Cargo delivery to adhering myoblast cells from liposome-containing poly(dopamine) composite coatings†
Martin E. Lyngea,
Boon M. Teoa,
Marie Baekgaard Laursena,
Yan Zhangab and
Brigitte Städler*a
aiNANO Interdisciplinary Nanoscience Centre, Aarhus University, Gustav Wieds Vej 14, Aarhus, Denmark. E-mail: bstadler@inano.au.dk; Fax: +45 8715 4041; Tel: +45 8715 6668
bState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Material Science and Engineering, Donghua University, Shanghai, People's Republic of China
First published on 5th July 2013
Abstract
Designing surfaces to deliver therapeutic compounds to adhering cells is of paramount importance for both implantable devices and tissue engineering. We report the assembly of composite films consisting of liposomes as drug deposits in a poly(dopamine) matrix. We monitor the film assembly using a quartz crystal microbalance with dissipation. We assess the response of adhering myoblast cells to these films containing fluorescent lipids in terms of uptake efficiency and cell mean fluorescence using flow cytometry. The viability of adhering myoblast cells, when the hydrophobic cytotoxic compound thiocoraline is entrapped in the lipid membrane, is assessed for different films. Coatings with one or two liposome deposition steps are considered. Further, the effect of the polymer separation layers between the liposome layers is determined. We found that it is possible to use the different nano-engineered composite coatings to impose a corresponding cellular response, e.g., a higher amount of embedded liposomes leads to higher uptake efficiency of the fluorescent lipids and cell mean fluorescence or a higher reduction in the viability of the adhering cells. Assessment of the uptake efficiency and cell mean fluorescence over time reveals a decrease in both parameters over 48 h. Our results demonstrate the ability to affect the cell response depending on the properties of the films, opening up a variety of opportunities for biomedical applications in substrate-mediated drug delivery.
Introduction
Functional surface coatings are a key aspect for many biomedical applications, from advanced tissue engineering scaffolds to drug eluting implantable devices. Employing multilayered thin films assembled via the sequential deposition of interacting polymers (LbL assembly) loaded with a diversity of cargo (small molecules, protein, DNA, etc.) have proven to be a promising approach to deposit such coatings for substrate-mediated drug delivery (SMDD).1,2 Coatings based on poly(dopamine) (PDA) assembled via the self-polymerization of dopamine at slightly basic pH are an alternative.3 PDA films for biomedical applications4 are particularly interesting since these biocompatible coatings form on virtually any surface of any shape and material. Further, it has been proven to render cytophobic substrates cytophilic for many different cell types,5,6 while allowing for subsequent modification of the films via amines or thiols.3,7–10 Also, dopamine can be mixed with a polymer7,11 or the reactants can be modified12–14 prior to the deposition. Both approaches yield mixed coatings with different properties compared to the plain PDA films. For a detailed overview of catechol chemistry and applications, the reader is referred to the recent comprehensive review by Sedo et al.15Drug deposits such as micelles,16 cyclodextrins,17 or hyaluronan-based assemblies containing hydrophobic nanodomains18 have been considered to deliver small hydrophobic compounds from thin coatings. In addition, we recently reported on the assembly of surface-adherent composite hydrogels based on poly(vinyl alcohol) and polymersomes, and the subsequent successful delivery of an active compound to adhering myoblast cells.5 Liposomes are used in many different biomedical applications from drug delivery19 to biosensing,20 and are particularly interesting subunits due to their potential to encapsulate hydrophobic and hydrophilic cargo, their biocompatibility or their fast way of assembly. Embedding liposomes into polymer coatings is an important step when assembling compartmentalized systems as advanced drug delivery systems,21 or as cell mimics.22 Their trapping into planar LbL films23–26 or PDA coatings27 has been considered. All these concepts yielded intact liposomes within a thin polymer coating involving multiple deposition steps. Further, examples of their use towards SMDD remain scarce. The uptake of fluorescent model cargo by mammalian cells has been reported.27,28 The use of active cargo is limited and includes the demonstration of antibacterial properties of substrates containing silver nitrate loaded liposomes deposited within poly(L-lysine)/hyaluronic acid multilayers,29 the assembly of antifungal surfaces employing PDA,30 or the delivery of a cytotoxic compound (thiocoraline) from liposome-containing polyelectrolyte multilayers.31
Here, we report the delivery of active cargo to myoblast cells from surfaces coated with liposome-containing PDA films (Scheme 1). Specifically, we (i) characterize the film assembly using a quartz crystal microbalance with dissipation monitoring (QCM-D), (ii) assess the uptake/association of fluorescent lipids with myoblasts depending on the dopamine (DA) concentration and the cell residence time using flow cytometry, (iii) incorporate a small hydrophobic cytotoxic compound into the liposomes and determine the viability of the adhering cells, (iv) evaluate the possibility to deposit a second liposome-containing layer using a QCM-D, (v) assess the ability of these coatings to deliver (active) cargo to adhering myoblast cells, (vi) monitor the second liposome deposition step depending on the polymer separation layers, (vii) characterize the myoblast response to these coatings depending on the polymer separation layers, and (viii) characterize the kinetics of fluorescent lipid uptake from the surface by myoblasts as well as the stability of the coatings. Taken together, our findings further evolve these hybrid coatings towards SMDD.
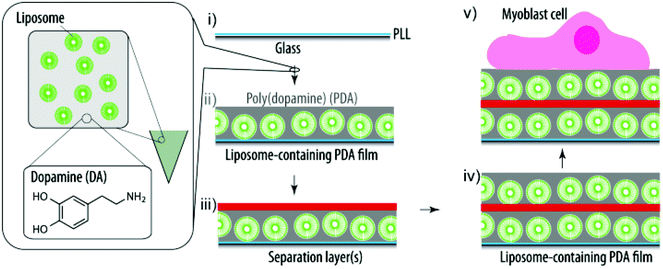 | ||
| Scheme 1 Schematic illustration of the assembly of a liposome-containing PDA film towards SMDD. PLL pre-coated glass slides (i) are exposed to a mixture of DA and liposomes and the DA self-polymerizes into PDA and entraps the liposomes during this process (ii). (Multiple) polymer separation layers ((m)SL) (iii) are required to allow for additional liposomes and PDA to be deposited (iv). Finally, myoblast cells are grown on these cargo-loaded coatings (v). | ||
Experimental
Materials
Poly(L-lysine) (PLL, Mw 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–60000 Da), 4-(2-hydroxyethyl)piperazine-1-ethane-sulfonic acid (HEPES), tris(hydroxymethyl)aminomethane (TRIS), boric acid, sodium chloride (NaCl), ethanol, chloroform (pty ≥ 99.5%), poly(allylamine hydrochloride) (PAH, Mw 17000 Da), poly(sodium 4-styrenesulfonate) (PSS, Mw 70000 Da), and dopamine hydrochloride (DA) were purchased from Sigma-Aldrich. Zwitterionic 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC, phase transition temperature −20 °C) and fluorescent lipids 1-oleoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl]-sn-glycero-3 phosphocholine (NBD-PC) were purchased from Avanti Polar Lipids, USA. Thiocoraline (TC) was isolated and purified by PharmaMar, S.A. (Colmenar Viejo, Madrid, Spain).
000–60000 Da), 4-(2-hydroxyethyl)piperazine-1-ethane-sulfonic acid (HEPES), tris(hydroxymethyl)aminomethane (TRIS), boric acid, sodium chloride (NaCl), ethanol, chloroform (pty ≥ 99.5%), poly(allylamine hydrochloride) (PAH, Mw 17000 Da), poly(sodium 4-styrenesulfonate) (PSS, Mw 70000 Da), and dopamine hydrochloride (DA) were purchased from Sigma-Aldrich. Zwitterionic 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC, phase transition temperature −20 °C) and fluorescent lipids 1-oleoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]hexanoyl]-sn-glycero-3 phosphocholine (NBD-PC) were purchased from Avanti Polar Lipids, USA. Thiocoraline (TC) was isolated and purified by PharmaMar, S.A. (Colmenar Viejo, Madrid, Spain).
Four types of buffers were used throughout all of the experiments: borate buffer consisting of 10 mM boric acid (pH 8.5), 10 mM phosphate buffer (pH 8.5), 10 mM TRIS buffer (pH 8.5), and HEPES buffer consisting of 10 mM HEPES and 150 mM NaCl (pH 7.4). The buffer solutions were made with ultrapure water (Milli-Q gradient A 10 system, resistance 18 MΩ cm, TOC < 4 ppb, Millipore Corporation, USA).
Unilamellar liposome stock solutions were prepared by evaporation of the chloroform of 2.5 mg lipids under vacuum for 1 h, followed by hydration using 1 mL of buffer solution and extrusion through 100 nm filters (11 times). For fluorescently labeled liposomes (FL), 1 wt% of NBD-PC was added to the lipid mixture.
Poly(methacrylic acid)-co-(cholesteryl methacrylate) (PMAc) was synthesized by following a previously published procedure23 with minor alterations.31
Quartz crystal microbalance with dissipation monitoring (QCM-D) measurements
QCM-D measurements (Q-Sense E4, Sweden) were used to analyze the adsorption behavior of DA–liposome mixtures onto surfaces pre-coated with PLL. Silica-coated crystals (QSX300, Q-Sense) were cleaned by immersion in a 2 wt% sodium dodecyl sulfate solution overnight and rinsing with ultrapure water. Afterwards, the crystals were blow-dried with N2 and treated with UV/ozone for 20 min before being mounted into the liquid exchange chambers of the instrument. The frequency and dissipation measurements were monitored at 22 ± 0.02 °C. When a stable baseline in buffer was achieved, PLL (1 mg mL−1 in HEPES buffer) was introduced into the measurement chamber and left to adsorb onto the crystal to form a PLL precursor layer, which is present for all the assembled coatings, but is not mentioned specifically from now on. After the surface was saturated, the chamber was rinsed with TRIS buffer to remove the excess polymer. All subsequent experiments were conducted using a flow rate of 50 μL min−1, and there was a washing step in the corresponding buffer between each deposition step.In order to test the film assembly in different buffer solutions, the PLL pre-coated crystal was exposed to DA (2.5 mg mL−1), a PDA–liposome mixture (10M2.5, 2.5 mg mL−1 DA with a 50 vol% liposome stock solution (L10)), or L10. Borate buffer, TRIS buffer and phosphate buffer were used for these experiments.
In order to compare the films assembly depending on the liposome and DA concentration, PLL pre-coated crystals were exposed to a freshly prepared DA–liposome solution (2.5 mg mL−1 DA stock solution mixed v/v = 10:90, 50:50, 75:25, and 95:5 with a liposome stock solution to a total volume of 1500 μL in TRIS buffer, or 0.5, 2.5, 5.0, and 7.1 mg mL−1 DA in TRIS buffer with 5 vol% (L1) liposome stock solution or L10) and left to incubate until the surface was saturated. The DA–liposome solution was replaced with HEPES buffer.
Coatings with a second liposome deposition step were assembled after the PDA–liposome mixture 10M2.5 was deposited onto a silica crystal using either no polymer separation layer (SL), PLL (1 mg mL−1 in HEPES buffer) or PLL/PMAc (0.25 mg mL−1 in HEPES buffer for PMAc) as SL. As a last step, the pre-coated crystals were exposed to either a freshly prepared DA solution (2.5 mg mL−1 DA in TRIS buffer, 30 min) or a 10M2.5 solution (freshly prepared) or L10. As a control, a bare silica crystal was exposed to a freshly prepared DA solution (2.5 mg mL−1, TRIS buffer). After rinsing with TRIS buffer, these steps were repeated twice.
Assembly of liposomes containing films with different types of multiple polymer SLs (mSL) were assembled using PLL/L10/PMAc pre-coated crystals. Alternating layers of PLL (1 mg mL−1 in HEPES buffer) and PMAc (0.25 mg mL−1 in HEPES buffer) or PAH (1 mg mL−1 in HEPES buffer) and PSS (1 mg mL−1 in HEPES buffer) were deposited, followed by the deposition of 10M2.5.
Substrate modification
18 × 18 mm2 (cell uptake experiments) or 9 mm diameter glass slides (cell viability experiments) were cleaned via sonication in ethanol for 10 min, rinsed with ultrapure water, dried under nitrogen flow, and exposure to UV/ozone for 20 min.PLL (1 mg mL−1, 10 min) was adsorbed as the precursor layer and rinsed with HEPES buffer. The PLL precursor layer is present for all the assembled coatings, but is not mentioned specifically from now on. The samples were then exposed to a freshly prepared DA–liposome solution of 0.5, 2.5, 5.0 and 7.1 mg mL−1 DA in TRIS buffer with L10 (10M0.5, 10M2.5, 10M5, and 10M7.1) for 30 min and rinsed with TRIS buffer followed by HEPES buffer.
For multilayered films, PLL was deposited (10 min, 1 mg mL−1 in HEPES buffer) onto 10M2.5 pre-coated glass slides, rinsed with HEPES buffer followed by exposure to a PMAc solution (10 min, 0.25 mg mL−1 in HEPES buffer) and washed with HEPES buffer and capped with either 10M2.5 (freshly prepared, 45 min) or with L10 (45 min), rinsed with HEPES buffer and followed by the deposition of DA (30 min, 2.5 mg mL−1 in TRIS buffer).
For substrates with mSLs, glass slides were pre-coated with PLL/L10/PMAc. Alternating layers of PLL (10 min, 1 mg mL−1 in HEPES buffer) and PMAc (10 min, 0.25 mg mL−1 in HEPES buffer) or PAH (10 min, 1 mg mL−1 in HEPES buffer) and PSS (10 min, 1 mg mL−1 in HEPES buffer) were deposited, followed by the deposition of 10M2.5 (45 min).
The coated substrates were stored in HEPES buffer prior to the cell experiments.
Cell work
The coated substrates were UV-sterilized for 30 min submerged in sterile PBS. The C2C12 mouse myoblast cell line (American Type Culture Collection) was used for all experiments. The cells (175000 cells per flask in 20 mL medium) were cultured in 75 cm2 culture flasks in a medium (Dulbecco's modified Eagle's Medium with Glutamax (DMEM) supplemented with 10% fetal bovine serum, 50 U mL−1 penicillin, 50 μg mL−1 streptomycin and 1 mM sodium pyruvate, all from Sigma) at 37 °C and 5% CO2. The cells were seeded onto the substrates at a density of 100000 cells per well in 1.5 mL medium in 6-well plates (cell uptake) or 15000 cells per well in 0.5 mL medium in 48-well plates (cell viability) and allowed to attach for different times at 37 °C and 5% CO2.
The results were statistically analyzed for significance using an unpaired 2-tail t-test (equal variances).
Flow cytometry
Prior to harvesting, bright field images of the live cells were obtained using an Olympus CKX41 microscope. For cell uptake experiments, the samples were transferred into new wells to ensure that only the cells grown on the substrate were considered in the subsequent analysis. The cells were washed 2× with 3 mL PBS. 200 μL trypsin was used to detach the cells from the surface for analysis by flow cytometry. The cells grown on substrates coated with fluorescently labeled liposomes were analyzed using a C6 Flow Cytometer (Accuri Cytometers Inc.) and an excitation wavelength of λ = 488 nm after 6 h, 24 h and 48 h. At least 2000 cells were analyzed. The auto-fluorescence of control cells grown on PDA (2.5 mg mL−1, 30 min) was subtracted, and the control cells have been gated out in all the presented results. All cell experiments were performed in at least three independent repeats.Cell viability
The amount of encapsulated TC in the liposomes was quantified prior to the film assembly. The liposome solutions were excited at a wavelength of 365 nm and the fluorescence intensity was recorded at an emission wavelength of 547 nm using a multiplate reader (PerkinElmer). The concentration of encapsulated TC was determined in correlation with a calibration curve.31 The TC containing liposome solution was diluted to yield a final concentration of 3.4 μg mL−1 TC in the dopamine solution which was added to the glass substrates during the sample preparation. The concentration of TC on the surface was estimated to be 9.4 ± 0.1 ng.31 Samples using liposomes without TC were prepared as controls: 10M0.5, 10M2.5, 10M5, 10M7.1, 10M2.5/PLL/PMAc/L10/PDA30, and 10M2.5/PLL/PMAc/10M2.5. The following cargo-loaded samples were prepared: 10_TCM0.5, 10_TCM2.5, 10_TCM5, 10_TCM7.1, 10_TCM2.5/PLL/PMAc/L10TC/PDA30, and 10_TCM2.5/PLL/PMAc/10_TCM2.5. The cells were let to adhere for 24 h and 48 h on the substrates. The cell viability was assessed using the Cell Counting Kit-8 (Doijindo) by adjusting the media volume per well to 200 μL and addition of a 20 μL assay solution per well followed by incubation for 2 h at 37 °C and 5% CO2 prior to the absorption measurements using a multiplate reader. The results were normalized to cells grown on bare PDA (2.5 mg mL−1, 30 min). The experiments were performed in at least three independent repeats.Results and discussion
Single liposome deposition step
First, we aimed to compare different buffering agents in their ability to support the PDA formation. The three buffer solutions all had an ionic strength of 10 mM at pH 8.5 and 2.5 mg mL−1 DA was used. Fig. 1 shows the PDA formation in these different buffers onto a PLL pre-coated silica crystal as assessed using a QCM-D. Borate buffer did not allow for the PDA assembly, likely due to the ester formation of the boric acid with the vicinal diol of DA.32,33 Phosphate and TRIS buffer allowed for the PDA formation, but Δf of the crystal in the latter case was larger, indicating a thicker deposited film. We also compared the deposition of a liposome–DA mixture (10M2.5, 50 vol% of the liposome stock solution (L10) and DA = 2.5 mg mL−1) and bare liposomes L10 using phosphate or TRIS buffer. As expected, larger Δf of the crystals was observed in TRIS buffer when depositing 10M2.5, while there was no difference in Δf depending on the buffer for L10. Therefore, TRIS buffer was used for all the subsequent experiments to ensure the largest amount of incorporated liposomes into the surface-adhering PDA film. While the PDA process is typically described as a self-oxidative polymerization at slightly basic pH, we demonstrate that TRIS is required for the optimal assembly. This supports the hypothesis recently brought forward that TRIS is incorporated into the film.34,35
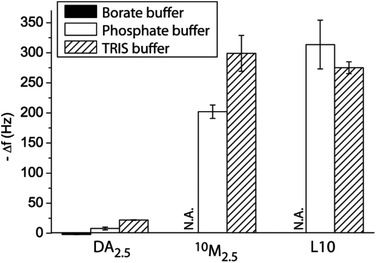 | ||
| Fig. 1 Film assembly. Change in frequency Δf of QCM-D crystals upon exposure to DA2.5, 10M2.5 and L10 using different buffer solutions (all 10 mM, pH 8.5). | ||
The next goal was to characterize the assembly of liposome–DA mixtures in more detail using QCM-D. First, we mixed different volumes of DA and liposomes prior to the deposition onto crystals, starting with a DA concentration of 2.5 mg mL−1 and the liposome stock solution (Fig. 2a and ESI Fig. S1a†). Increasing of the amounts of liposomes led to the increasing of the Δf and ΔD of the crystal, demonstrating the incorporation of an increasing amount of intact liposomes into the PDA film. The highest amount of liposomes was embedded into the film when liposomes between 25 vol% and 50 vol% were used, suggesting that in these cases more than just a monolayer of liposomes was adsorbed, since both Δf and ΔD were larger as compared to 100 vol% liposomes. The changes in Δf and ΔD were too large that they could only be attributed to the deposition of the PDA coating.
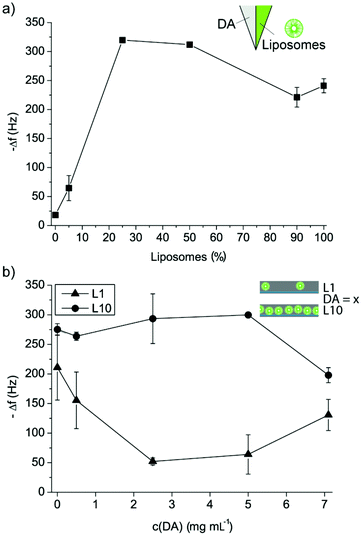 | ||
| Fig. 2 Liposome–PDA film assembly. (a) Change in frequency Δf of PLL pre-coated QCM-D crystals upon binding of different volume mixtures of DA–liposomes. (b) Change in frequency Δf of PLL pre-coated QCM-D crystals upon deposition of mixtures between L1 or L10 and different concentrations of DA in TRIS buffer. | ||
In a next step, we considered coatings assembled by keeping the liposome concentration constant and varying the DA concentration in the mixture (Fig. 2b and ESI Fig. S1b†). The DA concentration was varied between 0.5 and 7.1 mg mL−1 and two different liposome concentrations, namely L1 (5 vol% of the liposome stock solution) and L10, were used, and the film build-up was followed using QCM-D. As expected, mixtures with L1 yielded overall smaller Δf and ΔD of PLL pre-coated crystals as compared to mixtures with L10. Interestingly, with increasing the DA concentration, a decrease in Δf and ΔD of the crystals was measured for L1. This suggests that the few liposomes in solution were coated with PDA,36 and this coating was affecting their deposition. For the highest DA concentration, an increase in Δf and ΔD of the crystal was measured, but this was probably not due to liposome adsorption, but due to the deposition of PDA aggregates from the solution. Further, considering mixtures with L10, increasing Δf and ΔD of the crystals were measured for DA between 0 and 2.5 mg mL−1 suggesting that increasing amounts of liposomes were embedded into the PDA coatings. A concentration of DA = 5 mg mL−1 exhibited similar Δf and ΔD of the crystals as for DA = 2.5 mg mL−1 indicating a saturation of the film with liposomes and PDA. However, DA = 7.1 mg mL−1 yielded lower Δf and ΔD of the crystals, suggesting that at this high concentration, the DA was hindering the liposome deposition. It should be noted that in all cases, the surface got saturated and no continuous deposition of DA or DA–liposomes was observed despite flowing the solution over the crystal which was expected to provide sufficient DA and liposomes for continuous growth. This indicates that the interaction between the liposomes and the DA in solution, i.e. the coating of the liposomes with PDA,36 was affecting the film assembly with time, leading to saturation.
It was found that there was no significant difference in cell viability between these coatings (Fig. 3a), suggesting that the cell adhesion and initial proliferation were not affected by the presence of the liposomes in the PDA matrix. This is in agreement with our previous findings5,27 and those of others6,37 that PDA films in general supported cell adhesion. Fig. 3b shows representative microscopy images of myoblast cells allowed to adhere to 10M2.5 after 24 h to the different coatings. This demonstrates that the subsequent observed differences were due to different access possibilities and/or different amounts of lipids present on the surface and not due to a different number of cells.
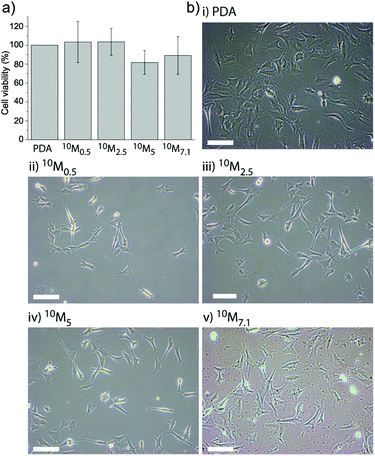 | ||
| Fig. 3 Myoblast cell adhesion to different liposome–PDA coatings. (a) The cell viability of myoblasts adhering to the four different coatings (10M0.5, 10M2.5, 10M5 and 10M7.1 corresponding to the different DA concentrations mixed with L10) as assessed after 24 h shows no significant difference when normalized to cells adhering to a PDA coating without liposomes. (b) Representative microscopy images of myoblast cells allowed adhering to different coatings for 24 h: (i) PDA, (ii) 10M0.5, (iii) 10M2.5, (iv) 10M5, and (v) 10M7.1. The scale bar is 100 μm. | ||
With the goal to determine the uptake/association efficiency of potential cargo by adhering myoblast cells, the amount of fluorescent lipid uptake by cells grown on coatings containing fluorescently labeled liposomes after 6 h and 24 h was assessed by flow cytometry.
Fig. 4a and 4b show the uptake efficiency and the mean fluorescence for cells adhering to the four different coatings (F_10M0.5, F_10M2.5, F_10M5 and F_10M7.1) with embedded fluorescent liposomes (FL), respectively. The amount of fluorescent cells was not significantly different for all the coatings after 6 h and 24 h adhesion time for most of the coatings, except for F_10M0.5, where a reduction after 24 h was observed. This suggests that this coating might be depleting the fluorescent lipids faster than the others, probably due to the lowest amount of DA used during the assembly. There was no significant difference in mean fluorescence neither for cells grown on different coatings after 6 h nor for cells grown on the same coatings for 6 h as compared to 24 h. This means that all the films were able to sustain the fluorescent lipid delivery to the adhering myoblast cells for at least 24 h. Previously, we have seen a decrease in cell mean fluorescence when the coatings were assembled by subsequently depositing liposomes and PDA onto PLL pre-coated glass slides.27 This shows that it is indeed possible to affect the cell response, i.e. the association with fluorescent lipids by myoblast cells depending on the way the coating has been assembled. Further, there was no significant difference in mean fluorescence between myoblasts adhering to most of the coatings after 24 h. The only exception, where a significantly higher mean fluorescence (and amount of fluorescent cells) was measured, was for cells adhering to F_10M5 as compared to F_10M0.5 after 24 h, demonstrating that it is feasible to control the biointerface even with just one liposome deposition step.
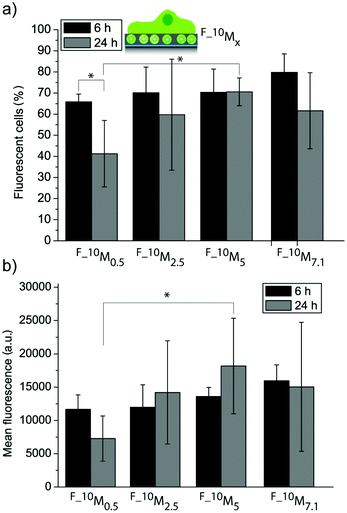 | ||
| Fig. 4 Myoblast cell interaction with liposome–PDA coatings. Uptake efficiency (a) and mean fluorescence (b) of myoblast cells let to adhere to four different coatings assembled from a mixture with L10 and four different DA concentrations (*p < 0.05, n = 3). | ||
The next goal was to entrap an active cargo into the liposomes and deliver it from the surface to the adhering myoblasts. We chose thiocoraline (TC), a small hydrophobic cytotoxic compound, for this report, since the detection of its activity is straightforward by assessing the cell viability. Fig. 5 shows the myoblast cell viability after 24 h and 48 h for cells adhering to two control coatings (C0: PDA30 (PDA coating with a coating time of 30 min), C1: 10M2.5) and four cargo-loaded coatings (S1: 10_TCM0.5, S2: 10_TCM2.5, S3: 10_TCM5, and S4: 10_TCM7.1) normalized to the cell viability measured for cells grown on a bare PDA30 coating. We have already shown that the four different coatings with empty liposomes were not affecting the cell viability (Fig. 3a) and these results are therefore omitted in this figure. It was found that for both time points, the cell viability was significantly lower when TC-loaded liposomes were embedded in the films compared to empty liposomes. Further, the cell viability for myoblasts grown on 10_TCM0.5 was significantly lower as compared to the other three TC-loaded liposome-containing films after 24 h. This again suggests that the cells had easier and faster access to the liposomes (and the TC) of the 10_TCM0.5 coating. After 48 h there was no significant difference in cell viability between cells adhering to the different liposome-containing coatings or compared to the 24 h time point. Although only cells grown on 10_TCM0.5 coatings showed a significant difference in viability after 24 h as compared to the other three TC loaded coatings, it nonetheless demonstrates the feasibility to control the cargo delivery with the film composition, and makes these coatings promising candidates in SMDD.
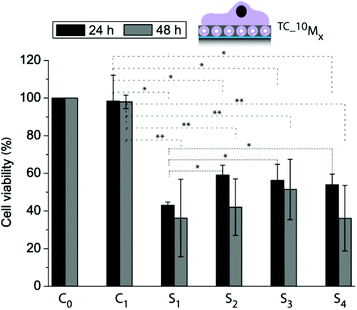 | ||
| Fig. 5 Cell viability of myoblasts allowed adhering to different films for 24 h and 48 h using control coatings: C0: PDA30, C1: 10M2.5, and four cargo-loaded coatings: S1: 10_TCM0.5, S2: 10_TCM2.5, S3: 10_TCM5, and S4: 10_TCM7.1 (*p < 0.05, **p < 0.005, n = 3). | ||
Two liposome deposition steps
Exposing crystal pre-coated with 10M2.5 to DA, L10 or 10M2.5 did not lead to the expected large Δf and ΔD when high numbers of liposomes were deposited (ESI Fig. S2a†). Therefore, in order to increase the amount of liposomes incorporated in the coating during the second deposition step, we employed polymer separation layers (SL) PLL (ESI Fig. S2b†) or PLL/poly(methacrylic acid)-co-(cholesteryl methacrylate) (PMAc) (Fig. 6), a strategy which had previously shown to be successful in assembling multiple liposome layers onto colloidal templates using different polymers than PDA for immobilization.38,39 Cholesterol40 has previously been proven to be a successful non-covalent anchor for liposomes into polymer films and is therefore employed here by using the cholesterol modified polymer PMAc. An intermediate PLL layer (ΔfPLL = −38 ± 11 Hz) was required to allow the subsequent deposition of a small amount of liposomes to a 10M2.5 pre-coated crystal as suggested by the larger ΔD for 10M2.5 than DA. Surprisingly, there was no binding for L10 to 10M2.5/PLL pre-coated crystals observed. On the other hand, when PLL/PMAc (ΔfPMAc = −115 ± 16 Hz) were used as SLs, large Δf and ΔD of 10M2.5/PLL/PMAc pre-coated crystals upon exposure to both 10M2.5 and L10 were measured, proposing the deposition of a large amount of intact liposomes. Similar changes in ΔD suggested similar amounts of deposited liposomes, while larger Δf for 10M2.5 indicated higher contribution of the PDA to the deposited film. Further, this demonstrates that only electrostatic interactions (PLL as SL) were insufficient to immobilize a second layer of liposomes, but cholesterol, as a non-covalent anchor between the polymer and the lipids, was required to deposit zwitterionic liposomes.
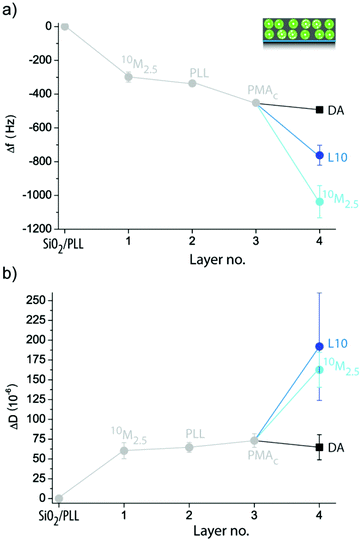
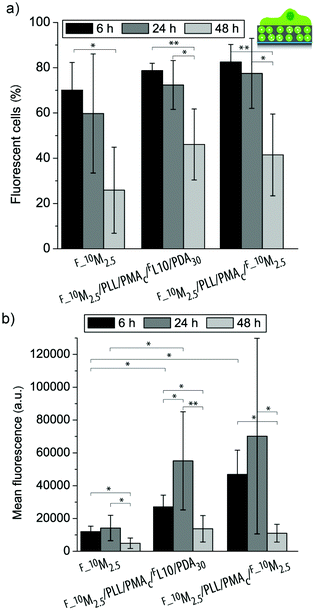 | ||
| Fig. 7 Uptake efficiency (a) and mean fluorescence (b) of myoblast cells let to adhere to three different coatings (F_10M2.5, F_10M2.5/PLL/PMAc/FL/PDA30, and F_10M2.5/PLL/PMAc/F_10M2.5) for 6 h, 24 h and 48 h (**p < 0.005, *p < 0.05, n = 6). | ||
With the goal to assess whether different functional coatings give different cell responses, the liposomes were loaded with TC and the cell viability of adhering myoblast cells was determined after 24 h and 48 h and normalized to cells grown on a PDA30 coating only (Fig. 8). There was no significant difference in viability for cells grown on 10M2.5 as compared to 10M2.5/PLL/PMAc/L10/PDA30 or 10M2.5/PLL/PMAc/10M2.5 after 24 h and 48 h with the exception of 10M2.5 vs. 10M2.5/PLL/PMAc/10M2.5 after 48 h. This suggests that the initial adhesion and proliferation of the cells was largely unaffected by the presence of the high amount of liposomes in the coatings. The viability of cells adhering to coatings with TC loaded liposomes was significantly reduced as compared to films with empty liposomes after 24 h and 48 h. The decrease in cell viability was significantly lower for 10_TCM2.5/PLL/PMAc/LTC/PDA30 as compared to 10_TCM2.5 after 24 h, while there was no significant difference between the different coatings after 48 h. Importantly, when the same amount of TC (9.4 ng) as for one liposome deposition step was delivered from solution, the viability was only reduced by ∼5–10%, demonstrating that the delivery in a substrate-mediated way was more effective, a finding that has previously been reported for the same5,31 or other cargo/coating.41,42
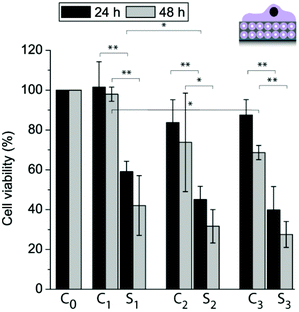 | ||
| Fig. 8 Myoblast cell viability allowed adhering to different coatings for 24 h and 48 h: C0: PDA30, C1: 10M2.5, S1: 10_TCM2.5, C2: 10M2.5/PLL/PMAc/L10/PDA30, S2: 10_TCM2.5/PLL/PMAc/L10TC/PDA30, C3: 10M2.5/PLL/PMAc/10M2.5 and S3: 10_TCM2.5/PLL/PMAc/10_TCM2.5 (*p < 0.05, **p < 0.005, n = 3). | ||
Effect of the polymer separation layers
We assessed the amount of 10M2.5 adsorbed to a PLL/L10/PMAc/(mSL)x pre-coated crystal using a QDM-D depending on the number of PLL/PMAc mSLs. The assembly of PLL/L10/PMAc/(mSL)x is plotted in ESI Fig. S4,† showing that the films were assembling and that neither PLL/PMAc nor PAH/PSS as mSLs were rupturing or displacing the initially deposited L10. With two exceptions, the obtained Δf and ΔD for 10M2.5 were similar with a slight trend to increase with increasing the number of mSL, indicating the deposition of an equivalent of an additional liposome monolayer (Fig. 9 and ESI Fig. S5†). The by far highest Δf was observed for 10M2.5 adsorbing to PLL/L10/PMAc/(PLL/PMAc)1. Higher Δf was observed for 10M2.5 adsorbing to PLL/L10/PMAc/(PAH/PSS)4 corresponding to ca. two monolayers of additionally deposited liposomes. (PAH/PSS) mSL were chosen for comparison to (PLL/PMAc) since they have been demonstrated to provide dense barriers blocking access to the underlying compounds.44
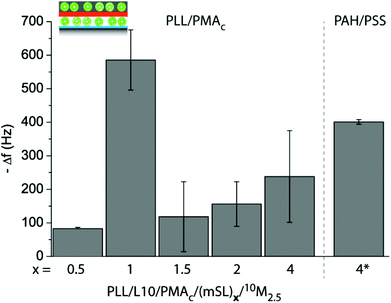 | ||
| Fig. 9 Assembly of coatings using two liposome deposition steps, first L10 and 10M2.5 in the second one, varying the number and type of adsorbed mSL, (PLL/PMAc)x or (PAH/PSS)4. The frequency change Δf of crystals pre-coated with PLL/L10/PMAc/(mSL)x upon exposure to 10M2.5 is shown. | ||
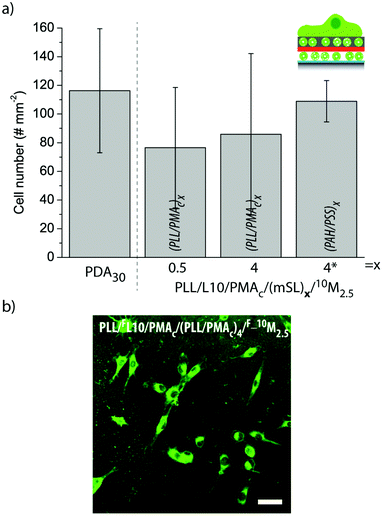 | ||
| Fig. 10 (a) The number of myoblasts adhering to different liposome-containing coatings. The number of cells per 0.915 mm2 is shown. (b) Representative CLSM image of myoblast cells adhering to PLL/FL10/PMAc/(PLL/PMAc)4/F_10M2.5 for 6 h. The scale bar is 50 μm. | ||
Fig. 10b shows a representative CLSM image of myoblast cells adhering to PLL/FL10/PMAc/(PLL/PMAc)4/F_10M2.5 for 6 h. The cells were spread homogeneously across the substrate. As previously observed for this cell type,27,31 the cells internalized the fluorescent marker with its accumulation in the proximity of the nuclei.
In a next step, we aimed to understand to what extent the mSLs are affecting the uptake of the fluorescent lipids from these coatings by the adhering myoblast cells in a more quantitative manner. The uptake efficiency (Fig. 11a) and the mean fluorescence (Fig. 11b) of myoblast cells adhering to PLL/FL10/PMAc/(mSL)x/F_10M2.5 for 6 h or 24 h were assessed by flow cytometry. When comparing (PLL/PMAc)x mSL after 6 h and 24 h, the uptake efficiency was similarly high, while the cell mean fluorescence was significantly higher for (PLL/PMAc)1 as compared to (PLL/PMAc)0.5 or (PLL/PMAc)1.5. This is in good agreement with the QCM-D results where (PLL/PMAc)1 was found to allow for the highest deposition of 10M2.5. Surprisingly though, coatings with (PLL/PMAc)1 did not exhibit significantly higher cell mean fluorescence compared to (PLL/PMAc)2 and (PLL/PMAc)4 as one would expect from the QCM-D results. This might indicate that there is a maximum amount of fluorescent lipids the adhering cells can internalize within this time frame. With the exception of (PLL/PMAc)1, it seemed that increasing the number of mSL (terminating with PMAc) led to higher cell mean fluorescence, which is in agreement with the QCM-D experiments, where higher deposition of 10M2.5 was observed with increasing the number of mSL. Interestingly, there was no significant difference between the two tested time points, indicating that the coatings were able to sustain the delivery of the fluorescent lipids to the adhering cells over this time period. When comparing different types of mSLs, (PAH/PSS)4 vs. (PLL/PMAc)4, the amount of fluorescent cells and their mean fluorescence were only significantly lower after 24 h in the former case, but not after 6 h. This indicates that the access to the underlying FL10 was probably partly hindered by (PAH/PSS)4. Since the QDM-D results showed similar layer assembly for both types of mSLs, faster depleting of the surface of the lipids by the cells for (PAH/PSS)4 is a less likely option.
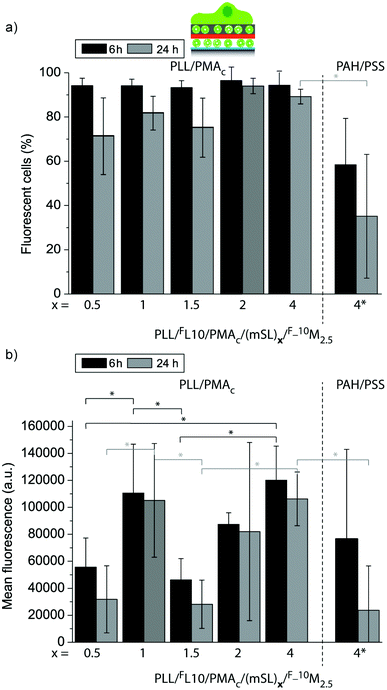 | ||
| Fig. 11 Uptake efficiency (a) and mean fluorescence (b) of myoblast cells let to adhere for 6 h or 24 h to PLL/FL10/PMAc/(mSL)x/F_10M2.5 coatings using different numbers of polymer mSLs between the liposome deposition steps (*p < 0.05, n = 3). | ||
With the goal to get a more detailed kinetics curve of the fluorescent lipid uptake by the adhering myoblasts, cells were let to adhere for different time points to PLL/FL10/PMAc/(PLL/PMAc)0.5/F_10M2.5, and the evolution of the uptake efficiency (left) and mean fluorescence (right) was monitored by flow cytometry (Fig. 12a). The number of fluorescent cells reached a maximum after 6 h, followed by a decrease to ∼40% after 48 h. The cell mean fluorescence decreased by 75% over 48 h. However, it is important to note that the measured intensity was still high (∼25000), indicating that the coatings could provide fluorescent lipids to the cells over this time. This is in contrast to our previous results, where a much more drastic decrease in cell mean fluorescence using liposomes capped with PDA30 over shorter times was observed.27
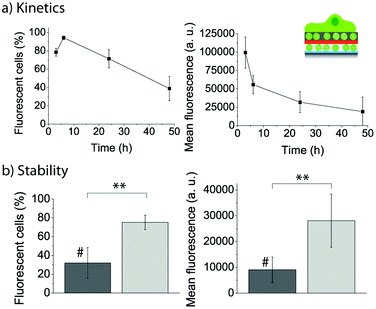 | ||
| Fig. 12 (a) Evolution of the uptake efficiency (left) and mean fluorescence (right) of myoblast cells let to adhere for different time points to PLL/FL10/PMAc/(PLL/PMAc)0.5/F_10M2.5. (b) Comparison of the uptake efficiency (left) and mean fluorescence (right) of myoblast cells let to adhere for 24 h to PLL/FL10/PMAc/(PLL/PMAc)3/F_10M2.5 with (#) and without 24 h pre-incubation in cell media (**p < 0.005, n = 3). | ||
Assessing the stability of the assembled coatings is essential when aiming to use them for biomedical applications. To this end, we pre-incubated PLL/FL10/PMAc/(PLL/PMAc)3/F_10M2.5 coatings for 24 h in cell media (containing 10% FBS) prior to the adhesion of the myoblasts for 24 h. The uptake efficiency of the fluorescent lipids by the cells and their mean fluorescence were compared to cells adhering to the same coating without the pre-incubation step (Fig. 12b). Both parameters were found to be significantly lower for cells adhering to the pre-incubated coatings. This finding indicated that the liposomes (or the polymer) were affected by the presence of the proteins in solution. While this fact is a challenge when SMDD using adhering cells are the aim, it might turn out to be useful when drug elution to cells in suspension is required. Also, we have previously shown that polymer capping layers were able to protect the embedded liposomes with no differences observed between pre-incubated and fresh coatings.31 Therefore, this aspect could be addressed/controlled by depositing additional polymer layers.
Conclusions
Herein, we report the assembly of composite coatings for SMDD using liposomes as embedded drug deposits in a PDA matrix. We characterize the film deposition by exposing substrates to different DA–liposome mixtures. The mean fluorescence and cell viability of myoblast cells adhering to the composite films loaded with fluorescently labeled liposomes or TC loaded liposomes, respectively, can be affected by the DA concentration used to assemble the film. Further, we demonstrate that a second (DA–)liposome deposition step was feasible when the appropriate SLs (i.e. PLL/PMAc) were used. The mean fluorescence and cell viability of myoblast cells adhering were higher or lower, respectively, for specific coatings depending on the amount of liposomal drug deposits present in the film. We showed that the 10M2.5 adsorption to PLL/L10/PMAc/(mSL)x coatings was affected by the number of mSLs with the highest amount of 10M2.5 deposited onto PLL/L10/PMAc/(PLL/PMAc)1 films. When comparing the uptake efficiency and the cell mean fluorescence for myoblasts adhering to PLL/FL10/PMAc/(mSL)x/F_10M2.5 after 6 h and 24 h depending on mSLs, it was found that there was no significant difference between the two time points for the same coating, and mSL terminating in PMAc led to higher cell mean fluorescence. The difference between (PAH/PSS)4 and (PLL/PMAc)4 was only significant after 24 h. Using PLL/FL10/PMAc/(PLL/PMAc)0.5/F_10M2.5 as an example coating, the cell mean fluorescence and uptake efficiency of fluorescent lipids by myoblasts decreased with time, and both parameters were reduced when cells were adhering to coatings pre-incubated in a protein solution.Taken together, we report an in-depth analysis of the assembly of multilayered liposome-containing PDA coatings and demonstrate that the response of adhering myoblasts can be guided by their composition, which makes them promising candidate films for SMDD.
Acknowledgements
This work was supported by a Sapere Aude Starting Grant from the Danish Council for Independent Research, Technology and Production Sciences, Denmark. We gratefully acknowledge Pharmamar S.A. and Prof. Fernando Albericio (Universitat de Barcelona, Spain) for providing the compound Thiocoraline, and Assistant Prof. Alexander N. Zelikin (Aarhus University, Denmark) for access to the flow cytometer and the multiplate reader.Notes and references
- V. Gribova, R. Auzely-Velty and C. Picart, Chem. Mater., 2012, 24, 854 CrossRef CAS
.
- A. N. Zelikin, ACS Nano, 2010, 4, 2494 CrossRef CAS
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426 CrossRef CAS
- M. E. Lynge, R. van der Westen, A. Postma and B. Städler, Nanoscale, 2011, 3, 4916 RSC
- L. Hosta-Rigau, B. E. B. Jensen, K. S. Fjeldsø, A. Postma, G. Guoxin Li, K. N. Goldie, F. Albericio, A. N. Zelikin and B. Städler, Adv. Health. Mater., 2012, 1, 791 CrossRef CAS
- S. H. Ku, J. Ryu, S. K. Hong, H. Lee and C. B. Park, Biomaterials, 2010, 31, 2535 CrossRef CAS
- W. B. Tsai, C. Y. Chien, H. Thissen and J. Y. Lai, Acta Biomater., 2011, 7, 2518 CrossRef CAS
- C. K. Poh, Z. L. Shi, T. Y. Lim, K. G. Neoh and W. Wang, Biomaterials, 2010, 31, 1578 CrossRef CAS
- R. Ogaki, D. T. Bennetsen, I. Bald and M. Foss, Langmuir, 2012, 28, 8594 CrossRef CAS
- M. Lai, K. Y. Cai, L. Zhao, X. Y. Chen, Y. H. Hou and Z. X. Yang, Biomacromolecules, 2011, 12, 1097 CrossRef CAS
- Y. Zhang, B. Thingholm, K. N. Goldie, R. Ogaki and B. Städler, Langmuir, 2012, 28, 17585 CrossRef CAS
- C. J. Ochs, T. Hong, G. K. Such, J. Cui, A. Postma and F. Caruso, Chem. Mater., 2011, 23, 3141 CrossRef CAS
- J. H. An, N. T. Huynh, Y. Sil Jeon and J.-H. Kim, Polym. Int., 2011, 60, 1581 CrossRef CAS
- J. Saiz-Poseu, J. Sedó, B. García, C. Benaiges, T. Parella, R. Alibés, J. Hernando, F. Busqué and D. Ruiz-Molina, Adv. Mater., 2013, 25, 2066 CrossRef CAS
- J. Sedó, J. Saiz-Poseu, F. Busqué and D. Ruiz-Molina, Adv. Mater., 2013, 25, 653 CrossRef
- B. S. Kim, S. W. Park and P. T. Hammond, ACS Nano, 2008, 2, 386 CrossRef CAS
- R. C. Smith, M. Riollano, A. Leung and P. T. Hammond, Angew. Chem., Int. Ed., 2009, 48, 8974 CrossRef CAS
- T. Boudou, P. Kharkar, J. Jing, R. Guillot, I. Pignot-Paintrand, R. Auzely-Velty and C. Picart, J. Control. Release, 2012, 159, 409 CrossRef
- R. R. Sawant and V. P. Torchilin, Soft Matter, 2010, 6, 4026 RSC
- M. Bally, K. Bailey, K. Sugihara, D. Grieshaber, J. Voros and B. Städler, Small, 2010, 6, 2481 CrossRef CAS
- L. Hosta-Rigau, R. Chandrawati, E. Saveriades, P. D. Odermatt, A. Postma, F. Ercole, K. Breheney, K. L. Wark, B. Städler and F. Caruso, Biomacromolecules, 2010, 11, 3548 CrossRef CAS
- B. Städler, A. D. Price, R. Chandrawati, L. Hosta-Rigau, A. N. Zelikin and F. Caruso, Nanoscale, 2009, 1, 68 RSC
- R. Chandrawati, B. Städler, A. Postma, L. A. Connal, S. F. Chong, A. N. Zelikin and F. Caruso, Biomaterials, 2009, 30, 5988 CrossRef CAS
- M. Michel, Y. Arntz, G. Fleith, J. Toquant, Y. Haikel, J. C. Voegel, P. Schaaf and V. Ball, Langmuir, 2006, 22, 2358 CrossRef CAS
- D. V. Volodkin, P. Schaaf, H. Mohwald, J. C. Voegel and V. Ball, Soft Matter, 2009, 5, 1394 RSC
- N. Graf, E. Thomasson, A. Tanno, J. Voros and T. Zambelli, J. Phys. Chem. B, 2011, 115, 12386 CrossRef CAS
- M. E. Lynge, R. Ogaki, A. O. Laursen, J. Lovmand, D. S. Sutherland and B. Städler, ACS Appl. Mater. Interfaces, 2011, 3, 2142 CAS
- N. Graf, A. Tanno, A. Dochter, N. Rothfuchs, J. Voros and T. Zambelli, Soft Matter, 2012, 8, 3641 RSC
- M. Malcher, D. Volodkin, B. Heurtault, P. Andre, P. Schaaf, H. Mohwald, J. C. Voegel, A. Sokolowski, V. Ball, F. Boulmedais and B. Frisch, Langmuir, 2008, 24, 10209 CrossRef CAS
- C. S. O. Paulo, M. Vidal and L. S. Ferreira, Biomacromolecules, 2010, 11, 2810 CrossRef CAS
- M. E. Lynge, M. B. Laursen, L. Hosta-Rigau, B. E. B. Jensen, R. Ogaki, A. A. A. Smith, A. N. Zelikin and B. Städler, ACS Appl. Mater. Interfaces, 2013, 5, 2967 CAS
- Q. Cui, M. M. Ward Muscatello and S. A. Asher, Analyst, 2009, 134, 875 RSC
- J. D. Larkin, K. A. Frimat, T. M. Fyles, S. E. Flower and T. D. James, New J. Chem., 2010, 34, 2922 RSC
- F. Bernsmann, V. Ball, F. D. R. Addiego, A. Ponche, M. Michel, J. J. D. A. Gracio, V. R. Toniazzo and D. Ruch, Langmuir, 2011, 27, 2819 CrossRef CAS
- N. F. Della Vecchia, R. Avolio, M. Alfè, M. E. Errico, A. Napolitano and M. d'Ischia, Adv. Funct. Mater., 2013, 23, 1331 CrossRef CAS
- R. van der Westen, L. Hosta-Rigau, D. S. Sutherland, K. N. Goldie, F. Albericio, A. Postma and B. Städler, Biointerphases, 2012, 7, 8 CAS
- S. H. Ku, J. S. Lee and C. B. Park, Langmuir, 2010, 26, 15104 CrossRef CAS
- R. Chandrawati, L. Hosta-Rigau, D. Vanderstraaten, S. A. Lokuliyana, B. Städler, F. Albericio and F. Caruso, ACS Nano, 2010, 4, 1351 CrossRef CAS
- L. Hosta-Rigau, B. Städler, Y. Yan, E. C. Nice, J. K. Heath, F. Albericio and F. Caruso, Adv. Funct. Mater., 2010, 20, 59 CrossRef CAS
- L. Hosta-Rigau, Y. Zhang, B. M. Teo, A. Postma and B. Städler, Nanoscale, 2013, 5, 89 RSC
- T. Crouzier, K. Ren, C. Nicolas, C. Roy and C. Picart, Small, 2009, 5, 598 CrossRef CAS
- M. Dimitrova, C. Affolter, F. Meyer, I. Nguyen, D. G. Richard, C. Schuster, R. Bartenschlager, J. C. Voegel, J. Ogier and T. F. Baumert, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 16320 CrossRef CAS
- B. Städler, R. Chandrawati, K. Goldie and F. Caruso, Langmuir, 2009, 25, 6725 CrossRef
- D. Mertz, J. Hemmerle, F. Boulmedais, J.-C. Voegel, P. Lavalle and P. Schaaf, Soft Matter, 2007, 3, 1413 RSC
Footnote |
| † Electronic supplementary information (ESI) available: QCM-D results for the film assembly and cell image. See DOI: 10.1039/c3bm60107b |
| This journal is © The Royal Society of Chemistry 2013 |