One-step fabrication of core–shell structured alginate–PLGA/PLLA microparticles as a novel drug delivery system for water soluble drugs†
Ming Pin Alan
Lim
a,
Wei Li
Lee
a,
Effendi
Widjaja
b and
Say Chye Joachim
Loo
*a
aSchool of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, 639798, Singapore. E-mail: joachimloo@ntu.edu.sg
bProcess Science and Modeling, Institute of Chemical and Engineering Sciences, 1 Pesek Road, Jurong Island, 627833, Singapore
First published on 11th February 2013
Abstract
Current focus on particulate drug delivery entails the need for increased drug loading and sustained release of water soluble drugs. Commonly studied biodegradable polyesters, such as poly(lactide-co-glycolide) (PLGA) and poly(L-lactide) (PLLA), are lacking in terms of loading efficiency of these drugs and a stable encapsulation environment for proteins. While hydrogels could enable higher loading of hydrophilic drugs, they are limited in terms of controlled and sustained release. With this in mind, the aim was to develop microparticles with a hydrophilic drug-loaded hydrogel core encapsulated within a biodegradable polyester shell that can improve hydrophilic drug loading, while providing controlled and sustained release. Herein, we report a single step method of fabricating microparticles via a concurrent ionotropic gelation and solvent extraction. Microparticles fabricated possess a core–shell structure of alginate, encapsulated in a shell constructed of either PLGA or PLLA. The cross-sectional morphology of particles was evaluated via scanning electron microscopy, calcium alginate core dissolution, FT-IR microscopy and Raman mapping. The incorporation of alginate within PLGA or PLLA was shown to increase encapsulation efficiency of a model hydrophilic drug metoclopramide HCl (MCA). The findings showed that the shell served as a membrane in controlling the release of drugs. Such gel-core hydrophobic-shell microparticles thus allow for improved loading and release of water soluble drugs.
Introduction
In the field of pharmaceutics, recent research has tended towards controlled release formulations and drug delivery systems, which are devised to achieve controlled release of therapeutic drugs .1,2 Controlled drug delivery systems would reduce institutional healthcare load caused by long term disease management, and increase patient compliance for diseases requiring extended treatment and monitoring regimes. There is an increased research focus on the delivery of water soluble drugs, including antigens and growth factors, antibiotics and anti-cancer drugs.3,4Several systems studied for drug delivery have been reported, including mesoporous nanoparticles like silica dioxide5–7 and layer by layer polymeric capsules that are capable of functional release behaviour triggered by environmental stimuli.8–11 However, fabrication of such systems requires multiple steps that are complex and thus extended production time.8 On the other hand, the use of biodegradable polyesters, such as poly(lactide-co-glycolide) (PLGA) and poly(L-lactide) (PLLA), was well studied due to their biodegradability and biocompatibility, and their ability to sustain release over a long period of time in vivo.12 Fabrication techniques of such polymer based drug delivery systems are also well established, which include emulsion solvent evaporation, nanoprecipitation and coacervation.4,13,14 However, these polyesters are unable to load hydrophilic drugs at higher encapsulation efficiencies due to their relative hydrophobicities.3 Burst release from such systems is also an issue, due to the localization of drugs on particulate surfaces or pores formed within the microparticles.15,16 Furthermore, emulsion-based solvent evaporation techniques commonly employed to produce hydrophilic drug-loaded microparticles exacerbate the leaching of these drugs into the aqueous continuous phase during fabrication, thus resulting in low drug encapsulation efficiency.17 In view of this, hydrogel-based drug delivery systems have been used, as they potentially allow for improved loading of hydrophilic drugs18 and are also favoured as a friendly environment for protein encapsulation.19 Examples of such hydrogel systems include alginate and chitosan, which can be physically gelled using multivalent ions such as Ca2+.20 However, sustained release from such systems is limited, due to susceptibility of these hydrogels to swell in water when immersed in an aqueous environment.21
Composite hydrogel–PLGA particulate systems were thus developed to provide sustained release and increased loading of hydrophilic drugs.22–24 These particulate systems feature gel particulates dispersed within an internal matrix23 or on the surface of PLGA microparticles.22 However, these techniques require the prefabrication of the hydrogel component in an additional step, requiring additional time and effort during fabrication.
In this study, we report on a single step based fabrication technique to produce alginate–PLGA microparticles (Alg–PLGA MP) and alginate–PLLA microparticles (Alg–PLLA MP) with a gel-core/hydrophobic polymer-shell. It was hypothesized that such a core–shell system would allow for higher encapsulation efficiencies of hydrophilic drugs, while providing controlled and sustained release. Drug-loaded microparticles of a core–shell structure were previously shown to have better control over drug release when compared to blended composite microparticles.25 As such, the release profiles can be tuned according to diffusion kinetics and degradation rates of the polymer used. In addition, the objectives include studying how certain key synthesis parameters would result in the formation of a core–shell structure, and the mechanisms leading to the formation of core–shell microparticles. Metoclopramide hydrochloride (MCA), a model hydrophilic drug, was used, and the release of this drug was compared between this core–shell particulate system against a pure system of calcium alginate (CaAlg) beads. As a therapeutic agent, MCA is used as an antiemetic and a gastroprokinetic agent for the treatment of nausea and gastric stasis. It is also used as a radiosensitizer/chemosensitizer for lung carcinoma treatment.26
Materials and methods
Materials
Poly(DL-lactide/glycolide) 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (intrinsic viscosity, IV, 1.03) and poly(L-lactide) (IV 2.4) were obtained from Purac Biomaterials; alginic acid sodium salt, from brown algae; Span® 80, poly(vinyl alcohol) (PVA) (molecular weight 30–70 kDa); trisodium citrate; trifluoroacetic acid; calcium chloride and metoclopramide HCl (MCA) were obtained from Sigma-Aldrich. High performance liquid chromatography (HPLC) grade dichloromethane (DCM) and acetonitrile were from Tedia Co. Ltd. Sodium chloride was obtained from J.T. Baker Ltd. Phosphate buffer saline (PBS) (pH 7.4) was obtained from OHME Scientific Pte Ltd, Singapore. All items were used as received.
:50 (intrinsic viscosity, IV, 1.03) and poly(L-lactide) (IV 2.4) were obtained from Purac Biomaterials; alginic acid sodium salt, from brown algae; Span® 80, poly(vinyl alcohol) (PVA) (molecular weight 30–70 kDa); trisodium citrate; trifluoroacetic acid; calcium chloride and metoclopramide HCl (MCA) were obtained from Sigma-Aldrich. High performance liquid chromatography (HPLC) grade dichloromethane (DCM) and acetonitrile were from Tedia Co. Ltd. Sodium chloride was obtained from J.T. Baker Ltd. Phosphate buffer saline (PBS) (pH 7.4) was obtained from OHME Scientific Pte Ltd, Singapore. All items were used as received.
Fabrication of microparticles
To fabricate Alg–PLGA MP with a core–shell structure, a double emulsion based solvent evaporation method was used. Microparticles of different types were synthesized by preparing polymer solutions, as tabulated in Table 1. Essentially, two different polymer solutions were separately prepared, one of which was a 10% (w/v) PLGA solution prepared by dissolving 400 mg of PLGA in DCM. Span 80 (1% (w/v)) was also added in the PLGA/DCM solution to stabilize the primary water-in-oil emulsion. The other polymer solution prepared was a 4.5% (w/v) sodium alginate (NaAlg) aqueous solution, made by dissolving 45 mg of sodium alginate in water. Sodium chloride (1.35 M) was also added as an osmolyte for the double emulsion. For drug-loaded microparticles, MCA was dissolved within the internal water phase for a theoretical drug loading of 20% (w/w).| Fabricated microparticles | Oil phase | Internal aqueous phase | External aqueous phase | |
|---|---|---|---|---|
| Polymer type | NaCl | CaCl2 | NaCl | |
| Alg–PLGA MP | PLGA | 1.35 M | 50 mM | 0.6 M |
| Alg–PLLA MP | PLLA | 1.35 M | 50 mM | 0.6 M |
| MCA-loaded | PLGA | — | 50 mM | — |
| Alg–PLGA MP | ||||
| MCA-loaded | PLLA | — | 50 mM | — |
| Alg–PLLA MP | ||||
| MCA-loaded | PLGA | — | — | — |
| Neat-PLGA MP | ||||
| MCA-loaded | PLLA | — | — | — |
| Neat-PLLA MP | ||||
An overview schematic of the fabrication process is shown in Fig. 1. The alginate solution was first emulsified in the PLGA/DCM solution under magnetic stirring for the formation of the primary water-in-oil (W/O) emulsion. This W/O emulsion was then further dispersed into a 100 ml aqueous solution of 0.5% (w/v) PVA, 50 mM CaCl2 and 0.6 M NaCl to form a double water-oil-water (W/O/W) emulsion, with an overhead stirrer (Calframo BDC1850-220). The stirrer was operated at 400 rpm for 3 hours to concurrently initiate the extraction of DCM and ionotropic gelation of sodium alginate. The resultant microparticles were then recovered via centrifugation, rinsed with deionised water, lyophilized and then stored in a desiccator for characterization.
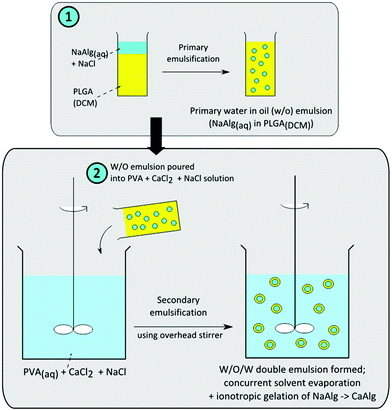 | ||
| Fig. 1 Schematic for the fabrication of alginate-polymer core–shell microparticles. | ||
Monolithic MCA-loaded neat PLGA and PLLA microparticles were also fabricated as a reference to MCA-loaded Alg-polymer core–shell microparticles. Sodium alginate was not included in the preparation of the internal aqueous phase.
Formation of calcium alginate (CaAlg) beads
CaAlg beads were fabricated as a control sample for comparison with Alg–PLGA MP. The beads were formed by extruding 4 ml of 4.5% (w/v) NaAlg solution through a syringe needle into a 5 ml cross-linking bath of 50 mM CaCl2 and 0.6 M NaCl (i.e. the same concentration for salts used in the external phase when fabricating neat Alg–PLGA MP). Beads were then left to gel in the solution for 3 hours before being lyophilized and used for Fourier transform infra-red (FT-IR) characterization. MCA-loaded CaAlg beads were similarly formed, with a 4 ml of 4.5% (w/v) alginate solution dissolved with 90 mg MCA prior to extrusion into a 5 ml of 50 mM CaCl2 cross linking solution. Drug-loaded beads were stored in deionised water at 4 °C.Characterization
Fourier transform infra-red (FT-IR) microscopy
To facilitate visual identification of the different chemical components in the Alg–PLGA MP formed, a Thermo Electron Nicolet Continuum FT-IR microscope was used. The core and the shell of the cross-sectioned particle were separated from each other using tweezers and placed on a gold coated glass slide. FT-IR spectra were then collected using the FT-IR microscope in reflectance mode, from 4000 cm−1 to 650 cm−1. FT-IR spectra of CaAlg beads and raw PLGA were also captured for comparison with the spectra of the microparticle core and shell.Raman mapping
Raman mapping was used to determine the polymer and drug distribution within the microparticles. Microparticles were first cross-sectioned as described above for SEM imaging, after which it is placed under a microscope objective of a laser power of up to approximately 20 mW. Raman measurements were performed on an area of 400 × 200 μm with a step size interval of 5 μm to form a grid map, using an In-Via Reflex, Renishaw Raman microscope equipped with a near infrared enhanced deep depleted thermoelectrically Peltier-cooled CCD detector array (576 × 38 g pixels) and a high grade Leica microscope. The sample was irradiated with a 785 nm near infrared diode laser, and the back scattered light was collected by a 20× objective. Measurement scans were collected through a static 1800 groove per mm dispersive grating from 300 to 1900 cm−1 and each spectrum acquisition time for each was approximately 35 s.Spectral pre-processing, including the removal of spikes due to cosmic rays, was carried out before the collected Raman spectra were subjected to the band target entropy minimization (BTEM) algorithm analysis. The BTEM algorithm was used to reconstruct pure component spectral estimates.28,29 When the entire normalized pure component spectra of underlying constituents had been reconstructed, the relative contributions of each measured point of these signals were calculated by projecting them back onto the baseline corrected and normalized data set. The colour-coded scale represents the intensities of each component recovered as a score image, in which the summation of the intensities (colour-coded scale) of all components at each particular grid pixel is equal to unity. These score images are then used to show the spatial distribution and the semi-quantitative content for all observed components in the microparticles.
Encapsulation efficiency
Actual drug loading and encapsulation efficiency of the microparticles fabricated were defined below:| Actual |
| drug |
| loading % (w/w) = 100% × (mass of |
| drug |
| loaded/total polymer mass) |
| Encapsulation efficiency (%) = 100% × (actual |
| drug |
| loading/theoretical drug loading) |
To calculate the actual drug loading of the microparticles, approximately 5 mg of microparticles were weighed and digested in 1 M NaOH by ultrasonication, for 10 min in an 80 °C water bath. The resultant solution was then neutralized with 1 M HCl and filtered. The drug concentration of the solution was measured using a reverse phase high performance liquid chromatography (HPLC) method, using the Agilent 1100 HPLC system with an XDB-C18 column. The analysis was done in gradient mode, with a varied proportion of 0.1% (v/v) trifluoroacetic acid and acetonitrile as the mobile phase. The amount of MCA was quantified using a detection wavelength of 309 nm at room temperature.
Drug release study
Microparticles (5 mg) were immersed in vials of 5 ml PBS each in triplicate, placed in a shaking incubator operating at 37 °C. At pre-determined time intervals, 1 ml of the release medium was extracted from the vial and analyzed for concentration of MCA released. A UV-VIS spectrophotometer (Shimadzu UV-2501) was used to measure the MCA concentration, at a detection wavelength of 309 nm. The vials were then replenished with fresh PBS of the same volume.Release data were fitted with the Peppas equation i.e. the power law, defined by the below equation:
| Mt/M∞ = ktn |
Results
Non drug-loaded Alg–PLGA MP and Alg–PLLA MP
The SEM cross-sectional view of an Alg–PLGA MP is shown in Fig. 2a. Cross-sectioned microparticles revealed a core and a shell within a particle. After immersion in trisodium citrate, the same cross-sectioned particle was again viewed under the SEM, whereby this time the core portion was shown to have dissolved (Fig. 2b). This suggests that the layer removed was calcium alginate, as the citrate ion is known to be a chelating agent that can sequester Ca2+ ions from calcium alginate gel, causing the dissolution of the gel structure.27 | ||
| Fig. 2 SEM image (a) of a cross-sectioned alginate–PLGA particle, and (b) the same microparticle subjected to trisodium citrate treatment (length denoted by scale bar is 100 μm). | ||
Fig. 3a shows the respective IR spectra of the microparticle shell and the core, with the representative spectra of pure PLGA and CaAlg for comparison. The spectrum of the core was different from the shell, with the presence of a strong broad peak at around 3500–3000 cm−1 arising from the core. This is characteristic of the O–H stretching of repeat –COOH and –OH group units on the alginate polymer chain.32 In addition, a COO− stretching absorption peak was also observed at around 1608 cm−1 for the alginate core.32,33 The doublet peak observed between 1790 and 1720 cm−1 for the shell, on the other hand, is representative of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch of the lactide and glycolide groups arising from PLGA. In essence, the IR spectra of the core and the shell of the microparticle match with the reference spectra of alginate and PLGA, respectively. In Fig. 3b, Raman mapping of a cross-sectioned microparticle and its associated pure component BTEM spectra estimates are shown, indicating the localization of each polymeric component. The core of the cross-sectioned microparticle again proved consistent to be alginate, which can be visually distinguished from the surrounding PLGA shell. The use of trisodium citrate to dissolve the alginate gel core shows that the shell left behind consists of PLGA. This is in agreement with the IR analysis and thus it is concluded that the formed microparticle has core–shell morphology of alginate and PLGA, respectively.
O stretch of the lactide and glycolide groups arising from PLGA. In essence, the IR spectra of the core and the shell of the microparticle match with the reference spectra of alginate and PLGA, respectively. In Fig. 3b, Raman mapping of a cross-sectioned microparticle and its associated pure component BTEM spectra estimates are shown, indicating the localization of each polymeric component. The core of the cross-sectioned microparticle again proved consistent to be alginate, which can be visually distinguished from the surrounding PLGA shell. The use of trisodium citrate to dissolve the alginate gel core shows that the shell left behind consists of PLGA. This is in agreement with the IR analysis and thus it is concluded that the formed microparticle has core–shell morphology of alginate and PLGA, respectively.
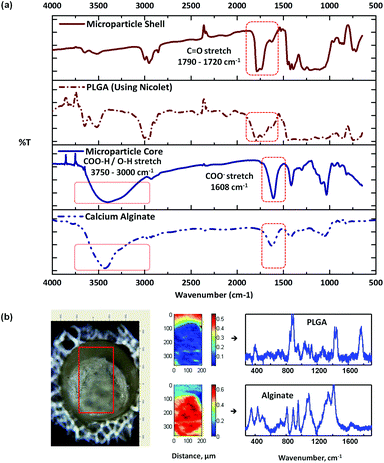 | ||
| Fig. 3 FT-IR spectra (a) of a typical alginate–PLGA microparticle, (b) optical image of a cross-sectioned alginate-PLGA microparticle, with the respective Raman mapping score images and BTEM components derived from the mapped enclosed rectangular area shown in the optical image. | ||
Similar to Alg–PLGA MP, Alg–PLLA MP exhibited a core–shell structure, as shown in Fig. 4a. Similarly, the alginate core was removed when cross-sectioned particles were immersed in citrate solution (Fig. 4b). This is in concordance with the Raman mapping analysis in Fig. 5, whereby the core and the shell were verified to be alginate and PLLA, respectively. Carbon was also detected, as the particles were mounted on carbon tape during Raman mapping.
 | ||
| Fig. 4 SEM image (a) of a cross-sectioned alginate–PLLA particle, and (b) the same microparticle subjected to trisodium citrate treatment (length denoted by scale bar is 100 μm). | ||
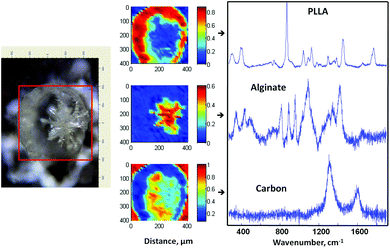 | ||
| Fig. 5 Optical image of a cross-sectioned alginate–PLLA microparticle, with the respective Raman mapping score images and BTEM components derived from the mapped enclosed rectangular area shown in the optical image. | ||
MCA-loaded Alg–PLGA MP and Alg–PLLA MP
To fabricate MCA-loaded microparticles, MCA was dissolved in the inner aqueous alginate phase prior to emulsification during the fabrication of drug-loaded Alg–PLGA MP. Similarly, a core–shell structure was obtained (Fig. 6a), and Raman mapping showed a strong MCA signal from the core and PLGA in the shell (Fig. 7). This time, the Raman spectrum of alginate was however not recovered due to a greater Raman scattering from the MCA component, hence resulting in a relatively strong signal that may have overshadowed any alginate present. Citrate treatment of this set of MCA-loaded microparticles further confirmed that the core was CaAlg (Fig. 6b). This same core–shell structure was also reproduced when PLGA was replaced with PLLA to give MCA-loaded Alg–PLLA MP (see ESI†). | ||
| Fig. 6 SEM image (a) of a cross sectioned metoclopramide HCl loaded alginate–PLGA particle, and (b) the same microparticle subjected to trisodium citrate treatment (length denoted by scale bar is 100 μm). | ||
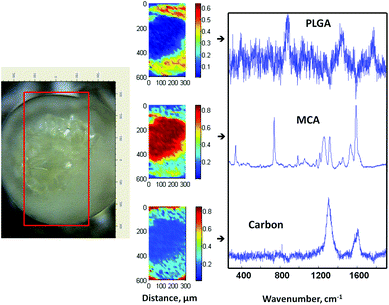 | ||
| Fig. 7 Optical image of a cross-sectioned alginate–PLGA microparticle loaded with metoclopramide HCl, with the respective Raman mapping score images and BTEM components derived from the mapped enclosed rectangular area shown in the optical image. | ||
Table 2 compares the encapsulation efficiency of MCA in PLGA and PLLA microparticles, fabricated with and without the inclusion of alginate. The encapsulation efficiency of MCA in Alg–PLGA MP and Alg–PLLA MP was almost double of neat PLGA and PLLA microparticles. This indicates that the incorporation of a hydrophilic polymer can significantly improve loading and encapsulation of water soluble drugs.
The release profiles for the Alg–PLGA MP, Alg–PLLA MP and CaAlg beads are shown in Fig. 8. Pure CaAlg beads showed a complete drug release within 6 hours, while both Alg–PLGA MP and Alg–PLLA MP exhibited a suppression of the initial burst release across the first 24 hours, followed by a sustained release of MCA up to 4 and 7 days, respectively. Alg–PLLA MP exhibited a more sustained release as compared to Alg–PLGA MP.
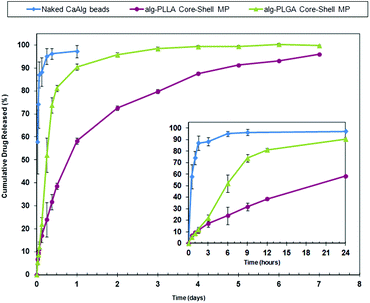 | ||
| Fig. 8 Release profile of metoclopramide HCl from Alg–PLGA MP, Alg–PLLA MP vs. naked calcium alginate beads across 7 days (main plot), and a closeup of the initial 24 hours (inset). | ||
The values of the release exponent n and the correlation coefficient R2 after fitting with the power law are shown in Table 3. This higher value of n for Alg–PLGA MP as compared to Alg–PLLA MP further suggests that the difference in release behaviour is influenced by a difference in the type of polymer used. This further leads to inference that the choice of polymer constructed for the shell could influence drug release profile and kinetics.
| Particle type | Release exponent n | Linear correlation coefficient R2 |
|---|---|---|
| Alg–PLGA MP | 0.8194 | 0.9686 |
| Alg–PLLA MP | 0.5104 | 0.9911 |
Discussion
The various microparticles fabricated in this study possess a core–shell structure of a hydrophobic polymeric shell and a calcium alginate core. Elucidating the geometry of the particulate structure required the use of a BTEM algorithm/Raman mapping to resolve the Raman signals combined from the various components, as shown in Fig. 3, 5 and7. This aided in the mapping out of the spatial distribution of the different components present, which is otherwise difficult to identify based on microscopy imaging alone. In addition, the use of trisodium citrate to sequester calcium ions dissolves the gel structure in Fig. 2, 4 and 6, which further confirms that the particle core is indeed alginate gelled by calcium ions. This allows the conclusive confirmation of the core–shell structure present.Alginate-polymer microparticles were fabricated through a W/O/W double emulsion solvent evaporation based technique, as shown in Fig. 1. This first involves the formation of a primary W/O emulsion, by emulsifying an aqueous solution containing NaAlg and NaCl into an oil phase (i.e. PLGA dissolved in DCM). The W/O emulsion was then subsequently dispersed into an external water phase with PVA, CaCl2 and NaCl dissolved. To achieve a stable W/O/W double emulsion, two different surfactants were used to disperse the primary W/O emulsion and the secondary double emulsion droplets in the external water phase.34 In this case, a hydrophobic Span 80 surfactant and a hydrophilic PVA surfactant were selected respectively to achieve this purpose. This subsequently forms the secondary double emulsion, as depicted in Fig. 9, and initiates two concurrent processes resulting in the formation of the core–shell microparticle. One is the extraction of a DCM solvent from the double emulsion droplet, which resulted in the precipitation of PLGA, and leading to the formation of the PLGA shell. The second concurrent process is the ionotropic gelation of the aqueous NaAlg phase within the inner aqueous droplet. Alginate can be gelled, i.e. physically cross-linked in the presence of divalent ions such as Ca2+ to form CaAlg hydrogels.35 The influx of Ca2+ ions from the external water phase into the internal alginate phase in this process causes the gelation of the dissolved alginate. In this manner, microparticles with an alginate-PLGA core–shell structure can be formed via this single fabrication step, whereby the water insoluble CaAlg core is gelled in situ within the hardening PLGA shell.
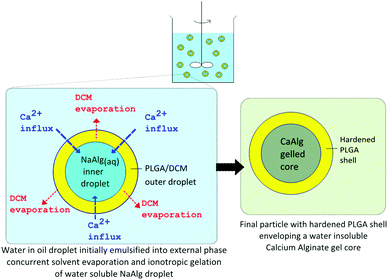 | ||
| Fig. 9 Principles of alginate–PLGA core shell microparticle formation. | ||
With the use of a double emulsion based solvent evaporation technique, changes to the formulation can be made to tailor the type of microparticle formed. For instance, the shell material of the microparticle can be varied by simply using a different polymer dissolved in a compatible solvent, which is in turn removed via a solvent extraction process. In this study, the fabrication of Alg–PLLA MP was achieved by simply dissolving PLLA instead of PLGA, for the oil phase polymer solution. Also, the loading of drugs can be done by dissolving the drugs either in the aqueous alginate phase or the oil (polymer) phase prior to emulsification. For instance, MCA was loaded prior to emulsification by dissolving the drug crystals in the alginate aqueous phase.
Given that the technique is based on the formation of a W/O/W double emulsion, the presence of large osmotic pressure differences between the two aqueous phases in the emulsion can effect a large movement of water between both phases. This has an effect on the emulsion stability, which can lead to the rupture of the DCM/PLGA oil layer, causing the double emulsion droplets to break.34 Preliminary studies showed that microparticles fabricated without NaCl dissolved in the internal aqueous phase resulted in a large number of broken microparticles fabricated, which indicates the likelihood of rupture of the double emulsion during fabrication (see ESI†). At the same time, the contents of the internal aqueous phase (i.e. alginate and MCA) can also leach into the external phase due to molecular migration.36 Leaching of alginate may result in an inconsistent gel core formation or formation of inhomogeneous alginate gel structures due to alginate partitioning effects seen in confined low volumes of alginate.37 To overcome these issues, NaCl, as an osmolyte, was therefore dissolved in both inner and outer water phases of the double emulsion. The intent was to regulate the osmotic pressure between both aqueous phases of the double emulsion, while at the same time, reduce alginate leaching.38,39 The use of NaCl in the external phase also acts as a non-gelling ion in increasing the homogeneity of the gel formed.40,41
Interestingly, when MCA was loaded into the microparticles, NaCl was no longer required in the formulation. Given that alginate can form strong complexes with poly-cations such as chitosan and poly-L-lysine,27 it is deduced that the dissolved MCA in drug-loaded formulations could also form a similar complex with alginate in the same way poly-cations do, possibly due to the presence of amine/amide groups in the drug. Such interactions could possibly slow down and disrupt the coupled diffusion of the sodium alginate polymer to the gelling front, resulting in a reduced partitioning rate of alginate and leading to the formation of a more homogeneous gel.40,41 At the same time, the hydration of MCA in the internal water phase may also have resulted in an increased osmotic pressure in the double emulsion.42 As such, this would reduce the alginate leaching and increase the retention of alginate within the double emulsion droplet. A drug like MCA can therefore replace NaCl, as an osmotic agent, to produce drug-loaded Alg–PLGA MP.
In terms of drug loading efficiency, an overall increased loading of MCA was observed for the core–shell MP as compared to the monolithic polymer particles. Given that the encapsulation efficiency is affected by the drug loss to the external phase in emulsion solvent evaporation methods,15 the presence of a hydrophilic polymer like alginate can better associate with water soluble drugs, as compared to a more hydrophobic PLGA. This would subsequently increase retention of drugs within the emulsion droplets and reduce out-flux of water soluble drugs during fabrication.
Compared to naked CaAlg beads that exhibited complete burst release within one day, both Alg–PLGA MP and Alg–PLLA MP demonstrated sustained release over 4 and 7 days respectively (Fig. 8). This reduced burst was due to the shell of the microparticles serving as a protective encapsulating envelope, which acts as a membrane to regulate the release of drugs. While hydrogels are known to release their contents rapidly,3,21 the enveloping PLGA or PLLA shell limits the rate of water influx and acts as a rate-limiting membrane for drug diffusion.
The use of a more crystalline and hydrophobic polymer as the shell such as PLLA can further reduce the release rate. This is apparent in the difference of release behaviour between Alg–PLGA MP and Alg–PLLA MP, whereby the latter exhibited a slower gradual release instead of the faster dual phase release exhibited by Alg–PLGA MP. Also, by fitting the release data with the power law, a lower n exponent value for Alg–PLLA MP was obtained, as compared to Alg–PLGA MP. This difference suggests that the semi-crystallinity of PLLA retards the release of drug, as compared to the amorphous PLGA with a more open polymer network.43 With the above observations, it is hence envisaged that release kinetics of these core–shell microparticles can be regulated and tailored through appropriate selection of a polymer as the shell material.
With improved drug loading and the reduced burst release from core–shell alginate–PLGA/PLLA microparticles as compared to CaAlg, these findings would pave the way towards protein encapsulation using the same particulate architecture. Future work would entail investigation into the release behaviour and bioactivity of proteins from such microparticles. As such, this new particulate drug delivery platform would be beneficial in areas such as vaccines or peptide delivery.
Conclusion
In this study, a novel microparticulate drug delivery system was fabricated with an alginate hydrogel core and encapsulated by a biodegradable hydrophobic polymeric shell. These microparticles were fabricated through a single step method, via concurrent emulsion solvent evaporation and ionotropic gelation processes. The incorporation of alginate within PLGA or PLLA was shown to increase encapsulation efficiency when MCA, a model hydrophilic drug, was loaded in the particles and compared to PLGA and PLLA microparticles fabricated without alginate. The shell formed was able to serve as a physical barrier between the MCA-loaded hydrogel core and the PBS medium during release. Thus, these gel-core hydrophobic-shell microparticles would allow for the improved loading and release of water soluble drugs, and potentially for protein loading.Acknowledgements
The authors would like to thank Prof. Richard David Webster for kindly allowing the usage of the Thermo Nicolet FT-IR microscope for the study of the fabrication of the composite core–shell microparticles. The authors would also like to acknowledge the financial support from the Agency for Science, Technology and Research (A*STAR) (Project No. 102 129 0098), Singapore Centre on Environmental Life Sciences Engineering (SCELSE), and Nanyang Technological University (NTU).Notes and references
- E. R. Balmayor, H. S. Azevedo and R. L. Reis, Pharm. Res., 2011, 28, 1241–1258 Search PubMed.
- K. J. Whittleseya and L. D. Sheaab, Exp. Neurol., 2004, 190, 1–16 CrossRef CAS.
- S. Vrignaudabc, J.-P. Benoitabcd and P. Saulnierab, Biomaterials, 2011, 32, 8593–8604 Search PubMed.
- R. C. Mundargi, V. R. Babu, V. Rangaswamy, P. Patel and T. M. Aminabhavi, J. Controlled Release, 2008, 125, 193–209 CrossRef CAS.
- F. Zhang, G. B. Braun, A. Pallaoro, Y. Zhang, Y. Shi, D. Cui, M. Moskovits, D. Zhao and G. D. Stucky, Nano Lett., 2011, 12, 61–67 Search PubMed.
- M. Ma, H. Chen, Y. Chen, X. Wang, F. Chen, X. Cui and J. Shi, Biomaterials, 2012, 33, 989–998 CrossRef CAS.
- Q. He and J. Shi, J. Mater. Chem., 2011, 21, 5845–5855 RSC.
- S. De Koker, R. Hoogenboom and B. G. De Geest, Chem. Soc. Rev., 2012, 41, 2867–2884 RSC.
- B. M. Wohl and J. F. J. Engbersen, J. Controlled Release, 2012, 158, 2–14 CrossRef CAS.
- M. Delcea, H. Möhwald and A. G. Skirtach, Adv. Drug Delivery Rev., 2011, 63, 730–747 CrossRef CAS.
- A. G. Skirtach, A. M. Yashchenok and H. Mohwald, Chem. Commun., 2011, 47, 12736–12746 RSC.
- H. Tamberab, P. Johansenac, H. P. Merklea and B. Gander, Adv. Drug Delivery Rev., 2005, 57, 357–376 CrossRef CAS.
- H. K. Makadia and S. J. Siegel, Polymer, 2011, 3, 1377–1397 Search PubMed.
- W. L. Lee, E. Widjaja and S. C. J. Loo, Small, 2010, 6, 1003–1011 Search PubMed.
- Y. Yeo and K. Park, Arch. Pharmacal Res., 2004, 27 Search PubMed.
- M. Ye, S. Kim and K. Park, J. Controlled Release, 2010, 146, 241–260 CrossRef CAS.
- P. B. O'Donnell and J. W. McGinity, Adv. Drug Delivery Rev., 1997, 28, 25–42 CrossRef CAS.
- T. Vermonden, R. Censi and W. E. Hennink, Chem. Rev., 2012, 112, 2853–2888 CrossRef CAS.
- Y. Capan, G. Jiang, S. Giovagnoli, K.-H. Na and P. DeLuca, AAPS PharmSciTech, 2003, 4, 147–156 Search PubMed.
- Z. Liua, Y. Jiaoa, Y. Wangb, C. Zhoua and Z. Zhanga, Adv. Drug Delivery Rev., 2008, 60, 1650–1662 CrossRef CAS.
- T. R. H. Vitae and D. S. K. Vitae, Polymer, 2008, 49, 1993–2007 CrossRef CAS.
- C.-H. Zheng, J.-Q. Gao, Y.-P. Zhang and W.-Q. Liang, Biochem. Biophys. Res. Commun., 2004, 323, 1321–1327 Search PubMed.
- A. Schoubben, P. Blasi, S. Giovagnoli, L. Perioli, C. Rossi and M. Ricci, Eur. J. Pharm. Sci., 2009, 36, 226–234 Search PubMed.
- X. L. Zheng, Y. Z. Huang, C. H. Zheng, S. Y. Dong and W. Q. Liang, AAPS J., 2010, 12, 519–524 Search PubMed.
- E. J. Pollauf, K. K. Kim and D. W. Pack, J. Pharm. Sci., 2005, 94, 2013–2022 CrossRef CAS.
- R. W. Pero, A. Olsson, M. Simanaitis, A. Amiri and I. Andersen, Pharmacol. Toxicol., 1997, 80, 231–239 Search PubMed.
- O. Smidsrød and G. Skjåk-Bræk, Trends Biotechnol., 1990, 8, 71–78 CrossRef CAS.
- E. Widjaja, N. Crane, T.-C. Chen, M. D. Morris, M. A. Ignelzi and B. R. McCreadie, Appl. Spectrosc., 2003, 57, 1353–1362 CrossRef CAS.
- E. Widjaja, W. L. Lee and S. C. J. Loo, Anal. Chem., 2010, 82, 1277–1282 Search PubMed.
- J. Siepmann and F. Siepmann, Int. J. Pharm., 2008, 364, 328–343 CrossRef CAS.
- J. Siepmann and N. A. Peppas, Adv. Drug Delivery Rev., 2001, 48, 139–157 CrossRef CAS.
- C. Sartori, D. S. Finch, B. Ralph and K. Gilding, Polymer, 1997, 38, 43–51 CrossRef.
- E. Pretsch, P. Bühlmann and M. Badertscher, in Structure Determination of Organic Compounds, Springer, Berlin, Heidelberg, 2009, pp. 1–67 Search PubMed.
- T. Florence Alexander and D. Whitehill, in Macro- and Microemulsions, American Chemical Society, 1985, pp. 359–380 Search PubMed.
- I. Donati and S. Paoletti, in Alginates: Biology and Applications, ed. H. A. R. Bernd, Springer, Verlag, 2009, p. 1 Search PubMed.
- J. Jiao and D. J. Burgess, Multiple Emulsions, John Wiley & Sons, Inc., 2007, pp. 1–27 Search PubMed.
- G. Skjåk-Bræk, H. Grasdalen and O. Smidsrød, Carbohydr. Polym., 1989, 10, 31–54 CrossRef.
- S. Magdassi and N. Garti, Colloids Surf., 1984, 12, 367–373 Search PubMed.
- N. Garti, A. Romano-Pariente and A. Aserin, Colloids Surf., 1987, 24, 83–94 Search PubMed.
- B. L. Strand, Y. A. Mørch, T. Espevik and G. Skjåk-Bræk, Biotechnol. Bioeng., 2003, 82, 386–394 CrossRef CAS.
- B. Thu, O. Gåserød, D. Paus, A. Mikkelsen, G. Skjåk-Bræk, R. Toffanin, F. Vittur and R. Rizzo, Biopolymers, 2000, 53, 60–71 CrossRef CAS.
- W. L. Lee, C. Loei, E. Widjaja and S. C. J. Loo, J. Controlled Release, 2011, 151, 229–238 Search PubMed.
- V. Shrivastava and U. K. Jain, Int. J. Pharm. Sci. Nanotechnol., 2010, 3, 1075–1084 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm00175j |
| This journal is © The Royal Society of Chemistry 2013 |