)Ru or (cym)Os; aa = His or Met). Interestingly, different reaction pathways were observed for 1a–c. 1c transiently formed Cys adducts, which disappeared again after 24 h. A two-step binding process of amino acids was detected involving initial mono-dentate coordination of an amino acid which induces the labilisation of the O,O-chelate and leads ultimately to cleavage of lapachol. In contrast to 1c, 1a directly formed His and Met adducts and no other adducts were detected. For 1b, Cys adducts are stable for more than 24 h and were observed besides His and Met adducts (Table S2, ESI†). The extent of His or Met adduct formation seems to be pH dependent (Fig. S2, ESI†), which is of relevance in certain slightly more acidic solid tumours due to hypoxia or upregulated glycolysis.12 In such an environment, the organometallic RhIII, RuII and OsII compounds seem to favour thioether over imine binding. In addition, the reactivity of 1a–c toward the model protein ubiquitin (ub) was investigated using ESI-TOF-MS. Incubation of 1a–c with ub (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for 24 h yielded primarily monoadducts accompanied by lapachol release from the metal centre (Fig. 2; Table S3, ESI†). These monoadducts were then subjected to in-source collision-induced dissociation (ISCID), in order to obtain information on the binding site of the metal ion on the protein. Detection of Ru(cym)A1 and Os(cym)B1 fragments suggest Met1 as the primary binding site for 2a and 2b, as did the fragment (Cp*)RhB3 for 2c (Table S4, ESI†). To the best of our knowledge, this is the first report on binding site determination of a Rh-metallodrug on a protein in a top-down approach.
:1) for 24 h yielded primarily monoadducts accompanied by lapachol release from the metal centre (Fig. 2; Table S3, ESI†). These monoadducts were then subjected to in-source collision-induced dissociation (ISCID), in order to obtain information on the binding site of the metal ion on the protein. Detection of Ru(cym)A1 and Os(cym)B1 fragments suggest Met1 as the primary binding site for 2a and 2b, as did the fragment (Cp*)RhB3 for 2c (Table S4, ESI†). To the best of our knowledge, this is the first report on binding site determination of a Rh-metallodrug on a protein in a top-down approach.
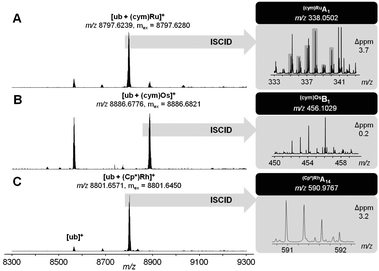 | ||
| Fig. 2 Ultra-high resolution ESI-TOF mass spectra of 2:1 reaction mixtures containing 1a (A), 1b (B) or 1c (C) and after 24 h (left). ISCID top-down signals of the respective mono-adduct revealed Met1 as the primary metallation site (right). | ||
The cytotoxicity of the organometallic complexes 1a–c was determined by means of the colorimetric MTT assay in the human cancer cell lines CH1 (ovarian carcinoma), SW480 (colon carcinoma), A549 (non-small cell lung carcinoma), HCT-116 (colon carcinoma) and HL60 (acute promyelocytic leukemia) and was compared to lapachol and cisplatin (Table 1 and Fig. S3, ESI†). Complexes 1a–c exhibit antitumour activity in the low μM range. In general, the activity of 1b was widely similar to that of lapachol (L), indicating that the ligand was the cytotoxicity-determining moiety of the compound. This may be related to ligand release in the presence of biomolecules as shown by the MS studies. The rhodium complex 1c was less cytotoxic than lapachol and more stable under physiological conditions. The organoruthenium compound 1a was the most potent compound of the series, especially in the otherwise less sensitive A549 and HCT-116 cells, where IC50 values suggest a synergistic effect of the metal ion. The complexes induced moderate but significant levels of apoptosis as determined by means of the annexin V assay in SW480 cells. The amount of annexin V/PI positive cells increased significantly after addition especially of 1a and 1c, where a more than 2-fold increase of apoptotic cells compared to the control was observed. In the cases of 1a and 1c but not 1b apoptosis induction was significantly enhanced as compared to L (Fig. S4, ESI†).
| IC50/μM | |||||
|---|---|---|---|---|---|
| CH1 | SW480 | A549 | HCT-116 | HL60 | |
| a 96 h exposure. b Taken from ref. 13. c Taken from ref. 14. | |||||
| L | 3.3 ± 0.2 | 5.5 ± 0.6 | 42 ± 14 | 92 ± 1.0 | 18 ± 5 |
| 1a | 4.1 ± 0.6 | 4.1 ± 1.5 | 20 ± 5 | 19 ± 0.1 | 25 ± 0.3 |
| 1b | 4.2 ± 0.8 | 9.0 ± 1.1 | 46 ± 8 | >100 | 18 ± 2.4 |
| 1c | 7.3 ± 1.5 | 39 ± 12 | 91 ± 19 | 93 ± 0.1 | 32 ± 0.5 |
| Cisplatin | 0.14 ± 0.03b | 3.3 ± 0.4b | 1.3 ± 0.4b | 2.7 ± 0.7c | — |
The cytotoxic activity of lapachol is related to the generation of ROS and interaction with nucleic acids.7,15 Thus the potential of 1a–c to induce oxidative stress through ROS was investigated by means of the DCFH-DA assay in HL60 cells. 1a was found to generate ROS to a higher extent than the free ligand and the Os and Rh compounds (Fig. 3). The induction of oxidative stress and apoptosis was accompanied by increased phosphorylation of the stress kinase p38 and enhanced levels of p53 in HCT-116 cells (Fig. 4). These observations confirm a synergistic effect of the organoruthenium coordination to the bioactive quinone. In addition, the impact of L and 1a–c on the cell cycle was investigated by FACS analysis in CH1, SW480 and HCT-116 cells (Fig. S5 and S6, ESI†). Treatment of the more resistant HCT-116 cells caused a significant arrest in the G2/M phase, especially for the ruthenium complex 1a. In the case of CH1 and SW480 cells, a substantial S phase arrest was observed in the IC50 range (Fig. S5 and S6, ESI†). The G1/S checkpoint is partially regulated by p53 in response to DNA damage.16 SW480 cells have a mutated p53 in contrast to HCT-116 cells, explaining the different cell cycle arrests after treatment with L and 1a–c. Furthermore, our data are comparable with lapachone induced S phase arrest in p53 mutated cell models.17 As a consequence of the cell cycle data, the dose-dependent expression changes in cyclins and the phosphorylation of cdc2 after 24 hours were elucidated by Western blot analyses in HCT-116 cells (Fig. 4). The G2/M arrest was accompanied by expression changes of cell cycle-related proteins, such as an increase in p21, cyclin B1 and E, while cyclin D1 expression was especially enhanced at lower drug concentrations. These data indicate that 1a interferes with cell cycle progression and induces apoptosis involving a p53 response.
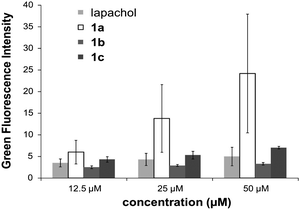 | ||
| Fig. 3 Determination of the ROS level induced by lapachol and 1a–c by the DCFH-DA-assay. | ||
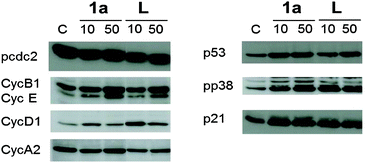 | ||
| Fig. 4 Western blot analysis of HCT-116 cells incubated with 1a and lapachol (L) for 24 h. | ||
In summary, organometallic lapachol complexes were prepared in high yields, which are activated by fast hydrolysis to the corresponding aqua species in aqueous solution. They are able to interact with biomolecules and show antiproliferative activity in the low μM range in human tumour cell lines. The first investigations of the underlying mechanisms showed an enhanced anticancer activity, especially in case of 1a, based on ROS-induced apoptosis and cell cycle arrest. Further investigations will elucidate the mode of action in more detail. Overall, the Ru complex 1a induced apoptosis to a higher degree compared to lapachol and its Rh and Os analogues, demonstrating a synergistic effect of the Ru centre and the bioactive ligand.
We thank the University of Vienna, the Austrian Science Fund (FWF), the Johanna Mahlke née Obermann Foundation, COST D39, CM0902 and CM1105 for financial support. We gratefully acknowledge Prof. Vladimir B. Arion for the refinement of the X-ray diffraction data and Anton A. Legin for instructions to the ROS assay.
Notes and references
- G. R. Zimmermann, J. Lehár and C. T. Keith, Drug Discovery Today, 2007, 12, 34–42 CrossRef CAS.
- S. Chatterjee, I. Biondi, P. Dyson and A. Bhattacharyya, J. Biol. Inorg. Chem., 2011, 16, 715–724 CrossRef CAS.
- A. Kurzwernhart, W. Kandioller, C. Bartel, S. Bächler, R. Trondl, G. Mühlgassner, M. A. Jakupec, V. B. Arion, D. Marko, B. K. Keppler and C. G. Hartinger, Chem. Commun., 2012, 48, 4839–4841 RSC.
- L. K. Filak, G. Mühlgassner, M. A. Jakupec, P. Heffeter, W. Berger, V. B. Arion and B. K. Keppler, J. Biol. Inorg. Chem., 2010, 15, 903–918 CrossRef CAS.
- W. Kandioller, A. Kurzwernhart, M. Hanif, S. M. Meier, H. Henke, B. K. Keppler and C. G. Hartinger, J. Organomet. Chem., 2011, 696, 999–1010 CrossRef CAS.
- E. Rodrigues de Almeida, Open Nat. Prod. J., 2009, 2, 42–47 CrossRef CAS.
- K. H. K. Hussain, V. U. Ahmad, G. A. Miana and I. R. Green, ARKIVOC, 2007, 2, 42–47 Search PubMed.
- G. Parrilha, R. Vieira, P. Campos, G. Silva, L. Duarte, S. Andrade and H. Beraldo, BioMetals, 2012, 25, 55–62 CrossRef CAS.
- L. G. de Oliveira, M. M. Silva, F. C. S. de Paula, E. C. Pereira-Maia, C. L. Donnici, C. A. de Simone, F. Frézard, E. N. da Silva Júnior and C. Demicheli, Molecules, 2011, 16, 10314–10323 CrossRef CAS.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS.
- G. Süss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- R. A. Gatenby and R. J. Gillies, Nat. Rev. Cancer, 2004, 4, 891–899 CrossRef CAS.
- Y. Y. Scaffidi-Domianello, A. A. Legin, M. A. Jakupec, V. B. Arion, V. Y. Kukushkin, M. Galanski and B. K. Keppler, Inorg. Chem., 2011, 50, 10673–10681 CrossRef CAS.
- M. R. Reithofer, S. M. Valiahdi, M. Galanski, M. A. Jakupec, V. B. Arion and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2160–2170 CAS.
- E. A. Hillard, F. C. de Abreu, D. C. M. Ferreira, G. Jaouen, M. O. F. Goulart and C. Amatore, Chem. Commun., 2008, 2612–2628 RSC.
- G. Damia and M. Broggini, Cell Cycle, 2004, 3, 46–50 CrossRef CAS.
- L. Huang and A. B. Pardee, Mol. Med., 1999, 5, 711–720 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures, X-ray diffraction data, experimental procedures to mass spectrometry and biological investigations. CCDC 918728 and 918729. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3cc40432c |
| ‡ Crystallographic details: 1a: C25H27ClO3Ru, Mr = 511.19, 0.60 × 0.10 × 0.10 mm, monoclinic, P21/n, a = 12.8956(5) Å, b = 11.8913(4) Å, c = 14.2756(5) Å, α = 90°, β = 98.504(2)°, γ = 90°, V = 2165.05(13) Å3, Z = 4, ρcalcd = 1.571 mg m−3, μ = 0.872 mm−1, T = 150(2) K, 50326 measured independent reflections, Rint = 0.0382, R1 = 0.0225, wR2 = 0.0595, GOF = 1.000; 1b: C25H27ClO3Os, Mr = 601.12, 0.12 × 0.10 × 0.02 mm, monoclinic, P21/n, a = 13.4764(4) Å, b = 8.4120(2) Å, c = 21.0225(5) Å, α = 90°, β = 107.9860(10)°, γ = 90°, V = 2266.72(10) Å3, Z = 4, ρcalcd = 1.761 mg m−3, μ = 5.767 mm−1, T = 150(2) K, 17682 measured independent reflections, Rint = 0.0426, R1 = 0.0247, wR2 = 0.0507, GOF = 0.997; description of data collection and refinement see ESI;† CCDC 918728 and 918729. |
| This journal is © The Royal Society of Chemistry 2013 |