DOI:
10.1039/C3BM60087D
(Paper)
Biomater. Sci., 2013,
1, 965-974
Preparation and biological characterization of hollow magnetic Fe3O4@C nanoparticles as drug carriers with high drug loading capability, pH-control drug release and MRI properties†
Received
8th April 2013
, Accepted 12th May 2013
First published on 19th June 2013
Abstract
Fe3O4@C nanocapsules were synthesized via a sacrificial-template method by coating SiO2 nanospheres with an Fe3O4@C double-shell structure, followed by dissolving SiO2 nanospheres through ammonia water under hydrothermal conditions. The nanocapsules show a loading capacity as high as 1300 mg g−1 for doxorubicin (DOX), and the DOX loaded on the surface of the carbon shells displays pH-sensitive release behavior. Drug release experiments were carried out at pH 7.4, 6.2 and 5.0. It was found that the drug release rate at pH 6.2 was about two times as fast as that at pH 7.4, and even faster at pH 5.0. A MTT assay was used to test the cytotoxicity of the DOX, nanocapsules and DOX–nanocapsules, which indicated the low cytotoxicity of the nanocapsules towards cells. The DOX–nanocapsules can be taken up by cancer cells through endocytosis, releasing DOX into the cytoplasm, which was observed by both transmission electron microscopy and confocal microscopy. In vitro magnetic resonance imaging (MRI) experiments validated the potential use of these nanocapsules as MRI contrast agents.
1. Introduction
In the past few years numerous nanoparticles have been used for biomedical applications.1–4 For example, polymeric nanoparticles,5 liposomes,6,7 gold nanoparticles,8–10 magnetic nanoparticles (MNPs)11–13 and quantum dots (QDs)14,15 continue to be used in biomedical research to realize the imaging, diagnosis and therapy of some diseases, especially cancer. The design of nanomaterials combining more than one function including drug delivery,16 magnetic resonance imaging,17 fluorescence imaging,18 two-photon imaging19–21 and cell targeting22,23 into a single system has become the trend of this realm. For example, Li et al. synthesized dendrimer-modified QDs and developed a class of Apt-conjugated nanoprobes that realized the targeting and imaging of U251 glioblastoma cells in vitro.15 Nanoparticles have been used in many drug delivery systems because of their excellent propensity to overcome multidrug resistance (MDR),24,25 accumulating in tumor tissue by the enhanced permeability and retention (EPR) effect26 and pH-targeting of the drug.27 For example, Wang et al. developed a drug delivery system by tethering doxorubicin onto the surface of AuNPs via an acid labile linkage and these AuNPs show high efficiency of cellular uptake by endocytosis and realize subsequent acid responsive release in cells which can overcome multidrug resistance.8 Among the multifunctional nanocarriers currently under investigation, magnetic nanoparticles, capable of theragnosis, drug delivery and monitoring of therapeutic response, are expected to play a significant role in the development of personalized medicine. For example, multifunctional drug delivery systems based on Fe3O4 nanoparticles have attracted much attention due to their merit of biocompatibility, magnetic targeting,28 hyperthermia applications,29 and MRI imaging.30
Fe3O4 nanoparticles have been synthesized through numerous ways including the co-precipitation of ferric and ferrous salts in water,31 the oxidation of ferrocene by hydrogen peroxide under solvothermal conditions,32 the thermal decomposition of iron pentacarbonyl and iron acetylacetonate in organic solvents,33etc. However, traditional Fe3O4 nanoparticles always need complex surface modification to form groups that can connect with drugs and they usually show low drug loading capability due to their relatively low surface area. For example Chen et al. coated Fe3O4 nanoparticles with SiO2 and then modified the surface with carboxyl groups which could be used to connect doxorubicin, yet their loading capability only reached 144.2 mg g−1.34 In order to solve the problem porous hollow Fe3O4 nanoparticles were developed as drug delivery systems.35 The inner cavity can not only provide potential space for drug loading, but also reduce the drug loss during circulation in the blood. Additionally, both hydrophilic drugs (such as doxorubicin) and hydrophobic drugs (such as cisplatin) can be loaded in the cavity. For example, Fuchigami et al. reported porous FePt nanocapsules with large pore sizes and large inner cavities and realized the loading and magnetic guidance of doxorubicin.36 Sun and coworkers developed porous hollow Fe3O4 nanoparticles by the controlled oxidation and etching of Fe nanoparticles which are ideal delivery systems for cisplatin, realizing a drug delivery capability as high as 280 mg g−1 (calculated from the Pt/Fe ratio of 25 wt%).37
In addition, it would be highly desirable if triggered drug release could be realized in response to the specific micro-environmental conditions within tumor cells, the design of drug delivery systems that can respond to the acid environment of tumor tissues has become the focus of this realm. In traditional chemotherapy drug distribution in tissue is non-specific and this leads to inevitable systemic side effects and limits the use of high dose strategies.38,39 In order to overcome the drawback of traditional chemotherapy many pH-sensitive drug delivery systems have been developed to realize the selective accumulation of drugs in tumor tissue due to the low pH-value of both the extracellular and endosomal environments of cancer cells.40,41 For example, Bae's group has developed many pH-targeting micelles, TAT shield–deshield nanosystems, virus-like infectious nanogels, and pop-up micelles as drug delivery systems to overcome systemic drug toxicity and multidrug resistance.42–44 Sun et al. realized the pH-controlled release of anticancer 5-FU by means of ZIF-8 owing to its pH-sensitive dissolution property.45 However, they were faced with the problems of a complex synthetic route, complicated surface modification or a low loading capability (less than 1000 mg g−1).
Therefore, developing a multifunctional drug delivery system with high drug loading capability, magnetic resonance imaging property, good biocompatibility and controlled drug release rate has become significant and desirable. As mentioned above, hollow structures can be used to improve the drug loading capability due to their large inner volume and high surface area, while Fe3O4 nanoparticles are traditional contrast agents for MRI. Thus, herein, magnetic Fe3O4 hollow nanostructures were prepared to realize large drug loading capability and magnetic resonance imaging. To further reduce the cytotoxicity of Fe3O4 to normal cells46 and enhance the suspension stability of Fe3O4 nanoparticles, amorphous carbon layers with carboxyl groups need to be coated on the surface of Fe3O4 nanoparticles, forming Fe3O4@C nanocapsules with a good aqueous dispersion. In order to prepare these Fe3O4@C nanocapsules, a procedure based on the solvothermal decomposition of ferrocene on SiO2 nanospheres,47 followed by the etching of the SiO2 ‘core’ was used, carbon layers can be further modified with carboxyl groups. It was found that these nanoparticles showed pH-sensitive zeta potential variation,48 which can be used to change the adsorption properties of the surface layer, and then realize the pH-controlled release of drugs.
2. Experimental section
2.1 Reagents and materials
Ferrocene (Fe(C5H5)2, 98%), hydrogen peroxide (H2O2, 30 wt%), acetone (C3H6O, 99%), tetraethyl silicate (TEOS, 98%), ammonia water (NH3·H2O, 25 wt%) were of analytic grade (AR) from the Shanghai Chemical Factory, China. Doxorubicin hydrochloride was purchased from Sangon Biotech (Shanghai) Co. Ltd. All chemicals were used as received without further purification.
2.2 Preparation of Fe3O4@C nanocapsules
The synthesis of the Fe3O4@C nanocapsules was carried out by coating the SiO2 nanospheres with Fe3O4@C double shells through the solvothermal decomposition of ferrocene that we reported before32,47,48 and is summarized in Scheme 1. Firstly, monodispersed SiO2 nanoparticles with a size of 80 nm were prepared through the traditional Stöber method. Typically, 75 mL ethanol was mixed with 6.0 mL ammonia water (30 wt%) and the mixture was heated to 55 °C with continuous agitation, then 0.8 mL tetraethyl orthosilicate (TEOS) was gradually added to the mixture. After 4 hours of agitation the solution became oyster white in colour, the resulting SiO2 nanoparticles were separated and washed with ethanol and water several times, respectively. Then 25 mg SiO2 nanoparticles and 0.1 g ferrocene were dispersed in 30 mL acetone through intense sonication. After that 2.0 mL hydrogen peroxide (30 wt%) was added drop by drop and the mixture was vigorously stirred for 2 h. Following that the mixture was transferred to a Teflon-lined stainless steel autoclave, then heated to 210 °C and kept at that temperature for 48 hours. The resulting SiO2@Fe3O4@C nanoparticles were gathered through an external magnetic field. Lastly, the inner SiO2 was etched by ammonia water under hydrothermal conditions. In a typical process, 50 mg SiO2@Fe3O4@C nanoparticles were mixed with 15 mL ammonia water and 30 mL deionized water and then transferred to a Teflon-lined stainless steel autoclave, heated to and maintained at 160 °C for 6 hours. The as-prepared hollow Fe3O4@C nanocapsules were gathered through an external magnetic field and washed with deionized water several times.
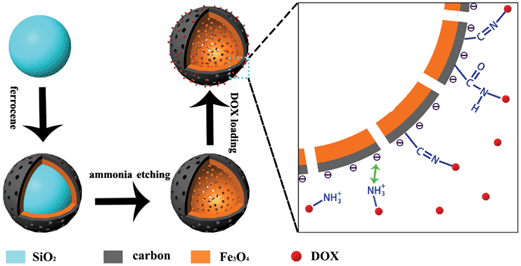 |
| | Scheme 1 A schematic diagram of the preparation and drug loading of the porous hollow Fe3O4@C nanocapsules. Firstly, the SiO2 nanospheres were coated with a Fe3O4@C shell through the decomposition of ferrocene and then the SiO2 was etched by ammonia under hydrothermal conditions. The loading of DOX on to the HMNPs can be realized through the electrostatic interactions of the carbon shell and chemical interactions. | |
2.3 Drug uptake
10 mg DOX was mixed with 5 mg as-prepared Fe3O4@C nanocapsules (HMNPs) which were dissolved in 10 mL phosphate buffer (0.0067 M, pH 7.4). After 24 hours of agitation in a dark environment under room temperature the anti-cancer drug DOX was loaded onto the nanocapsules to form DOX-loaded hollow magnetic nanoparticles (DOX–HMNPs), then the particles were separated through using a magnet (Fig. 1). The concentration of unloaded DOX was determined through UV-vis absorption spectroscopy by detecting the peak at 480 nm. Then the DOX loading content was calculated using eqn (1).| | 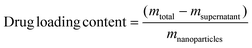 | (1) |
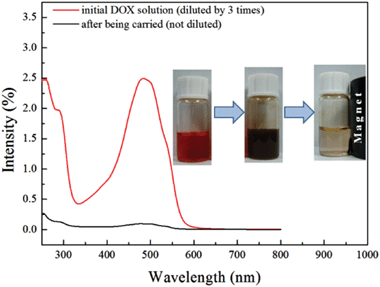 |
| | Fig. 1 UV-vis absorption spectra of DOX in water solution before (red line) and after (black line) loading. The inset pictures show the process of the loading of DOX to the Fe3O4@C nanocapsules: the water solution of DOX was mixed with the Fe3O4@C nanocapsules forming a homogeneous colloid solution, and then the nanocapsules were separated from the solution under an external magnetic field. It can be seen from the pictures that almost all the DOX in water was loaded onto the Fe3O4@C nanocapsules. | |
2.4 Drug release in vitro
Firstly, a dialysis membrane (3500 Da) was prepared by treating the dialysis with boiled EDTA solution (0.02 M) and NaHCO3 (0.02 M) solution. Then the dialysis membrane was washed with phosphate buffer (0.0067 M, pH 7.4) several times to remove the ions adsorbed on the surface of it during the treatment of the dialysis membrane. Subsequently, 5 mg DOX–HMNPs and 10 mL phosphate buffer were added to the dialysis membrane tubing and the tubing was immersed in a 15 mL phosphate buffer. At the same time intervals the outer phosphate buffer was collected (at the same time the identical volume of phosphate buffer was added to replace it) and the concentration of the DOX was determined by UV-vis adsorption spectroscopy. The ratio between Na2HPO4 and NaH2PO4 in PBS was changed to adjust the pH-value of the phosphate buffer and different pH-value (pH = 7.4, 6.2, 5.0) buffers were used to perform the experiments.
2.5 Cell culture and viability assay
MCF-7 cells, from American Type Culture Collection (Rockville, MD) were used to determine the cytotoxicity of the nanoparticles by a tetrazolium dye (MTT) method. Cells were seeded in a 96-well plate and maintained as subconfluent monolayers in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) with 10% fetal bovine serum (FBS, Hyclone) and 100 units per mL penicillin plus 100 g mL−1 streptomycin (Invitrogen) at 37 °C with 8% CO2. After being cultured for 24 or 48 h, the medium was replaced with nanoparticles at different concentrations. For contrast, a medium containing different concentrations of DOX-loaded nanoparticles was also employed. After incubation, MTT solution was put into each well for another 4 h incubation, which allowed the viable cells to reduce the yellow MTT into dark blue formazan crystals. Then an ELISA reader was used to measure the absorbance of each well.
2.6 Confocal microscopy observation
MCF-7 cells were seeded onto sterile, acid-treated, 12 mm coverslips in 24-well plates (Corning Glass Works) and maintained as subconfluent monolayers in DMEM. In general, 24 hours after incubation with nanoparticles, the MCF-7 cells were rinsed three times in PBS and fixed in freshly prepared 4% paraformaldehyde in PBS and then rinsed three times in PBS. Cells on the coverslips were blocked with 0.05% Tween 20 in PBS (TPBS) with 1% bovine serum albumin (Sigma). These cells were incubated with various primary antibodies in a humidified chamber for 1 h and then washed three times in TPBS. Filamentous actin was labeled with Alexa Fluor 488 Phalloidin (Invitrogen) while DNA was stained with DAPI (Sigma) for verification of internalization. Coverslips were supported on slides by grease pencil markings and mounted in Vectashield (Vector Laboratories). Images were taken with a laser scanning microscope (Zeiss LSM 710) using a 63 × 1.3 numerical aperture Plan Apo objective. Figures were constructed using Adobe Photoshop. In the test for assessing the NIR improving the drug release rate, the cells were seeded and cultured in the same manner except that the nanoparticles were replaced by DOX-loaded nanocarriers.
2.7 Bio-transmission electron microscopy observation
MCF-7 cells were seeded onto sterile, acid-treated 6-well plates (Corning Glass Works) and maintained as subconfluent monolayers in DMEM. After incubation with nanoparticles for 24 h, cells were collected and fixed in PBS plus 2.5% glutaraldehyde (Sigma). After dehydration through an ethanol series twice (70% for 15 min, 90% for 15 min, and 100% for 15 min), cells were embedded in Epon Araldite resin (polymerization at 65 °C for 15 h). Thin sections containing the cells were then cut, placed on copper grids, and stained with 4% uranyl acetate and 0.2% Reynolds lead citrate (water). The sections were examined by a Tecnai F20 cryoEM (FEI).
2.8
In vitro MRI
The in vitro MRI images (T2-weighted) and T2 measurements were performed using a MRI imager (Bruker BIOSPEC-47/30 MRI imager at 3.0 T). A multislice and multiecho (MSME) sequence with a frequency of 200 MHz, repetition time of 3000 ms, echo time from 11 ms to 330 ms, matrix size 128 × 128 mm2, slice thickness of 1.6 mm, was used for all the T2-weighted imaging. The products were dispersed in PBS buffer (pH = 7.4) of various concentrations of Fe (0.5 mg mL−1, 1.25 mg mL−1, 2.5 mg mL−1, 5 mg mL−1, 10 mg mL−1 and 20 mg mL−1), and loaded in NMR tubes (Norell, Quality NMR sample tubes 502-8, USA).
2.9 Characterization
Powder X-ray diffraction (XRD) patterns were collected on a Japan Rigaku D/MAX-γA X-ray diffractometer equipped with Cu Kα radiation (λ = 1.54178 Å) over the 2θ range 10–70°. Transmission electron microscopy (TEM) images were obtained on a Hitachi H-800 transmission electron microscope, using an accelerating voltage of 200 kV. Scanning electron microscopy (SEM) images were taken on a JSM-6700F (JEOL). A superconducting quantum interference device (SQUID) magnetometer (Quantum Design MPMS XL-7) was used to measure the magnetic properties of the as-prepared samples. The concentration of doxorubicin was measured by means of spectrophotometry on a SolidSpec-3700 (SHIMADZA). The surface area and pore volume were determined by accelerated surface area and porosimetry (ASAP 2020 M+C). The FTIR spectrum was measured on a Nicolet 8700 Spectrometer. The optically counted particle size distribution was revealed on a Nanosight LM10-HSB. Surface charge was measured by a Particle Analyzer (Delsa Nano C) from Beckman Coulter.
3. Results and discussion
3.1 Property of magnetic nanocapsules
Fig. 2 is the XRD pattern of the sample, the appearance of peaks (311), (440) and (511) indicate the existence of Fe3O4 (JCPDS file 19-0629, magnetite, or JCPDS file 39-1346, maghemite), in accordance with the XPS analysis (Fig. S1†). The binding energies near 710 and 724 eV indicate the presence of Fe(II) and Fe(III), and the peaks around 531 and 285 eV are the characteristic peaks of O1s and C1s. The TEM and SEM images indicate that all the nanoparticles in the process of synthesis are uniformly distributed, including SiO2 nanoparticles, SiO2@Fe3O4@C nanoparticles and Fe3O4@C nanocapsules. The TEM images (Fig. 3a–c) show that the particle sizes of these three types of particles are 80 nm, 120 nm and 120 nm, respectively. From Fig. 3c, a 80 nm cavity in the center of the nanocapsules can be observed, in accordance with the size of the SiO2 templates. The SEM image of the Fe3O4@C nanocapsules (Fig. 3f) indicates that the surface of the nanocapsules is much rougher than that of the SiO2@Fe3O4@C nanoparticles. This phenomenon is caused by the etching of the ammonia water to the carbon layer under hydrothermal conditions, which also helps to increase the surface area of the nanocapsules and enhances the loading capability of doxorubicin.
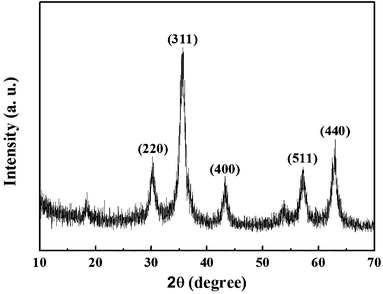 |
| | Fig. 2 XRD pattern of Fe3O4@C nanocapsules. The pattern indicates the existence of Fe3O4. | |
The nitrogen adsorption–desorption isotherm of the Fe3O4@C nanocapsules (Fig. S2†) belongs to the type-IV isotherm, which indicates the presence of mesopores on the nanocapsules. The isotherm also reveals a surface area of about 159.84 m2 g−1. Fig. S2b† is the pore distribution of Fe3O4@C, calculated by the BJH method. A sharp peak near 5 nm can be observed, corresponding to the mesopores on the surface of the carbon shell. The relatively high surface area, the mesoporous structure and the inner cavity suggest the high drug loading capacity of these nanocapsules.
The magnetic hysteresis loop (Fig. S3†) of these nanocapsules was measured at room temperature (300 K) in an applied magnetic field of up to 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Oe. The saturation magnetization is as high as 55.93 emu g−1, and the inserted figure displays the superparamagnetic characteristic of the sample, showing the potential application of these nanoparticles in magnetic resonance imaging.
000 Oe. The saturation magnetization is as high as 55.93 emu g−1, and the inserted figure displays the superparamagnetic characteristic of the sample, showing the potential application of these nanoparticles in magnetic resonance imaging.
3.2 Drug uptake and pH-control release
DOX was first dissolved in distilled water to form a red solution and then the HMNPs were added. After several hours of agitation, DOX can be loaded on the HMNPs. A large part of DOX will be loaded on the outer surface of the HMNPs due to the greater absorption capability of the amorphous carbon. Then the DOX–HMNPs can be separated from solution under an external magnetic field. 1 mol mL−1 DOX and 5 mol mL−1 HMNPs were used in this process, and Fig. 1 (inserted) shows that the solution is totally clear and without any color, suggesting nearly 100% uptake efficiency of HMNPs. Meanwhile, the UV-vis absorption spectrum in Fig. 1 also proves this result. The characteristic peak of DOX at 480 nm is very high in the original DOX solution, while after being treated with HMNPs, the peak at 480 nm is almost negligible which indicates that all the DOX has been loaded on the HMNPs. However, the drug loading capability of the HMNPs is finite and an excessive amount of DOX was used in this experiment to determine the maximum loading capability of the HMNPs. According to our experiment the drug loading capability of these kinds of HMNPs is about 1300 mg g−1 calculated by eqn (1). The loading capability is much higher than the drug delivery systems, especially the Fe3O4 drug delivery systems, reported before which only realized a drug loading capability of 144 mg g−134 or even did not reach 100 mg g−1.49 The drug loading capability of the HMNPs is also higher than that of SiO2@Fe3O4@C which is below 400 mg g−1. This high loading capability was attributed to the greater adsorption capability of the outer carbon layer and the inner cavity. On the one hand the carbon layer provides various organic functional groups to form chemical bonds with DOX. On the other hand the inner cavity lowers the weight of the nanoparticles and increases the surface area.
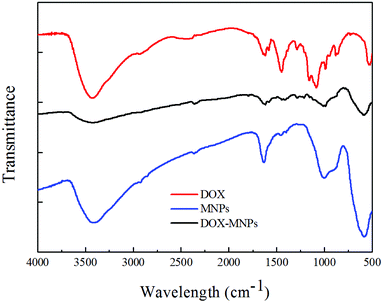
The FTIR (Fig. 4) and Raman (Fig. S4†) spectra for the HMNPs indicate the existence of carbonyl, carboxyl and hydroxyl groups on the outer surface of the HMNPs, these groups can form chemical bonds with the functional groups of DOX. For example, the amino and hydroxyl groups of DOX can interact with the carbonyl and carboxyl groups to form hydrazone and amido link, respectively. Moreover, both the hydrolytic action of hydrazone8 and amidation34 are pH-dependent, therefore, the drug release rate of DOX can be controlled through pH variation, as shown in Fig. 5. The drug release at pH 6.2 in 30 hours is about three times as much as that at pH 7.4, and the release rate is even faster at pH 5.0. The drug release rate of this system is much more sensitive to pH than many other drug delivery systems. For example, the drug release percentage of mesoporous silica only doubled when the pH dropped from 7.4 to 4.0 in one hour.50 There is an equilibrium between the DOX loaded on our delivery system and the DOX in PBS. With the release of DOX, the loaded DOX decreases while the DOX in PBS increases and the DOX in PBS will restrain the release rate. That is the reason why the release does not follow a straight line. It is worth noting that even though the release rate is slow after a long time, the release does not reach a plateau, if you refresh the PBS in the release experiment the drug release will go on. Besides the sensitivity of hydrazone and amidation to pH, the electrostatic interaction also contributes to the pH-dependent release rate. The zeta potential of the magnetic nanocapsules in different pH value solutions was also measured, which showed that the surface of the particles was negatively charged at a pH higher than 3, at pH 5.0–7.4, the ionization of the carboxyl groups on the HMNPs formed COO− and the amino groups of DOX combined with the hydrogen ions to form NH3+, the electrostatic interactions of COO− and NH3+ also contributed to the loading of DOX on the HMNPs. With the pH drop from 7.4 to 5.0 the zeta potential of the HMNPs rises which means that the surface of the HMNPs becomes less negative. The less negative the surface, the weaker the interactions between the HMNPs and DOX are, so DOX is more inclined to be released in low pH-value environments. The interactions between the HMNPs and DOX are summarized in Scheme 1.
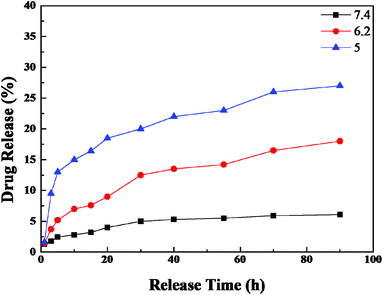 |
| | Fig. 5 The pH-dependent release of DOX from DOX–HMNPs. The DOX–HMNPs were cultured in PBS at pH 7.4, 6.2 or 5 at 37 °C. At each pH, the concentration of DOX was detected by UV-vis absorption spectroscopy. | |
3.3 pH controlled release of DOX–HMNPs in vitro
It has been reported that the pH-value of the intracellular and extracellular environment of normal cells and cancer cells are different.51 The pH-value of blood and a normal cells’ intracellular environment is about 7.4 and the intracellular pH of both cancer cells and normal cells is about 7.2 (a cancer cells’ intracellular pH is just a little bit lower than 7.2 because of their fast glycolysis52). The drug release experiment carried out at pH 7.4 can be used to simulate the behavior of DOX–HMNPs when they are injected into blood or in the intracellular environment or the cytoplasm of normal cells. The release rate at pH 7.4 is pretty low. This both reduces the drug loss in the process of transportation by blood and alleviate the damage of DOX to normal cells. Additionally, even though the intracellular pH of a cancer cell is around 7.2 the endosomes and lysosomes formed in the process of endocytosis contain much more acid than cytoplasm and their pH-value is about 5.0–6.5.41,53 The drug release experiment carried out at pH 5.0 corresponded to this condition. What's more, the accumulation of lactic acid and carbonic acid around cancer cells leads to the low pH-value (varying from 5.8 to 7.8) of the extracellular environment of cancer cells40 (corresponding to the drug release experiment carried out at pH 6.2). Thus when the DOX–HMNPs accumulate in the intracellular environment of cancer cells due to the enhanced permeability and retention (EPR) effect or get into cells through endocytosis (for our HMNPs the clathrin-mediation is more important than other subcategories of endocytosis because it will lead to the formation of primary endosomes which will form endosomes and lysosomes in the end41) they will be exposed to a low pH environment and the release of DOX will accelerate (Scheme 2). According to our previous work, about 10−9 mg DOX may be enough to kill one MCF-7 cancer cell.20 Based on our own experiment the half lethal dose of DOX to MCF-7 is about 10 mM.
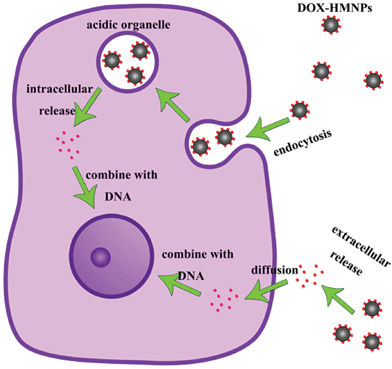 |
| | Scheme 2 The extracellular and intracellular drug release process of DOX–HMNPs in a cancer cell. | |
Except for the pH-controlled release property of the DOX–HMNPs, both the size and surface charge of the HMNPs is optimal for drug delivery. The size of the HMNPs is about 120 nm which is large enough to cause the enhanced permeability and retention (EPR) effect54 and the HMNPs can accumulate around the cancer cells at concentrations much higher than in normal tissues. What's more, generally the larger the nanoparticles are the easier they are to be cleaned by immune system55 and the nanoparticles around 100 nm also show ideal distribution and good clearance/accumulation behavior in the human body.56 Additionally, the polar functional groups on the surface of the HMNPs make it hydrophilic and the ionization of the carboxyl groups will lead to the surface being negatively charged. The zeta potential48 of the HMNPs measured at pH 6 is about −30 mV which prevents the HMNPs from aggregating when they are injected into blood.
3.4
In vitro cytotoxicity of DOX, HMNPs and DOX–HMNPs
The anticancer efficacy of the DOX–HMNPs was tested on MCF-7 cancer cells using a traditional MTT assay. Fig. 6 shows the cell viabilities after being cultured with DOX, HMNPs or DOX–HMNPs for 24 h. Fig. 6 reveals that the HMNPs show negligible cytotoxic effects and an apoptosis rate of only about 20% is observed when the concentration of the HMNPs is extremely high (1000 mg L−1). It has been reported that the cytotoxicity of Fe3O4 comes from ROS production and oxidative stress, liberation of toxic Fe2+ and the disturbance of the electronic or ion transport activity in the cell membrane.57,58 However, our Fe3O4 is coated with amorphous carbon which will protect the Fe3O4 being etched by extracellular and intracellular acid. Additionally, cell “vision” is also an important factor in nanotoxicology. Previously, Mahmoudi's group has reported that when considering the cell “vision” the cytotoxicity of superparamagnetic iron oxide nanoparticles will be lower than most cells.59 Thus, the cytotoxicity of our HMNPs may be less than that determined by the traditional MTT assays. Considering the fact that the HMNPs will not influence the cell viability in low concentrations, the apoptosis rate mainly comes from the toxicity of DOX instead of Fe3O4. The cytotoxic effects of both DOX and DOX–HMNPS were enhanced with the increase in the drug concentration. Additionally, it is worth noting that the DOX–HMNPs show less cytotoxicity than free DOX when the concentration of the drug is equal. This phenomenon comes from the fact that when DOX is loaded into HMNPs it needs a long time to release even in low pH-value environments. The extracellular pH of cancer cells is much lower than that of normal cells so it can be inferred that when the normal cells are cultured with DOX–HMNPs higher cell viability will be observed due to the slower drug release rate. This will protect the normal cell from being killed by DOX in the therapy.
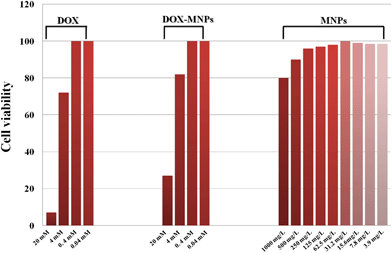 |
| | Fig. 6 Cell viabilities of MCF-7 cells cultured with free DOX, HMNPs and DOX–HMNPs. | |
3.5 Cell uptake
The main mechanism for nanoparticles to be taken up by cells is endocytosis, which includes several subcategories such as phagocytosis, pinocytosis, clathrin-dependent receptor-mediated and caveolae-dependent endocytosis. A previous paper8 indicated that caveolae and clathrin mediated endocytosis were crucial to the entrance of nanoparticles. For our DOX–HMNPs, clathrin-mediated endocytosis is the most important type of endocytosis not only because it is the main approach for a nanoparticle’s entrance but also because it leads to the formation of primary endosomes which will subsequently become acidic organelles. The low pH value in acidic organelles will accelerate the release of DOX from HMNPs, which will realize the accumulation of drugs to the level for cells to be killed rapidly.
Both transmission electron microscopy and confocal laser microscopy were used to instigate the mechanism of the intracellular drug release of the DOX–HMNPs. MCF-7 cells were used to conduct this experiment and the cells were cultured with DOX–HMNPs at 37 °C for 24 hours in physiological conditions. Fig. 7a and b are the TEM images of the cancer cells after 24 hours culture. These images show the intracellular distribution of the DOX–HMNPs and the nanoparticles were found mainly in the cytoplasm region of the cells. However, only a small amount of the DOX–HMNPs can be found in the nucleus due to the hindrance of the nuclear membrane to the nanoparticles. It should be mentioned that the DOX–HMNPs are most likely concentrated in the primary endosomes which are formed in the process of endocytosis, instead of evenly distributing in the cytoplasm. This phenomenon also confirms the fact that the uptake of DOX–HMNPs is carried out by endocytosis. It is worth noting that we replaced the media from fetal bovine serum, which contains rich protein, to PBS, which contains no protein, when we added HMNPs into the petri dish. So the protein corona that formed on the surface of our HMNPs will be different from that when cultured in human plasma or in the human body. Because the protein corona greatly influences the interaction between the nanoparticles and cells,60 our experiment here cannot predict the behavior of the HMNPs in the human body completely.
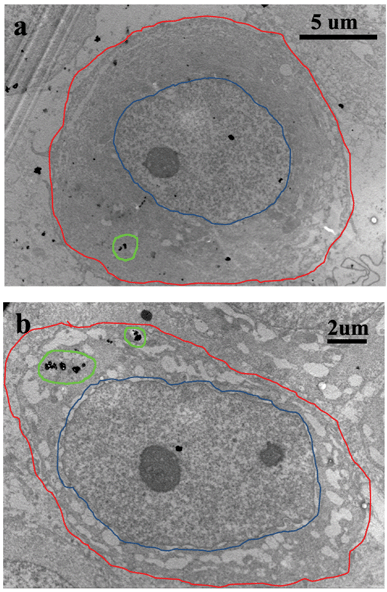 |
| | Fig. 7 TEM images of MCF-7 cells after they were cultured with DOX–HMNPs for 24 hours. The red line corresponds to the cell membrane, the blue line corresponds to the nuclear membrane. The distribution of HMNPs in the cytoplasm is marked with a green line. The nanocapsules mainly localized in the cytoplasm, small nanocapsules were found in cell nucleus. | |
In order to observe the release of DOX from the DOX–HMNPs to the nucleus, confocal microscopy images were also taken. In this experiment MCF-7 cancer cells were cultured with free DOX or DOX–HMNPs for 24 hours, and the red fluorescence of DOX was used to observe the distribution of DOX in the cells. Two differences between the DOX group and the DOX–HMNPs group can be observed. Firstly, after 24 hours culture (Fig. 8) the cancer cells cultured with DOX–HMNPs show more red fluorescence than the cells cultured with free DOX, this phenomenon indicates that DOX–HMNPs are more easily taken up by cells than free DOX.28 Secondly, the DOX mainly dispersed in the nucleus when the cells were cultured with free DOX. Yet the red fluorescence of DOX can be observed in both the nucleus and the cytoplasm when using the DOX–HMNPs. Additionally, the intensity of the fluorescence reveals that the concentration of DOX in the nucleus is slightly less than that in the cytoplasm (Fig. S5† also confirms this conclusion). The free DOX group shows negligible fluorescence in the cytoplasm because the DOX will quickly combine with the DNA in the nucleus when entering the cancer cells. However, when DOX was combined with HMNPs it cannot reach the nucleus due to the hindrance of the nuclear membrane and the release rate of DOX from the DOX–HMNPs is limited so some of the DOX is still kept in the cytoplasm when cultured with cells for 24 hours. In a nutshell, we can realize the accumulation and controlled release of DOX thorough cultured MCF-7 cancer cells with DOX–HMNPs.
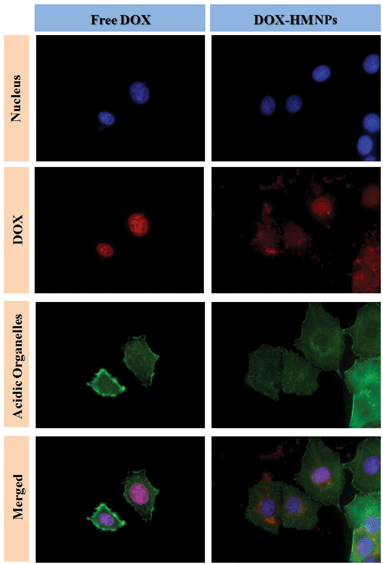 |
| | Fig. 8 Confocal laser microscopy observation of MCF-7 cells cultured with free DOX or DOX–HMNPs for 24 h. The dose of DOX or its equivalent was 5 μg mL−1 in the cell culture. The cells were counterstained with DAPI (blue) for the cell nucleus and Alexa Fluor 488 phalloidin (green) for the cell membrane. | |
3.6
In vitro MRI
Superparamagnetic Fe3O4 has been reported as an excellent contrast agent for MRI.61 In order to determine whether our Fe3O4@C nanocapsules can be used as contrast agents, the contrast effect of the nanocapsules in aqueous solution was measured at different Fe concentrations (determined by ICP-AES) on a clinical 3.0 T magnetic resonance system at room temperature. Fig. 9a is the T2-weighted MR imaging of Fe3O4@C nanocapsules. As shown in this figure, the image changes remarkably in signal intensity with the increase in Fe concentration. Fig. 9b show that the relaxivity R2(1/T2) increases linearly with the increase in Fe concentration which indicates that the Fe3O4@C nanocapsules can generate MRI contrasts on T2-weighed spin-echo sequences. From the plot in Fig. 9b we can calculate that the specific relaxivity of the Fe3O4@C nanocapsules is 54.55 Fe mM−1 s−1, showing the potential application of the HMNPs as MRI contrast agents. The crystallinity of the Fe3O4 synthesized through the hydrothermal decomposition of ferrocene is relatively low. This is also why our R2 only reaches 54.55 Fe mM−1 s−1. Our R2 value is lower than Feridex (160 s−1 mM−1, 72 nm) and a commercial MRI agent such as Resovist (164 s−1 mM−1, 65 nm).62 But our R2 values are comparable to the Fe3O4 synthesized through the thermal decomposition of iron acetylacetonate which is about 65 s−1 mM−1.63
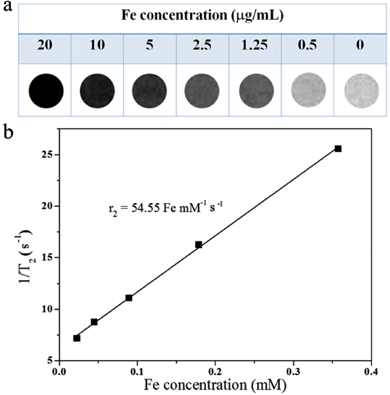 |
| | Fig. 9 (a) T2-weighted MR imaging of Fe3O4@C nanocapsules suspended in water at different Fe concentrations; (b) a plot of the T2 relaxation rate R2(1/T2) against the Fe concentration of Fe3O4@C nanocapsules. | |
4. Conclusions
In summary, we developed a drug delivery system of magnetic hollow Fe3O4@C nanoparticles. It was found that the DOX loading capability of the nanocapsules can reach 1300 mg g−1. The electrostatic interaction between the carbon shell and DOX, the chemical bonds between the surface functional groups and DOX and the inner volume of the nanocapsules all contribute to DOX loading. This drug delivery system shows the pH controlled release of DOX in acidic environments. Additionally, the outer carbon layer improves the biocompatibility and colloid stability of the nanocapsules, and the MTT assay showed the low cytotoxicity of the nanocapsules. TEM and confocal microscopy observations revealed that the DOX–HMNPs can be taken up by cancer cells through endocytosis and release DOX in cytoplasm. The in vitro MRI measurements indicated the potential use of the nanocapsules as contrast agents. Thus the Fe3O4 nanocapsules can realize the combination of pH controlled release, drug delivery and magnetic resonance imaging to form a multifunctional drug delivery system.
Acknowledgements
This work was supported by the National Natural Science Foundation (NSFC, 21071137) (Q. W. Chen), 31100992 (Z. Guo), Anhui Province Project Grant 11040606Q54, 08040102005 (Z. Guo), Doctoral Fund of the Ministry of Education of China 20113402130010 (Z. Guo).
References
- M. Al Kobiasi, B. Y. Chua, D. Tonkin, D. C. Jackson and D. E. Mainwaring, J. Biomed. Mater. Res., Part A, 2012, 100A, 1859–1867 CrossRef CAS.
- J. Z. Zhang, J. Phys. Chem. Lett., 2010, 1, 686–695 CrossRef CAS.
- K.-S. Lee and M. A. El-Sayed, J. Phys. Chem. B, 2006, 110, 19220–19225 CrossRef CAS.
- C. C. Lee, J. A. MacKay, J. M. J. Frechet and F. C. Szoka, Nat. Biotechnol., 2005, 23, 1517–1526 CrossRef CAS.
- L. E. van Vlerken, Z. Duan, M. V. Seiden and M. M. Amiji, Cancer Res., 2007, 67, 4843–4850 CrossRef CAS.
- P. Pradhan, J. Giri, F. Rieken, C. Koch, O. Mykhaylyk, M. Doeblinger, R. Banerjee, D. Bahadur and C. Plank, J. Controlled Release, 2010, 142, 108–121 CrossRef CAS.
- A. Schroeder, J. Kost and Y. Barenholz, Chem. Phys. Lipids, 2009, 162, 1–16 CrossRef CAS.
- F. Wang, Y.-C. Wang, S. Dou, M.-H. Xiong, T.-M. Sun and J. Wang, ACS Nano, 2011, 5, 3679–3692 CrossRef CAS.
- C. H. J. Choi, C. A. Alabi, P. Webster and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 1235–1240 CrossRef CAS.
- J. Nam, N. Won, H. Jin, H. Chung and S. Kim, J. Am. Chem. Soc., 2009, 131, 13639–13645 CrossRef CAS.
- Y. Chen, H. Chen, S. Zhang, F. Chen, S. Sun, Q. He, M. Ma, X. Wang, H. Wu, L. Zhang, L. Zhang and J. Shi, Biomaterials, 2012, 33, 2388–2398 CrossRef CAS.
- H. Wu, S. Zhang, J. Zhang, G. Liu, J. Shi, L. Zhang, X. Cui, M. Ruan, Q. He and W. Bu, Adv. Funct. Mater., 2011, 21, 1850–1862 CrossRef CAS.
- H. Zhou, J. Chen, E. Sutter, M. Feygenson, M. C. Aronson and S. S. Wong, Small, 2010, 6, 412–420 CrossRef CAS.
- C. Walther, K. Meyer, R. Rennert and I. Neundorf, Bioconjugate Chem., 2008, 19, 2346–2356 CrossRef CAS.
- Z. Li, P. Huang, R. He, J. Lin, S. Yang, X. Zhang, Q. Ren and D. Cui, Mater. Lett., 2010, 64, 375–378 CrossRef CAS.
- J. Ren, S. Shen, D. Wang, Z. Xi, L. Guo, Z. Pang, Y. Qian, X. Sun and X. Jiang, Biomaterials, 2012, 33, 3324–3333 CrossRef CAS.
- C. Zhang, X. Xie, S. Liang, M. Li, Y. Liu and H. Gu, Nanomed.: Nanotechnol., Biol. Med., 2012, 8, 996–1006 CrossRef CAS.
- Y. Wei, Q. Chen, B. Wu, A. Zhou and D. Xing, Nanoscale, 2012, 4, 3901–3909 RSC.
- J. Qian, D. Wang, F. Cai, Q. Zhan, Y. Wang and S. He, Biomaterials, 2012, 33, 4851–4860 CrossRef CAS.
- J. Chen, Z. Guo, H.-B. Wang, M. Gong, X.-K. Kong, P. Xia and Q.-W. Chen, Biomaterials, 2013, 34, 571–581 CrossRef CAS.
- Y.-M. Zhou, M. Gong, Z. Sun, K. Cheng, Z. Guo and Q. Chen, Dalton Trans., 2013 10.1039/C3DT50789K.
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Adv. Drug Delivery Rev., 2008, 60, 1615–1626 CrossRef CAS.
- X. Shi, S. H. Wang, M. E. Van Antwerp, X. Chen and J. R. Baker, Jr., Analyst, 2009, 134, 1373–1379 RSC.
- Q. Yin, J. Shen, L. Chen, Z. Zhang, W. Gu and Y. Li, Biomaterials, 2012, 33, 6495–6506 CrossRef CAS.
- D. Vergara, C. Bellomo, X. Zhang, V. Vergaro, A. Tinelli, V. Lorusso, R. Rinaldi, Y. M. Lvov, S. Leporatti and M. Maffia, Nanomed.: Nanotechnol., Biol. Med., 2012, 8, 891–899 CrossRef CAS.
- Y. Yang, F. An, Z. Liu, X. Zhang, M. Zhou, W. Li, X. Hao, C.-S. Lee and X. Zhang, Biomaterials, 2012, 33, 7803–7809 CrossRef CAS.
- B.-X. Zhao, Y. Zhao, Y. Huang, L.-M. Luo, P. Song, X. Wang, S. Chen, K.-F. Yu, X. Zhang and Q. Zhang, Biomaterials, 2012, 33, 2508–2520 CrossRef CAS.
- Y. Chen, H. Chen, S. Zhang, F. Chen, L. Zhang, J. Zhang, M. Zhu, H. Wu, L. Guo, J. Feng and J. Shi, Adv. Funct. Mater., 2011, 21, 270–278 CrossRef CAS.
- G. Bealle, R. Di Corato, J. Kolosnjaj-Tabi, V. Dupuis, O. Clement, F. Gazeau, C. Wilhelm and C. Menager, Langmuir, 2012, 28, 11834–11851 CrossRef CAS.
- D. K. Kim, M. Mikhaylova, F. H. Wang, J. Kehr, B. Bjelke, Y. Zhang, T. Tsakalakos and M. Muhammed, Chem. Mater., 2003, 15, 4343–4351 CrossRef CAS.
- M. Racuciu, D. E. Creanga and A. Airinei, Eur. Phys. J. E, 2006, 21, 117–121 CrossRef CAS.
- H. Wang, Y. Sun, Y. Yu, J. Chen, R. Li, K. Cheng and Q. Chen, Dalton Trans., 2012, 41, 346–350 RSC.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS.
- F. H. Chen, Q. Gao and J. Z. Ni, Nanotechnology, 2008, 19, 165103 CrossRef CAS.
- Q. He, Z. Wu and C. Huang, J. Nanosci. Nanotechnol., 2012, 12, 2943–2954 CrossRef CAS.
- T. Fuchigami, R. Kawamura, Y. Kitamoto, M. Nakagawa and Y. Namiki, Biomaterials, 2012, 33, 1682–1687 CrossRef CAS.
- K. Cheng, S. Peng, C. Xu and S. Sun, J. Am. Chem. Soc., 2009, 131, 10637–10644 CrossRef CAS.
- K. T. Oh, E. S. Lee, D. Kim and Y. H. Bae, Int. J. Pharm., 2008, 358, 177–183 CrossRef CAS.
- N. Carelle, E. Piotto, A. Bellanger, J. Germanaud, A. Thuillier and D. Khayat, Cancer, 2002, 95, 155–163 CrossRef.
- K. Engin, D. B. Leeper, J. R. Cater, A. J. Thistlethwaite, L. Tupchong and J. D. McFarlane, Int. J. Hyperthermia, 1995, 11, 211–216 CrossRef CAS.
- C. Brandenberger, C. Muehlfeld, Z. Ali, A.-G. Lenz, O. Schmid, W. J. Parak, P. Gehr and B. Rothen-Rutishauser, Small, 2010, 6, 1669–1678 CrossRef CAS.
- K. M. Huh, H. C. Kang, Y. J. Lee and Y. H. Bae, Macromol. Res., 2012, 20, 224–233 CrossRef CAS.
- M. D. Howard, A. Ponta, A. Eckman, M. Jay and Y. Bae, Pharm. Res., 2011, 28, 2435–2446 CrossRef CAS.
- A. Ponta and Y. Bae, Pharm. Res., 2010, 27, 2330–2342 CrossRef CAS.
- C.-Y. Sun, C. Qin, X.-L. Wang, G.-S. Yang, K.-Z. Shao, Y.-Q. Lan, Z.-M. Su, P. Huang, C.-G. Wang and E.-B. Wang, Dalton Trans., 2012, 41, 6906–6909 RSC.
- T. R. Pisanic II, J. D. Blackwell, V. I. Shubayev, R. R. Finones and S. Jin, Biomaterials, 2007, 28, 2572–2581 CrossRef.
- K. Cheng, Q. Chen, Z. Wu, M. Wang and H. Wang, CrystEngComm, 2011, 13, 5394–5400 RSC.
- K. Cheng, Y.-M. Zhou, Z.-Y. Sun, H.-B. Hu, H. Zhong, X.-K. Kong and Q.-W. Chen, Dalton Trans., 2012, 41, 5854–5861 RSC.
- J.-H. Park, G. von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Angew. Chem., Int. Ed., 2008, 47, 7284–7288 CrossRef CAS.
- F. Weijun, Y. Jing, G. Jiawei and Z. Nanfeng, Adv. Funct. Mater., 2012, 22, 842–848 CrossRef.
- E. S. Lee, Z. Gao and Y. H. Bae, J. Controlled Release, 2008, 132, 164–170 CrossRef CAS.
- I. F. Tannock and D. Rotin, Cancer Res., 1989, 49, 4373–4384 CAS.
- A. I. Minchinton and I. F. Tannock, Nat. Rev. Cancer, 2006, 6, 583–592 CrossRef CAS.
- H. Maeda, Bioconjugate Chem., 2010, 21, 797–802 CrossRef CAS.
- E. Gullotti and Y. Yeo, Mol. Pharmaceutics, 2009, 6, 1041–1051 CrossRef CAS.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharmaceutics, 2008, 5, 505–515 CrossRef CAS.
- T. K. Jain, M. A. Morales, S. K. Sahoo, D. L. Leslie-Pelecky and V. Labhasetwar, Mol. Pharmaceutics, 2005, 2, 194–205 CrossRef CAS.
- A. E. Nel, L. Maedler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS.
- S. Laurent, C. Burtea, C. Thirifays, U. O. Häfeli and M. Mahmoudi, PLoS One, 2012, 7, e29997 CAS.
- I. Lynch and K. A. Dawson, Nano Today, 2008, 3, 40–47 CrossRef CAS.
- L. Josephson, J. Lewis, P. Jacobs, P. F. Hahn and D. D. Stark, Magn. Reson. Imaging, 1988, 6, 647–653 CrossRef CAS.
- T. Allkemper, C. Bremer, L. Matuszewski, W. Ebert and P. Reimer, Radiology, 2002, 223, 432–438 CrossRef.
- J. Qin, S. Laurent, Y. S. Jo, A. Roch, M. Mikhaylova, Z. M. Bhujwalla, R. N. Muller and M. Muhammed, Adv. Mater., 2007, 19, 1874–1878 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: XPS spectrum, nitrogen adsorption–desorption isotherm and pore distribution, magnetic hysteresis loop, Raman spectra and confocal laser microscopic image (Fig. S1–S5). See DOI: 10.1039/c3bm60087d |
| ‡ These authors made an equal contribution to this work. |
|
| This journal is © The Royal Society of Chemistry 2013 |