 Open Access Article
Open Access ArticleA new class of NO-donor pro-drugs triggered by γ-glutamyl transpeptidase with potential for reno-selective vasodilatation†
Qingzhi
Zhang
*a,
Agnieszka
Kulczynska
a,
David J.
Webb
b,
Ian L.
Megson
*c and
Nigel P.
Botting‡
*a
aUniversity of St Andrews, EaStChem School of Chemistry and Centre for Biomolecular Sciences, North Haugh, St Andrews, Fife KY16 9ST, UK. E-mail: qz@st-andrews.ac.uk; Fax: +44 (0)1334 463808; Tel: +44 (0)1334 467274
bCentre for Cardiovascular Science, The Queen's Medical Research Institute, The University of Edinburgh, 47 Little France Crescent, Edinburgh, EH16 4TJ, UK
cFree Radical Research Facility, Department of Diabetes and Cardiovascular Science, The University of The Highlands & Islands, Centre for Health Science, Old Perth Road, Inverness, IV2 3JH, UK. E-mail: ian.megson@uhi.ac.uk; Fax: +44 (0)1463 711245; Tel: +44 (0)1463 279562
First published on 21st December 2012
Abstract
This communication describes the synthesis of a new class of N-hydroxyguanidine (NHG) pro-drugs which release nitric oxide (NO), triggered by the action of γ-glutamyl transpeptidase (γ-GT), and have potential for the treatment of acute renal injury/failure (ARI/ARF).
Acute renal injury (AKI), or failure (ARF), is a common complication that affects millions of people worldwide, particularly in intensive care units, where it is associated with a mortality rate of between 50% and 80%.1 There is no effective pharmaceutical therapy to date. One of the major causes of AKI is ischemia-reperfusion injury,2,3 following aortic ring cross-clamping during by-pass surgery, which can lead to renal ischemia.4 Reperfusion of ischemic renal tissue causes the generation of reactive oxygen species which induce renal cell injury5 and promote impairment of renal perfusion at least in part via inactivation of the vasodilator, nitric oxide (NO).6–8 Thus, a kidney selective vasodilator with antioxidant properties is attractive to maintain blood flow to offset AKI and scavenge the reactive oxygen species. Localisation of activity to the kidney would avoid a systemic reduction in blood pressure. Dopamine and fenoldopam, specific agonists of the dopamine-1 receptor, have been used clinically in an effort to reduce the risk of perioperative renal dysfunction, but the effectiveness of these agents is not clear.9,10 We hypothesised that an effective exogenous NO-donor, which selectively increases renal vasodilatation, would offer an alternative.
There are a wide range of NO-donor drugs in existence,11 including conventional organic nitrates and nitrites, S-nitrothiols, NONOates and N-hydroxyguanidines (NHGs).12–16 The NHGs 1 are analogues of Nω-hydroxy-L-arginine (NOHA), a biosynthetic intermediate involved in the generation of NO from L-arginine.11 Several enzymatically activated NHG pro-drugs have been reported such as peptidylglycine α-amidating monooxygenase (PAM)-active O-carboxymethyl N-hydroxyguanidines17 and N-β-galactosidases-active (β-D-galactopyranos-1-yl)oxyguanidine.18 Our approach aimed to mask the NO generating N-OH group with a γ-glutamyl residue to facilitate activation by the enzyme, γ-glutamyl transpeptidase (γ-GT). Given that γ-GT is primarily expressed in the kidney (5–10 fold higher than in the liver and pancreas),19 it was envisaged that this enzyme could be used to trigger reno-selective release of an NHG and subsequent in situ generation of NO (Scheme 1). A similar strategy has been described for reno-selective L-3,4-dihydroxyphenylalanine (L-DOPA), the Glu-DOPA.20,21
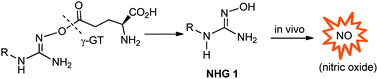 | ||
| Scheme 1 Approach to γ-GT triggered release of NHG 1 and the reno-selective release of nitric oxide. | ||
However, the direct coupling of NHGs with a γ-glutamyl residue was hampered by intramolecular cyclization and dehydration leading to a 1,2,4-oxadiazole ring; or alternatively lactamization and release of a pyroglutamic acid (Scheme 2, data not included).
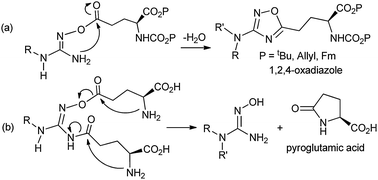 | ||
| Scheme 2 Cyclization of direct coupling of NHGs with γ-glutamyl residue(s). | ||
In an effort to prevent these modes of cyclization, we investigated the use of a bridge between the NHG and the γ-glutamyl group. Both γ-glutamyl itself and γ-aminobutanoyl (GABA)22 were explored as linkers. Thus 2a and 2b became synthesis targets (Scheme 3) and they were prepared via appropriately protected dipeptide intermediates (ESI;† Scheme S1). Unfortunately 2a gradually decomposed presumably due to the carboxylic acid moieties promoting autodegradation. On the other hand, 2b could be purified by preparative HPLC but was found to be resistant to γ-GT-mediated cleavage in vitro and was considered not to be a useful pro-drug. This prompted the preparation of 3 (Scheme 3), involving the conjugation of only one GABA-Glu dipeptide onto a hydroxamic acid, an alternative NO-donor.11 Compound 3 too, unfortunately, was found to be resistant to γ-GT mediated deacylation, suggesting that the GABA-Glu peptide linker is not suitable for γ-GT cleavage in this setting.
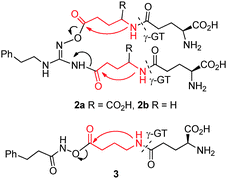 | ||
| Scheme 3 Design of Glu/Gaba linked γ-glutamyl NO-donor pro-drugs of NHG and hydroxamic acid. | ||
γ-Glutamyl anilines are known substrates for γ-GT23 and presented an alternative linker option. The success of such an approach would involve a 1,6-elimination following the action of γ-GT on N-γ-glutamylaminobenzyloxy-guanidine 4a–c, as illustrated in Scheme 4. Similar spacers have been employed previously in anticancer pro-drug design.24
![Design and synthesis of aminobenzyl linked γ-glutamyl NO-donor pro-drugs of NHG: (i) 4-aminobenzylalcohol, EEDQ, DCM, rt, 12 h, 85%; (ii) PBr3, THF, 0 °C, 2 h, 87%; (iii) BocNHOH, NaH, THF, 0 °C, 4 h, 83%; (iv) CF3CO2H, DCM, 92%; (v) 9a R = Ph or 9b R = PhCH2CH2 or 9c R = furfuryl, Et3N, DMAP, DCM, 38–53%; (vi) [Pd(PPh3)4], PhSiH3, DCM, 37–89%.](/image/article/2013/CC/c2cc38382a/c2cc38382a-s4.gif) | ||
| Scheme 4 Design and synthesis of aminobenzyl linked γ-glutamyl NO-donor pro-drugs of NHG: (i) 4-aminobenzylalcohol, EEDQ, DCM, rt, 12 h, 85%; (ii) PBr3, THF, 0 °C, 2 h, 87%; (iii) BocNHOH, NaH, THF, 0 °C, 4 h, 83%; (iv) CF3CO2H, DCM, 92%; (v) 9a R = Ph or 9b R = PhCH2CH2 or 9c R = furfuryl, Et3N, DMAP, DCM, 38–53%; (vi) [Pd(PPh3)4], PhSiH3, DCM, 37–89%. | ||
In the event, the synthesis of 4a–c was successfully accomplished through a six-step reaction sequence (Scheme 4). Firstly, γ-glutamylation of 4-aminobenzylalcohol with Alloc-L-glutamic acid 1-allyl ester (Alloc-Glu-OAll) (ESI;† Scheme S1) gave benzyl alcohol 5. Conversion of the benzylalcohol moiety to the corresponding bromide 6 followed by nucleophilic displacement with BocNHOH generated aminooxide 7, and then treatment with CF3COOH–DCM, gave the key intermediate 8 which was coupled with the required amino(alkyl/aryliminio)-methanesulfonate 9a–c to generate 10a–c. Finally the All/Alloc groups were removed under neutral conditions with ([Pd(PPh3)4]/PhSiH3) to give 4a–c.
The same aminobenzyl linker was also used for the γ-glutamylation of N-hydroxyformamidines (NHFs) (Scheme 5). N′-Hydroxy-N-(4-butyl-2-methylphenyl)formamidine25 and N′-hydroxy-N-(3-chloro-4-morpholin-4-ylphenyl)formamide26 have been documented as 20-hydroxyeicosatetraenoic acid (20-HETE) inhibitors. 20-HETE is a major metabolite of arachidonic acid and is a potent vasoconstrictor; localisation of an NHF would counter the effect of 20-HETE and induce a synergic vasodilation effect mediated by NO. Thus N′-hydroxyphenylethylformamidine 12 was prepared in this study and converted to pro-drug 14.
![Synthesis of N-hydroxyformamidine and its glutamyl pro-drug: (i) Me2NCH(OMe2), reflux, 2 h, quantitative; (ii) NH2OHHCl, MeOH, 63%; (iii) 8, THF, reflux, 29%; (iv) [Pd(PPh3)4], PhSiH3, DCM, rt, 6 h, 53%.](/image/article/2013/CC/c2cc38382a/c2cc38382a-s5.gif) | ||
| Scheme 5 Synthesis of N-hydroxyformamidine and its glutamyl pro-drug: (i) Me2NCH(OMe2), reflux, 2 h, quantitative; (ii) NH2OHHCl, MeOH, 63%; (iii) 8, THF, reflux, 29%; (iv) [Pd(PPh3)4], PhSiH3, DCM, rt, 6 h, 53%. | ||
Pro-drugs 4a–c and 14 were rapidly cleaved by γ-GT and they were completely deacylated after 1 h, as judged by LC-MS. Fig. 1(a) and (b) illustrates the LCMS trace of 4b and the conversion of 4b to deacylated intermediate 15 [M-Glu]+ by γ-GT. This was in clear contrast to the GABA-linked candidates 2b and 3, which proved to be resistant to the action of γ-GT. 1,6-Elimination and loss of the linker from 15 to generate the parent NHG 1b is significantly slower (trace amount of parent 1b was detected by selective ion monitoring at m/z 180) than the cleavage of the γ-glutamyl moiety. In preliminary experiments with animal tissue, LC-MS analysis revealed ∼90% conversion of 4b (100 μM) to 1b in a rat renal homogenate (37 °C; 45 min). In addition, 4b was found to induce substantial vasodilatation in rat isolated perfused kidney preparations (50% of maximum vasodilatation induced by ∼40 μM 4b). Details of the bioactivity of these pro-drugs will be reported elsewhere.
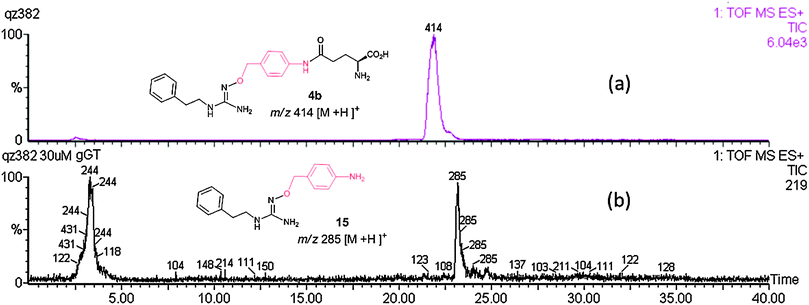 | ||
| Fig. 1 LCMS trace of 4b incubated in Krebs buffer at 37 °C for 1 h (a) without γ-GT and glutamyl acceptor Gly–gly, 4b is intact; (b) with γ-GT (100 mU mL−1) and glutamyl acceptor Gly–gly (5 mM), 4b is deglutamylated to give the species 15. | ||
In summary, several candidate NO-donor pro-drugs have been prepared, designed for activation by γ-GT. The pro-drugs comprise the parent NO-donor, a linker and a γ-glutamyl moiety. GABA-linked pro-drugs are not suitable substrates for γ-GT, but those linked by the aminobenzyl moiety proved to be good substrates for the enzyme. The γ-glutamyl group is cleaved rapidly, with a slower decomposition of the aminobenzyl linker. Improved design is now focussed on tuning the spacer to encourage a more rapid release of the parent NHG drug.
The authors are grateful to the Wellcome Trust (Catalyst Biomedica Development Award 063729/Z/01/Z) for financial support. Thanks go to Prof. David O'Hagan (University of St Andrews) for his input into manuscript preparation.
References
- R. W. Schrier, W. Wang, B. Poole and A. Mitra, J. Clin. Invest., 2004, 114, 5–14 CAS.
- J. V. Bonvebtre and J. M. Weinberg, J. Am. Soc. Nephrol., 2003, 14, 2199–2210 CrossRef.
- T. A. Sutton, C. J. Fisher and B. A. Molitoris, Kidney Int., 2002, 62, 1539–1549 CrossRef CAS.
- G. Mariscalco, R. Lorusso, C. Dominici, A. Renzulli and A. Sala, Ann. Thorac. Surg., 2011, 92, 1539–1547 CrossRef.
- P. A. Grace, Br. J. Surg., 1994, 81, 637–647 CrossRef CAS.
- M. L. Brocq, S. J. Leslie, P. Milliken and I. L. Megson, Antioxid. Redox Signaling, 2008, 10, 1631–1674 CrossRef.
- A. MacKenzie and W. Martin, Br. J. Pharmacol., 1998, 124, 710–728 CrossRef.
- M. Saran, C. Michel and W. Bors, Free Radical Res. Commun., 1990, 10, 221–226 CrossRef CAS.
- M. D. Denton, G. M. Chertow and H. R. Brady, Kidney Int., 1996, 49, 4–14 CrossRef.
- T. Bove, G. Landoni, M. G. Calabrò, G. Aletti, G. Marino, E. Cerchierini, G. Crescenzi and A. Zangrillo, Circulation, 2005, 111, 3230–3235 CrossRef CAS.
- P. G. Wang, T. B. Cai and N. Taniguchi, Nitric Oxide Donors: For Pharmaceutical and Biological Applications, Wiley-VCH, 2005, ISBN, 3-527-31015-0 Search PubMed.
- A. Renodon-Cornière, S. Dijols, C. Perollier, D. Lefevre-Groboillot, J. L. Boucher, R. Attias, M. A. Sari, D. Stuehr and D. Mansuy, J. Med. Chem., 2002, 45, 944–954 CrossRef.
- D. Mansuy and J. L. Boucher, Drug Metab. Rev., 2002, 34, 593–606 CrossRef CAS.
- M. Xian, X. P. Li, X. P. Tang, X. C. Chen, X. L. Zheng, J. J. Galligan, D. L. Kreulen and P. G. Wang, Bioorg. Med. Chem. Lett., 2001, 11, 2377–2380 CrossRef CAS.
- M. Xian, N. Fujiwara, T. Cai, Z. Wen, S. Kazuma, A. Janczuk, X. Tang, V. Telyatnikov, Y. Miyamoto, N. Taniguchi and P. G. Wang, Bioorg. Med. Chem., 2002, 10, 3049–3055 CrossRef CAS.
- Q. Jia, T. Cai, M. Huang, H. Li, M. Xian, T. L. Poulos and P. G. Wang, J. Med. Chem., 2003, 46, 2271–2274 CrossRef CAS.
- D. Schade, J. Kotthaus, H. Hungeling, J. Kotthaus and B. Clement, ChemMedChem, 2009, 4, 1595–1599 CrossRef CAS.
- D. Schade, J. Kotthaus, N. Klein, J. Kotthaus and B. Clement, Org. Biomol. Chem., 2011, 9, 5249–5259 CAS.
- J. P. Ward, Ann. R. Coll. Surg. Engl., 1975, 57, 248–261 CAS , Academic Press.
- S. Wilk, H. Mizoguchi and M. Orlowski, J. Pharmacol. Exp. Ther., 1978, 206, 227–232 CAS.
- M. Barthelmebs, A. Caillette, J. D. Ehrhardt, J. Velly and J. L. Imbs, Kidney Int., 1990, 37, 1414–1422 CrossRef CAS.
- R. G. G. Leenders, K. A. A. Gerrits, R. Ruijtenbeek and H. W. Scheeren, Tetrahedron Lett., 1995, 36, 1701–1704 CrossRef CAS.
- A. Ménard, R. Castonguay, C. Lherbet, C. Rivard, Y. Roupioz and J. W. Keillor, Biochemistry, 2001, 40, 12678–12685 CrossRef.
- B. E. Toki, C. G. Cerveny, A. F. Wahl and P. D. Senter, J. Org. Chem., 2002, 67, 1866–1872 CrossRef CAS.
- M. Sato, T. Ishii, Y. Kobayashi-Matsunaga, H. Amada, K. Taniguchi, N. Miyata and K. Kameo, Bioorg. Med. Chem. Lett., 2001, 11, 2993–2995 CrossRef CAS.
- N. Miyata, T. Seki, Y. Tanaka, T. Omura, K. Taniguchi, M. Doi, K. Bandou, S. Kametani, M. Sato, S. Okuyama, L. Cambj-Sapunar, D. R. Harder and R. J. Roman, J. Pharmacol. Exp. Ther., 2005, 314, 77–85 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cc38382a |
| ‡ N. P. Botting died on 4th June 2011. |
| This journal is © The Royal Society of Chemistry 2013 |