Comparative study of guanidine-based and lysine-based brush copolymers for plasmid delivery†
Peter M. Carlson, Joan G. Schellinger, Joshuel A. Pahang, Russell N. Johnson and Suzie H. Pun*
University of Washington and Department of Bioengineering and Molecular Engineering and Sciences Institute, William H. Foege Building, Box 355061, 3720 15th Ave NE, Seattle, WA 98195, USA. E-mail: spun@uw.edu; Fax: +1 206 543 3488; Tel: +1 206 685 3488
First published on 25th April 2013
Abstract
Polyethylenimine (PEI), one of the most frequently used polycations for non-viral nucleic acid delivery, exhibits good transfection efficiency to cultured cells but generally has to be used in restricted concentration ranges due to high cytotoxicity. We recently reported a family of HPMA-co-oligolysine brush copolymers that show nucleic acid delivery efficiencies approaching that of PEI. Guanidine-containing polymers have been reported in some systems to be more effective at cellular delivery of cargo than their primary-amine analogs. The goal of this work is to investigate the effect of guanidinylation on gene transfer ability of HPMA-co-oligolysine copolymers. Several parameters were evaluated: arginine versus homoarginine monomers, oligopeptide length, and charge density within the peptide. Using reversible addition-fragmentation chain transfer (RAFT) polymerization, a series of six copolymers were synthesized containing the cationic peptides K10, R10, K5, and (GK)5. Lysine-containing copolymers were functionalized with guanidine by reaction with O-methylisourea to generate an additional five homoarginine-based copolymers. All eleven copolymers readily condensed into small polyplexes (<250 nm) and remained stable in physiological salt conditions. The best performing copolymers provided more efficient gene transfection with less associated cytotoxicity than PEI. Reducing the number of charge centers (from 10 to 5) further reduced toxicity while retaining comparable transfection efficiency to PEI.
Introduction
The Tat peptide is a basic, arginine-rich domain of the 86 amino acid Tat protein from HIV-1 that is a critical motif for cellular uptake.1 The importance of the arginine groups, and more specifically the guanidine groups, in mediating intracellular delivery has been reported by the Sugiura and Wender groups.2,3 While these peptides were initially thought to be cell-penetrating by passing through the cell membrane directly into the cytoplasm, Richard et al. showed that arginine-rich peptides enter cells by endocytosis.4 There have been several arginine-based carriers developed for intracellular nucleic acid delivery in the past decade.5–9 These carriers have demonstrated efficient transfection of plasmid DNA and siRNA both in vitro and in vivo.Lysine is another amino acid often used in synthetic gene delivery systems due to its positive charge at neutral pH. Poly-(L)-lysine (PLL) was one of the earliest polycations used for non-viral gene transfer to mammalian cells.10 There have been several comparisons in the literature of cationic transfection reagents based on lysine versus arginine, and the relative transfection efficiency between the two types of carriers appears to be highly structure dependent. For example, Pouton et al. showed that polyarginine polymers transfect cells with plasmids much more poorly than polylysine polymers.11 In contrast, branched oligoarginine-based peptides generally delivered plasmid to K562 cells better than their branched oligolysine counterparts.12 In addition, poly(amido amine) (PAMAM) dendrimers modified with L-arginine show higher plasmid transfection efficiency than PAMAM dendrimers modified with L-lysine.13
Our group recently reported a family of peptide-based polycations synthesized by RAFT copolymerization of oligolysine macromonomers with HPMA (hydroxypropylmethacrylamide).14–16 The comb structure of these copolymers afforded much higher transfection efficiency than linear poly(L)lysine. We optimized the oligolysine polymers by varying the oligolysine monomer content, the oligolysine length, and the total molecular weight of the copolymers. We found that polymers with 20 mol% oligolysine, containing either K5 or K10, and of polymer with degree of polymerization (DP) of 150 or 190 provided high transfection efficiency.15 We have further demonstrated that multiple peptide entities, such as targeting ligands or endosomal release sequences, can be readily incorporated into these materials to further improve gene delivery efficiencies.14,17
In this work, we synthesize a panel of HPMA-co-peptide copolymers with comb-like architecture to test the effect of charge moiety (amine vs. guanidine) on cell transfection efficiency and cytotoxicity. Guanidinylated polymers were synthesized by both polymerization of oligoarginine monomers prepared by solid phase peptide synthesis (SPPS) and by post-polymerization guanidinylation of HPMA-co-oligolysine copolymers with O-methylisourea; the latter approach yields homoarginine copolymers. The effect of comb length (pentamer or decamer peptides), peptide density, and charge density in individual peptides on gene transfection efficiency and cytotoxicity was also investigated. From this work, several HPMA-co-peptide copolymers that surpass branched polyethylenimine (bPEI) in transfection efficiency without additional cytotoxicity were identified.
Materials and methods
Materials
N-(2-Hydroxypropyl)methacrylamide (HPMA) was purchased from Polysciences (Warrington, PA), VA-044 initiator from Wako Chemicals USA (Richmond, VA), reagents used in solid phase peptide synthesis from Apptec (Louisville, KY), O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate) from Advanced Chem Tech (Louisville, KY), N-succinimidyl methacrylate from TCI America (Portland, OR), and all other chemicals including O-methylisourea and bPEI (average Mw ∼ 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 by LS, average Mn ∼ 10000 by GPC) from Sigma-Aldrich (St. Louis, MO). The chain transfer agent ethyl cyanovalerictrithiocarbonate (ECT) was generously provided by Anthony Convertine (University of Washington). All cell culture reagents were purchased from Cellgro/Mediatech (Fisher Scientific, Pittsburgh PA). Endotoxin-free plasmid pCMV-Luc2 was isolated using the Qiagen Plasmid Giga Kit (Qiagen, Germany) according to the manufacturer's instructions. BCA protein quantification assay kits were obtained from Thermo Fischer Scientific (Waltham, MA), and luciferase expression quantification kits were obtained from Promega (Madison, WI).
000 by LS, average Mn ∼ 10000 by GPC) from Sigma-Aldrich (St. Louis, MO). The chain transfer agent ethyl cyanovalerictrithiocarbonate (ECT) was generously provided by Anthony Convertine (University of Washington). All cell culture reagents were purchased from Cellgro/Mediatech (Fisher Scientific, Pittsburgh PA). Endotoxin-free plasmid pCMV-Luc2 was isolated using the Qiagen Plasmid Giga Kit (Qiagen, Germany) according to the manufacturer's instructions. BCA protein quantification assay kits were obtained from Thermo Fischer Scientific (Waltham, MA), and luciferase expression quantification kits were obtained from Promega (Madison, WI).Synthesis of peptide monomers
A total of four methacrylamido-functionalized polycationic peptides containing a methacrylated (Ma) 6-aminohexanoic acid (Ahx) linker, MaAhxKKKKKKKKKK (MaAhxK10), MaAhxRRRRRRRRRR (MaAhxR10), MaAhxKKKKK (MaAhxK5), and MaAhxGKGKGKGKGK (MaAhx(GK)5), were synthesized using standard Fmoc/tBu SPPS techniques on an automated PS3 peptide synthesizer (Protein Technologies, Phoenix, AZ). Synthesized peptides were deprotected and cleaved from the solid phase resin supports using a solution of trifluoroacetic acid (TFA)–triisopropylsilane (TIPS)–1,3-dimethoxybenzene (DMB) (92.5:2.5:5, v/v/v) for 3 h (4 h in the case of MaAhxR10) under constant gentle mixing. The cleaved product was collected in three rounds of precipitation into cold ether, pelleting and dissolving in a minimum amount of methanol. After the third ether precipitation, the peptides were dried under vacuum, dissolved and frozen in a minimum volume of double distilled water (ddH2O), and lyophilized for long term storage. Each peptide was analyzed by RP-HPLC and shown to have greater than 95% purity. MALDI-TOF was used to confirm each synthesis reaction and all measured masses matched targeted masses. Measured mass for MaAhxK10 was 1480.11, expected 1479.7; measured mass for MaAhxR10 was 1760.4, expected 1759.4; measured mass for MaAhxK5 was 839.6, expected 838.8; and measured mass for MaAhx(GK)5 was 1124.6, expected 1124.1.:I molar ratio of 10. Each flask was capped and purged with nitrogen for 10 minutes before submerging in an oil bath preheated to 44 °C for 24 ± 0.5 h. Reaction solutions were then dialyzed (MWCO = 10000 Da) against ddH2O, changing dialysate every 18–24 hours, for three days. The resulting copolymer solution was transferred to a scintillation vial and lyophilized for long term storage.
| Copolymer (HPMA-co-X) | Target peptide (mol%) | Target Mwa (kDa) | Measured Mwb (kDa) | PDIb | Measured % peptidec |
|---|---|---|---|---|---|
| a Based on Mw = [Mo]/[CTAo] × conversion × FW with no counterions.b Determined by GPC.c Determined by amino acid analysis. | |||||
| X = MaAhxK10 | 20 | 78.3 | 77.6 | 1.18 | 20 |
| X = MaAhxR10 | 20 | 88.6 | 88.7 | 1.16 | 20 |
| X = MaAhx(hR)10 | 20 | 89.0 | 88.6 | 1.05 | 20 |
| X = MaAhxK5 | 20 | 45.8 | 47.2 | 1.15 | 16 |
| X = MaAhx(GK)5 | 20 | 61.1 | 61.2 | 1.07 | 21 |
| X = MaAhx(hR)5 | 20 | 58.4 | 57.5 | 1.29 | 16 |
| X = MaAhx(GhR)5 | 20 | 68.6 | 65.3 | 1.11 | 21 |
| X = MaAhxK5 | 40 | 75.8 | 73.4 | 1.11 | 42 |
| X = MaAhx(GK)5 | 40 | 96.4 | 67.3 | 1.01 | 43 |
| X = MaAhx(hR)5 | 40 | 91.0 | 71.5 | 1.05 | 42 |
| X = MaAhx(GhR)5 | 40 | 111.5 | 72.0 | 1.01 | 43 |
:1 (v/v) saturated Na2CO3 and ddH2O and fully dissolved before combining in a round bottom flask sealed with a rubber septum. Reactions were carried out at room temperature under constant stirring under a stirbar for 72 h, followed by dialysis against ddH2O (MWCO = 10000 Da) for an additional 72 h, replacing the dialysate every 18–24 hours and then lyophilized. Complete conversion of side chain primary amines to guanidine groups was confirmed by amino acid analysis of polymers.Characterization of copolymers
Characterization of polyplexes
Cell culture
Results and discussion
Synthesis and evaluation of HPMA-co-peptide copolymers based on decamers of lysine, arginine and homoarginine for plasmid delivery
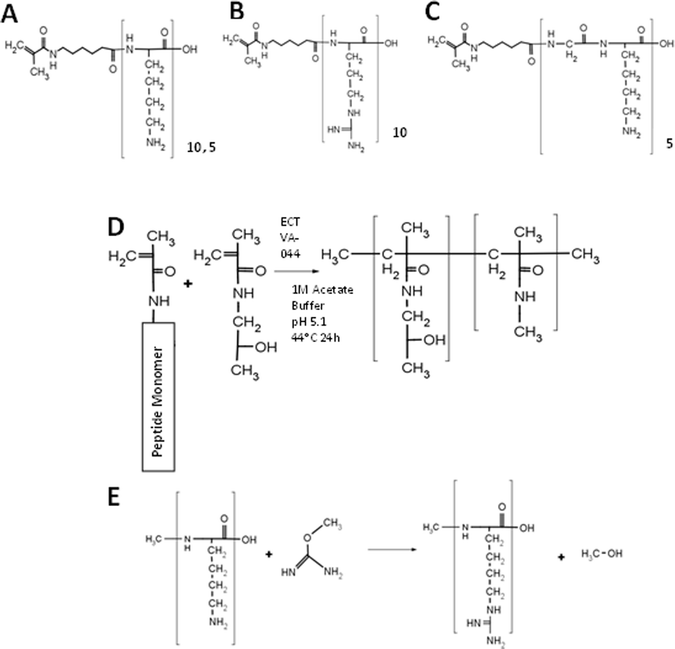 | ||
| Scheme 1 Polymer synthesis. (A–C) Structure of monomers: MaAhxK10 (A), MaAhxR10 (B), and MaAhx(GK5) (C). (D) Synthesis of peptide brush copolymers by RAFT copolymerization. (E) Guanidinylation reaction between a primary amine and O-methyl isourea to form homoarginine. | ||
Decamer peptide copolymers HPMA-co-MaAhxK10 and HPMA-co-MaAhxR10 were synthesized by RAFT copolymerization of HPMA with MaAhxK10 or MaAhxR10 monomers and HPMA-co-MaAhxhR10 polymers were synthesized by reaction of O-methylisourea with HPMA-co-MaAhxK10 copolymers. We showed previously that statistical peptide-based copolymers with good control over polymer composition are synthesized using this approach.14 Amino acid analysis confirmed that guanidinylation efficiency was >99%. The initial set of polymers was synthesized with ∼20 mol% peptide monomer content and with a target degree of polymerization (DP) of ∼180–190. Copolymer properties (molecular weight, polydispersity, and composition) are reported in Table 1. Measured molecular weights were within 1% of target molecular weights and polydispersity index (PDI) of polymers was below 1.2. Amino acid analysis confirmed that polymers contained ∼20% peptide.
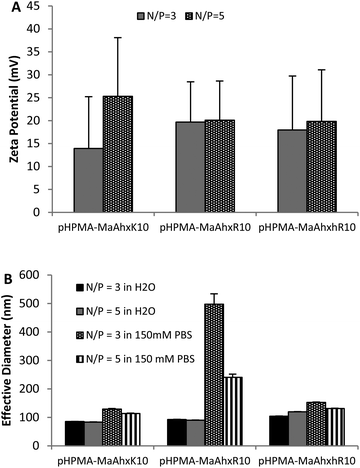 | ||
| Fig. 1 Zeta potential (A) and particle sizing (B) of decamer polymer polyplexes measured by dynamic light scattering (DLS). Polyplexes were formed in water and allowed to form for 5 minutes. After addition of either water or 150 mM PBS, mixtures were measured by DLS. Data is presented as mean ± SD, n = 3. | ||
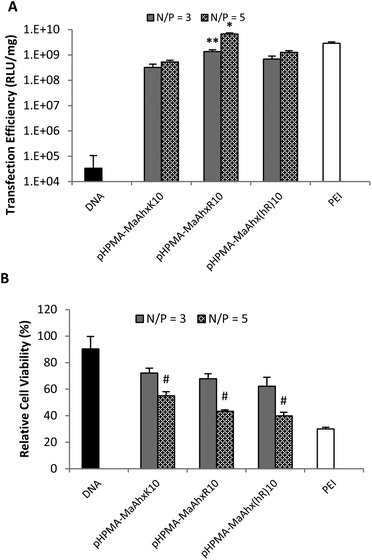 | ||
| Fig. 2 Luciferase plasmid transfection efficiency (A) and cytotoxicity (B) using polyplexes from HPMA-co-MaAhxK10, HPMA-co-MaAhxR10, and HPMA-co-MaAhxhR10 at N/P ratios of 3 and 5. Naked DNA and bPEI (25 kD) controls are included for comparison. Data presented as mean ± SD, n = 3. * greater transfection than PEI (p < 0.05). ** greater transfection than pHPMA-co-MaAhxK10 and pHPMA-co-MaAhx(hR)10 (p < 0.05). # higher cell viability than cells treated with PEI at the same N/P ratio (p < 0.05). | ||
Synthesis and evaluation of HPMA-co-peptide copolymers based on reduced positive charge density
Increased charge densities in polycations has been correlated with greater cytotoxicity.25 Therefore, we next attempted to decrease the cytotoxicity of the HPMA-co-MaAhxhR10 polymers by (i) decreasing the number of homoarginine residues per peptide monomer (from decamers to pentamers), (ii) decreasing the density of charge in peptide monomers by including glycine spacers between charges and (iii) changing the peptide monomer content in the copolymers. An additional eight HPMA-co-peptide polymers were synthesized using MaAhxK5 and MaAhx(GK)5 monomers and their guanidinylated, homoarginine derivatives at 20 and 40 mol% monomer incorporation (Table 1).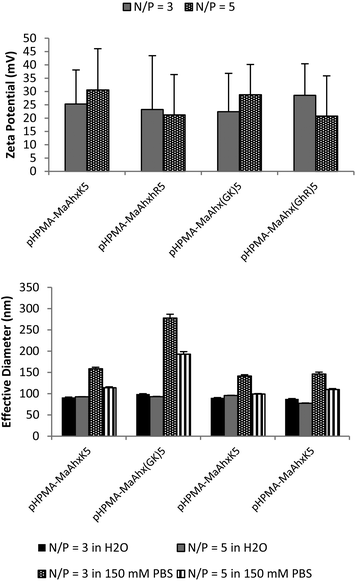 | ||
| Fig. 3 Zeta potential (A) and particle sizing (B) of 40 mol% pentamer polymer polyplexes measured by dynamic light scattering (DLS). Polyplexes were formed in water and allowed to form for 5 minutes. After addition of either water of 150 mM PBS, mixtures were measured by DLS. Data is presented as mean ± SD, n = 3. | ||
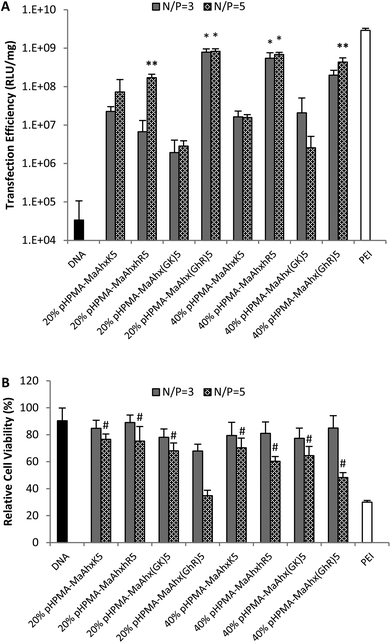 | ||
| Fig. 4 Luciferase transfection efficiency (A) and relative cytotoxicity (B) of the eight pentamer peptide copolymers. Data is presented as mean ± SD, n = 3. * higher transfection efficiency than all other copolymer with the same peptide incorporation (p < 0.05). # greater cell viability than cells treated with bPEI at the same N/P ratio (p < 0.05). | ||
Reducing the charge per peptide from 10 to 5 significantly decreased polymer cytotoxicity (Fig. 4B). For example, when comparing polymers containing the same charge centers per polymer (i.e. HPMA-co-MaAhxhR5 (40% peptide) vs. HPMA-co-MaAhxhR10 (20% peptide)) polymers synthesized using pentamers of charge were better tolerated than polymers synthesized using decamers of charge (60% cell survival vs. 43% cell survival at N/P = 5 in the aforementioned example). Furthermore, increasing peptide content correlated with increases in cell toxicity; for example, cells treated with HPMA-co-MaAhxhR5 (20% peptide) at N/P = 5 showed ∼75% cell survival compared with ∼60% cell survival for cells treated with HPMA-co-MaAhxhR5 (40% peptide). As with the decamer polymers, polyplexes were more toxic when formulated at higher charge ratios. Surprisingly, polymers synthesized with peptides containing glycine-spaced charge groups were more toxic than non-spaced peptides, although these polymers were still less toxic than the decamer analogues. One possible explanation is that the spacing increases the distance of charge from the polymer backbone, which may result in more non-specific protein interactions. Increased spacing between charge center and polymer backbone has been previously shown to correlate with higher polymer cytotoxicity for other polycation systems.26,27 Overall, the cell viability of HPMA-co-peptide formulations was better tolerated than the PEI control.
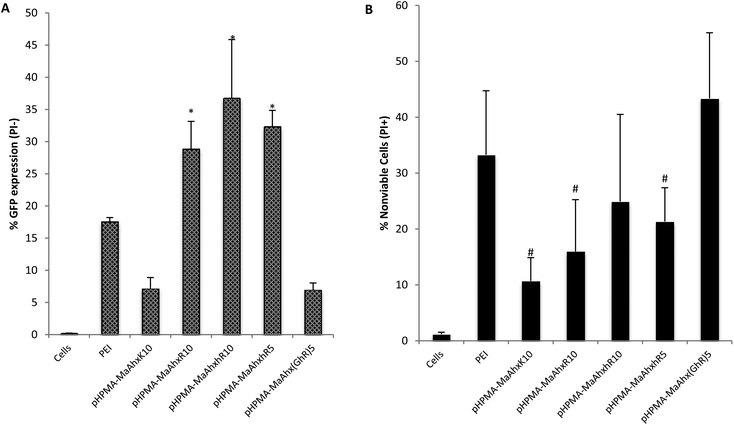 | ||
| Fig. 5 GFP transfection (A) and % of nonviable cells (B) of HeLa cells as measured by flow cytometry. Cells were transfected using the above copolymers at N/P = 5. Percent GFP+ is given as Mean ± SD for n = 4. * greater percentage of cells GFP+ compared to bPEI treatment (p < 0.05). # lower toxicity compared to PEI treated cells (p < 0.05). | ||
By EGFP analysis, the HPMA-co-MaAhxhR10 copolymer provided the highest transfection efficiency (36 ± 9% of live cells EGFP+), with the HPMA-co-MaAhxR10 copolymer transfecting comparably (28.9 ± 5% of live cells EGPF+). EGFP plasmid transfection efficiency for guanidine-based copolymers was up to 5-fold higher compared to lysine-based copolymers. Hashimoto and co-workers recently investigated polyarginine vs. polylysine as gene transfer vectors and demonstrated similar trends of transcription suppression after microinjection into cell nuclei and cytosol, although guanidinylated polymers were more efficient overall at gene transfer. They concluded that the guanidine-containing polymers are more efficient at endosomal escape compared to their primary-amine based analogues by analyzing the difference in transgene expression with and without chloroquine treatment.28 In this work, we further demonstrate that the three guanidinylated polymers showed higher transfection efficiency with less cytotoxicity (cell viability > 75%) compared to PEI, which is hypothesized to use proton sponge buffering for endosomal escape.29,30
Conclusion
In conclusion, we have synthesized a panel of guanidine-based brush copolymers by RAFT polymerization. All copolymers condensed plasmid DNA to salt-stable particles. Chemical guanidinylation was established as resulting in higher yield and greater purity copolymers than direct arginine incorporation while maintaining comparable transfection efficiencies and toxicity profiles. Guanidine-based copolymers exhibit higher transfection efficiency to cultured cells than analogous lysine-based polymers but with higher cytotoxicity. The cytotoxicity of arginine-based polymers could be mitigated by reducing the peptide length without much change in EGFP plasmid delivery efficiency. In contrast, cytotoxicity increased by reducing charge density in peptide brushes via glycine spacers. The copolymers developed and presented here provide a viable alternative to bPEI for in vitro plasmid transfection. Guanidinylation is therefore a straightforward modification that could be used to improve the in vitro and in vivo delivery efficiencies of other primary amine-based polycations used for nucleic acid delivery.Acknowledgements
This work was supported by NIH 1R01NS064404. PMC was supported by a Mary Gates Fellowship for this work. We are grateful to Profs Anthony Convertine and Patrick Stayton for generously providing the ECT chain transfer agent, to Prof. Kim Woodrow for use of the NanoSizer and to David Chu for helpful discussions.Notes and references
- E. Vives, P. Brodin and B. Lebleu, J. Biol. Chem., 1997, 272, 16010–16017 CrossRef CAS.
- S. Futaki, T. Suzuki, W. Ohashi, T. Yagami, S. Tanaka, K. Ueda and Y. Sugiura, J. Biol. Chem., 2001, 276, 5836–5840 CrossRef CAS.
- P. A. Wender, D. J. Mitchell, K. Pattabiraman, E. T. Pelkey, L. Steinman and J. B. Rothbard, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13003–13008 CrossRef CAS.
- J. P. Richard, K. Melikov, E. Vives, C. Ramos, B. Verbeure, M. J. Gait, L. V. Chernomordik and B. Lebleu, J. Biol. Chem., 2003, 278, 585–590 CrossRef CAS.
- S. Futaki, W. Ohashi, T. Suzuki, M. Niwa, S. Tanaka, K. Ueda, H. Harashima and Y. Sugiura, Bioconjugate Chem., 2001, 12, 1005–1011 CrossRef CAS.
- T. I. Kim, M. Lee and S. W. Kim, Biomaterials, 2010, 31, 1798–1804 CrossRef CAS.
- T. I. Kim, M. Ou, M. Lee and S. W. Kim, Biomaterials, 2009, 30, 658–664 CrossRef CAS.
- C. Rudolph, C. Plank, J. Lausier, U. Schillinger, R. H. Muller and J. Rosenecker, J. Biol. Chem., 2003, 278, 11411–11418 CrossRef CAS.
- Y. W. Won, S. M. Yoon, K. M. Lee and Y. H. Kim, Mol. Ther., 2011, 19, 372–380 CrossRef CAS.
- G. Y. Wu and C. H. Wu, J. Biol. Chem., 1987, 262, 4429–4432 CAS.
- C. W. Pouton, P. Lucas, B. J. Thomas, A. N. Uduehi, D. A. Milroy and S. H. Moss, J. Controlled Release, 1998, 53, 289–299 CrossRef CAS.
- C. Plank, M. X. Tang, A. R. Wolfe and F. C. Szoka, Hum. Gene Ther., 1999, 10, 319–332 CrossRef CAS.
- J. S. Choi, K. Nam, J. Park, J. B. Kim and J. K. Lee, J. Controlled Release, 2004, 99, 445–456 CrossRef CAS.
- R. N. Johnson, R. S. Burke, A. J. Convertine, A. S. Hoffman, P. S. Stayton and S. H. Pun, Biomacromolecules, 2010, 11, 3007–3013 CrossRef CAS.
- R. N. Johnson, D. S. H. Chu, J. Shi, J. G. Schellinger, P. M. Carlson and S. H. Pun, J. Controlled Release, 2011, 155, 303–311 CrossRef CAS.
- J. Shi, R. N. Johnson, J. G. Schellinger, P. M. Carlson and S. H. Pun, Int. J. Pharm., 2012, 427, 113–122 CrossRef CAS.
- J. G. Schellinger, J. Pahang, R. N. Johnson, D. S. H. Chu, D. L. Sellers, D. O. Maris, A. J. Convertine, P. S. Stayton, P. J. Horner and S. H. Pun, Biomaterials, 2013, 34, 2318–2326 CrossRef CAS.
- F. L. Brancia, S. G. Oliver and S. J. Gaskell, Rapid Commun. Mass Spectrom., 2000, 14, 2070–2073 CrossRef CAS.
- A. M. Funhoff, C. F. van Nostrum, M. C. Lok, M. M. Fretz, D. J. A. Crommelin and W. E. Hennink, Bioconjugate Chem., 2004, 15, 1212–1220 CrossRef CAS.
- V. P. Taori, H. Lu and T. M. Reineke, Biomacromolecules, 2011, 12, 2055–2063 CrossRef CAS.
- N. J. Treat, D. Smith, C. W. Teng, J. D. Flores, B. A. Abel, A. W. York, F. Q. Huang and C. L. McCormick, ACS Macro Lett., 2012, 1, 100–104 CrossRef CAS.
- H. Han, S. Nho, A. Lee and J. Kim, Bull. Korean Chem. Soc., 2010, 31, 1527–1534 CrossRef CAS.
- L. A. Tziveleka, A. M. G. Psarra, D. Tsiourvas and C. M. Paleos, J. Controlled Release, 2007, 117, 137–146 CrossRef CAS.
- S. Boeckle, K. von Gersdorff, S. van der Piepen, C. Culmsee, E. Wagner and M. Ogris, J. Gene Med., 2004, 6, 1102–1111 CrossRef CAS.
- S. Hwang, N. Bellocq and M. Davis, Bioconjugate Chem., 2001, 12, 280–290 CrossRef CAS.
- M. E. Davis, S. H. Pun, N. C. Bellocq, T. M. Reineke, S. R. Popielarski, S. Mishra and J. D. Heidel, Curr. Med. Chem., 2004, 11, 179–197 CrossRef CAS.
- T. M. Reineke and M. E. Davis, Bioconjugate Chem., 2003, 14, 247–254 CrossRef CAS.
- T. Hashimoto, T. Kawazu, T. Nagasaki, A. Murakami and T. Yamaoka, Sci. Technol. Adv. Mater., 2012, 13 Search PubMed.
- O. Boussif, F. Lezoualcl'h, M. Zanta, M. Mergny, D. Scherman, B. Demeneix and J.-P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS.
- N. D. Sonawane, F. C. Szoka and A. S. Verkman, J. Biol. Chem., 2003, 278, 44826–44831 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Included as separate formatted document. See DOI: 10.1039/c3bm60079c |
| This journal is © The Royal Society of Chemistry 2013 |