Advancements in nanosensors using plastic antibodies
Anna A.
Volkert
and
Amanda J.
Haes
*
University of Iowa, Department of Chemistry, 204 IATL, Iowa City, Iowa 52242, USA. E-mail: amanda-haes@uiowa.edu; Fax: +1 391-335-1270; Tel: +1 319-384-3695
First published on 25th October 2013
Abstract
Biosensors possess recognition elements that bind to target molecules which lead to detectable signals. Incorporation of noble metal nanomaterials into biosensors allows for rapid and simple biomolecule detection. Herein, recent developments in affinity nanosensors will be discussed. These sensors often include naturally occurring recognition elements such as antibodies and DNA. As samples become more complex, new recognition elements are sought. For instance, plastic antibodies provide alternative and more environmentally stable recognition elements than traditional antibodies. Molecular imprinted polymers, a class of plastic antibodies, promote biomolecule recognition and detection. The incorporation of noble metal nanomaterials into molecular imprinted polymer biosensors for real world applications will be explored. Further improvements in the design of artificial recognition agents are envisioned to facilitate new methods for complex biological and chemical analyses.
 Anna A. Volkert | Anna A. Volkert earned a B.S. in comprehensive chemistry from University of Wisconsin - Eau Claire in Eau Claire, Wisconsin and is presently a Ph.D. candidate in chemistry at the University of Iowa under the direction of Prof. Amanda J. Haes. Her research interests include quantifying the stability of size-tunable nanostructures in biologically-relevant solution conditions as well as the synthesis, fabrication, and integration of molecular imprinted polymers into devices to improve the selectivity of small molecule detection using Raman spectroscopy and SERS. |
 Amanda J. Haes | Amanda J. Haes is an Associate Professor in the Chemistry Department and Associate Director of the Nanoscience and Nanotechnology Institute at the University of Iowa. She earned her Ph.D. in Chemistry at Northwestern University with Richard P. Van Duyne. Before beginning her independent career, she was a National Research Council Research Associate with Greg E. Collins at the U.S. Naval Research Laboratory. Haes group research activities include designing and purifying novel nanomaterials for stable spectroscopic studies, improving detection limits of biological and environmental pathogens, and investigating how surface chemistry impacts nanomaterial function in separations and spectroscopy. |
1. Introduction
Biosensors contain immobilized receptors to detect biomolecules and are typically classified as affinity or affinity/catalytic devices.1–3 In the former, a ligand binds to a receptor which causes either a change in receptor charge, conformation, optical parameter, and/or in the temperature of the medium.1–3 In affinity/catalytic devices, initial ligand binding is followed by the production of a new reactive species that is then detected.1–3 This review will focus on recent developments in affinity biosensors. Affinity biosensors typically rely on specific interactions between natural recognition elements such as antibodies, aptamers, and/or DNA with a ligand.2,4 Signal transduction methods applied to biosensors include amperometry, potentiometry, and optical spectroscopy.3 Regardless of the method of signal transduction, biosensors are assessed based on figures of merit such as assay time, dissociation constant (Kd), limits of detection, and throughput.2,3Immunoassays are a class of affinity biosensors which use immobilized recognition elements for either competitive or non-competitive detection of target analytes.5 Competitive immunoassays typically employ an analyte and labeled analyte which competitively bind to an immobilized antibody.1,2,5 In competitive immunoassays, analyte detectability is limited by the affinity constant to the employed antibody.5 To improve detection limits, a non-competitive or two-site immunoassay (sandwich type) is employed where the analyte and a secondary labeled antibody are added sequentially to an immobilized capture antibody.2,5 Sandwich-type immunoassays allow for the quantification of proteins and chemical/biowarfare agents with sub-μM range detection limits.2 Typical assays times range anywhere from 2 to 60 minutes and additional pretreatment and washing steps which can take hours to days.2
To further improve the signal to noise of optical affinity biosensor responses, noble metal nanomaterials are often incorporated allowing for numerous “enhanced” detection methods such as such as colorimetry,6–10 surface enhanced Raman spectroscopy (SERS),11–16 and surface plasmon-based methods.17–25 Biosensors which incorporate noble metal nanomaterials were extensively reviewed elsewhere and specifically focused on localized and surface plasmon biosensors as well as SERS biosensors.17,18,26
Biosensors that employ naturally occurring recognition elements require “biocompatible” solution conditions to prevent antibody degradation. In contrast, plastic antibodies provide a more environmentally stable and cheaper alternative than these naturally occurring recognition elements.27 Molecular imprinted polymers (MIP), a class of plastic antibodies, promote biomolecule recognition and detection with affinities and selectivities that rival that of traditional antibody-antigen couples thereby allowing for specific detection of target analytes.28–32 Previously, molecular imprinting generated stable, specific recognition elements to detect drug molecules,33–37 proteins,38–46 and toxins47–52 for use in biological and chemical sensors,28,34–37,41,51–55 drug delivery,47,48,56,57 and separations.27,33,49,58,59 Other review articles summarized molecular imprinted polymer applications,27,54,60 biosensors,28,44 drug delivery,56 and synthesis.53,55,61
Herein, the first section of this review examines biosensors that include noble metal nanoparticles along with naturally occurring recognition elements for optical sensors. Specifically, nanomaterials and enhanced signal transduction in aggregation-based immunoassays, plasmonic biosensors, and SERS biosensors will be discussed. In the second section of the review, discussion of artificial recognition elements will focus on plastic antibody synthesis, limitations, and recent advances in plastic antibody technology. In the third and final section of this review, the advantages and methods of incorporating plastic antibodies into nanosensors for real world applications will be explored. Specifically, noble metal nanomaterial incorporation into electrochemical, fluorescence, SERS, and surface plasmon resonance (SPR) MIP sensors will be described. To our knowledge, this is the first review which focuses on noble metal nanomaterial incorporation into molecular imprinted polymers for real-world sensor applications for biological and environmental sample analysis. Future improvements in design of artificial recognition agents are envisioned to facilitate new methods for complex biological and chemical analyses.
2. Naturally occurring recognition elements for optical sensors
Biosensors contain biological elements that recognize target analytes and signal transduction elements for detection.4,22 The biological recognition element typically consists of naturally occurring recognition species such as antibodies, aptamers, and/or DNA.4 These molecules possess high selectivity and affinity for target biomolecules which lead to specific detection of biomarkers,16,19 proteins,6–13,15,20–23,25,62 and viruses.14,24 Improvement to optical biosensors can be made by incorporating noble metal nanomaterials for improved signal transduction allowing for numerous “enhanced” detection methods.2Biosensors which utilize noble metal (Ag, Au, Cu) nanoparticles exhibit novel size dependent properties and enhanced biological and chemical detectability. For instance, the extinction (absorption + scattering) spectrum of gold nanoparticles can be tuned throughout visible to near-infrared wavelengths by varying the local dielectric environment (i.e. the surrounding medium and/or surface modification), metal, shape, or size.63–65 Extinction spectra arise when the frequency of the electromagnetic field is in resonance with the oscillation of conduction band electrons. This phenomenon is known as the localized surface plasmon resonance (LSPR) and can be exploited for biosensor signal transduction.66 A typical extinction maximum wavelength for 13 nm diameter solution-phase spherical gold nanoparticles in water is 520 nm. When these particles agglomerate and/or aggregate, a new lower energy resonance forms at ∼650 nm.67 Relevant to biosensor developments, the exploitation of the controlled aggregation of antibody or aptamer functionalized nanomaterials allows for quantitative protein detection in complex matrices.6–10,62
Herein, noble metal nanomaterials and their method of signal transduction in aggregation-based immunoassays, plasmonic biosensors, and SERS will be described. Specifically, the signal transduction method, assay times, detection limits, and advantages of each biosensor will be thoroughly discussed and are briefly summarized in Table 1.
| Nanoparticle signal transduction method | Assay time | Detection limits | Advantages | |
|---|---|---|---|---|
| Aggregation-based immunoassays6–10,62 | Extinction magnitude between 600 and 650 nm | 1–6 hours | 0.2–20 nM | Simple instrumentation |
| Plasmonic biosensors19,20,22–25 | Refractive index changes | 30 min–3 hours | 1 aM–10 pM | High sensitivity |
| SERS biosensors11–14,16 | Enhanced Raman scattering | 20 min–24 hours | 0.2–200 pM | Multiplexed and rapid detection in complex matrices |
2.1 Aggregation-based immunoassays
The simplest aggregation-based immunoassays with noble metal nanomaterials involve adding a target antigen to an aptamer or antibody functionalized nanoparticle solution and monitoring nanoparticle aggregation using extinction spectroscopy.6,9,10 Thanh and Rosenzweig used an aggregation-based immunoassay to quantify anti-protein A in solution by aggregating protein A functionalized gold nanoparticles with 0–50 μg mL−1 of anti-protein A. As expected, the optical density at 620 nm increased systematically with increasing anti-protein A concentration yielding a detection limit of 1 μg mL−1. The detection limits are comparable to that observed using ELISA but required a total assay time of only 2 hours.9In another example, Aslan and coworkers determined that the aggregation of functionalized nanoparticles in the presence of a receptor depended on the affinity of the ligand/receptor binding as well as the number of collisions between ligand functionalized nanoparticles and receptors.7 Aggregation of biotinylated gold nanoparticles using 4–200 nM streptavidin was quantified by plotting extinction magnitude changes at 600 nm. The well-studied biotin streptavidin system exhibits a strong binding constant (∼1013 M−1)18,22 which allowed for the assumption that aggregation is dominated by the collision frequency of streptavidin and biotin functionalized gold nanoparticles (no longer depended on ligand/receptor binding constraints). The collision frequency was then correlated to the extent of aggregation and used to model nanoparticle aggregation kinetics for biotin functionalized gold nanoparticles. These findings led to a better understanding of nanoparticle aggregation and as a result, more reproducible responses in this class of immunoassays.
In the previous examples, the magnitude of aggregation depended on analyte concentration. To overcome this inherent limitation, catalytic amplification promoted nanoparticle aggregation assays are used to amplify signals.8,62 For instance, thrombin aptamer functionalized gold nanoparticles were aggregated in the presence of thrombin, separated from the nanoparticle solution, and used as seeds for catalytic growth in the presence of CTAB, HAuCl4, and a reducing agent (NADH). The thrombin detection limit was ∼20 nM, a 5-fold improvement when compared to traditional colorimetric thrombin detection methods. The detection of thrombin was further amplified using catalytic enlargement on glass surfaces where a thrombin aptamer was immobilized on a glass substrate and incubated in various concentrations of thrombin. The second binding site of thrombin was then available for binding with aptamer functionalized gold nanoparticles which were then enlarged using a growth solution (Fig. 1A).8 Absorbance spectra were collected after incubating the biosensor in thrombin concentrations varying from 0 to 160 nM (Fig. 1B).8 Specifically, the number of surface bound gold nanoparticles caused a systematic increase in absorbance at 650 nm as the thrombin concentration increased. A calibration curve was generated using the plasmon magnitude at 650 nm as a function of thrombin concentration (Fig. 1C).8 The detection limit improved by an order of magnitude for catalytically enhanced assays vs. solution-phase measurements. All in all, controlled amplification of nanoparticle aggregation after a specific recognition event yielded reproducibly amplified signals and therefore, more sensitive aggregation-based immunoassay responses.
 | ||
| Fig. 1 (A) Amplified detection of thrombin on surfaces by the catalytic enlargement of thrombin aptamer-functionalized gold nanoparticles. (B) Absorbance spectra of thiolated aptamer modified glass slides incubated in (a) 0, (b) 2, (c) 5, (d) 19, (e) 94, and (f) 167 nM thrombin using thiolated aptamer functionalized gold nanoparticles and the catalytic enlargement process. Slides were removed from the enhancement solution and rinsed prior to the recording of the spectra. (C) Calibration curve corresponding to the amplified optical detection of thrombin (reprinted with permission from ref. 8, Copyright 2004 American Chemical Society). | ||
2.2 Plasmonic biosensors
While aggregation-based immunoassays use simple instrumentation and nanomaterial design strategies, biosensor sensitivity is limited by the number of aggregation events and require long incubation times to ensure maximized signals (i.e. equilibrium responses). As a result, sensing platforms using surface plasmon resonance (SPR) and LSPR spectroscopy are used as these signal transduction methods rely on measuring small changes in refractive index that result from analyte binding to the recognition element at or near the surface of a noble metal thin film or nanoparticle.17,18,22 Molecular binding kinetics near the metal surface can be measured in real-time on the 10−1–103 s time scale with refractive index sensitivities on the order of 1 part in 105–106.17,22 For instance, a SPR sensor successfully quantified avian influenza virus by first immobilizing an avian influenza aptamer to a gold surface and measuring the refractive index through changes in the angle of incidence in 0–12.8 haemagglutinating units (HAU) avian influenza.24 The avian influenza virus SPR sensor was portable and shown to detect avian influenza in 1.5 hours which is ∼100× faster than current state of the art virus isolation and identification methods (i.e. ELISA).Similar to SPR sensors, LSPR sensors take advantage of small refractive index changes induced by recognition events near a nanoparticle surface by measuring extinction magnitude or wavelength shifts.18,22 Previously, a typical LSPR biosensor was constructed by (1) functionalizing surface immobilized triangular Ag nanoparticles with a self-assembled monolayer (SAM), (2) covalently attaching antibodies or aptamers to the SAM carboxylate groups, and (3) incubating the biosensor in a target biomolecule solution while monitoring shifts in the extinction maximum wavelength (Δλmax).19,20,22 The biotin/streptavidin binding couple was used to determine the mechanism of LSPR biosensor responses.20,22 Incubating a biotin functionalized nanosensor in 100 nM streptavidin resulted in a 27 nm red shift in the extinction maximum wavelength.22 Varying the streptavidin concentration from 10−15–10−6 M resulted in a systematic λmax shift yielding a surface-confined binding constant (Ka,surf) of 1011 M−1 and a streptavidin detection limit less than 1 pM.22 This work demonstrates the selectivity and specificity capabilities of LSPR biosensors.
Since this work, development of LSPR biosensor applications continued.19,20,22 The LSPR nanobiosensor response can be magnified by incorporating traditional “sandwich” bioassays using functionalized gold nanoparticles.19,20,22 For example, nanoparticle substrates were fabricated by functionalizing surface immobilized Ag nanoparticles with an octanethiol/11-mercaptundecanoic acid SAM layer and amine-conjugated biotin. After incubation in a biotin solution, anti-biotin functionalized gold (Au@Ab) nanoparticles were introduced to the biosensor. Extinction spectra were measured after the LSPR sensor was incubated with 100 nM unlabeled antibiotin (Δλmax = 11 nm). Alternatively, after incubation with the Au@Ab nanoparticles, a 42.7 nm redshift occurred, a ∼400% increase compared to the unlabeled antibiotin.20
Increased LSPR nanobiosensor sensitivity without the use of a sandwich assay can be achieved by altering the nanostructure employed in the biosensor.25 As an example, Balamurugan and coworkers observed that increased thrombin sensitivity was achieved with gold nanorod substrates vs. gold bipyramid substrates as a result of higher surface coverage of thrombin aptamer on the gold nanorod substrates.25 After the substrate-bound nanoparticles were functionalized with thrombin aptamer, thrombin solutions (concentration = 0–10−6 M) were incubated with the samples. Systematic λmax shifts for the gold nanorod substrates and gold bipyramid substrates are shown in Fig. 2A and B, respectively.25 Interestingly, the gold bipyramid substrate which exhibited a refractive index sensitivity twice that of gold nanorods revealed a ∼25% smaller response vs. the gold nanorod substrate.25 X-ray photoelectron spectroscopy indicated that the reduced detection signals for the gold bipyramid could be attributed to ∼1/2 the number of aptamer capture strands (i.e. surface density) on the bipyramids vs. the gold nanorods.25 This study demonstrates that careful control of aptamer surface coverage on as well as refractive index sensitivity of the plasmonic substrate is vital to biosensors design.
 | ||
| Fig. 2 Wavelength shift responses for 0 pM–1 μM thrombin for (a) gold nanorod substrates and (b) gold bipyramid substrates. The inset shows the UV-vis spectra obtained for increasing thrombin concentrations (reprinted with permission from ref. 25, Copyright 2013, Wiley). | ||
2.3 Surface-enhanced Raman scattering biosensors
While LSPR sensors exhibit high sensitivities (∼350 nm per RIU) and selectivities when receptors are used, multi-analyte detection is limited by non-specific refractive index changes resulting in universal signal transduction. One opportunity to overcome this limitation of refractive index sensors is to use SERS biosensors. SERS sensors combine natural recognition elements such as antibodies and antigens with large signal enhancements from gold and/or silver nanomaterials as well as with narrow and unique Raman vibrational bands for multiplexed detection.11–14,16 SERS biosensors are characterized as either label (indirect) or label-free (direct). Incorporating extrinsic Raman labels into sandwich-style immunoadsorbant assays for indirect measurements decreased assay time without sacrificing sensitive responses to biomarkers,16 proteins,13 and viruses.14For instance, extrinsic Raman labels (ERLs) can be synthesized by functionalizing gold nanoparticles with a Raman reporter molecule and antibody. When the target antigen is present, the ERLs bind to the target antigen, and the SERS signal of the reporter molecule is measured.13,14,16 For example, simultaneous multi-analyte identification capabilities are demonstrated in Fig. 3 and was achieved using three ERLs that were functionalized with 3-methoxybenzenethiol, 2-methoxybenzenethiol, or 4-nitrobenzenethiol and rabbit IgG, human IgG, and mouse IgG, respectively.13 The SERS spectra for a mixture of all three antibodies, combinations of two analytes, individual analytes, and a blank positively identified target antibodies and illustrated successful multi-analyte detection.13 Furthermore, quantitative detection of multiple antibodies was simultaneously achieved using ERLs.13 Possible applications of ERLs include clinical and industrial applications where rapid, multi-analyte quantification is often required.
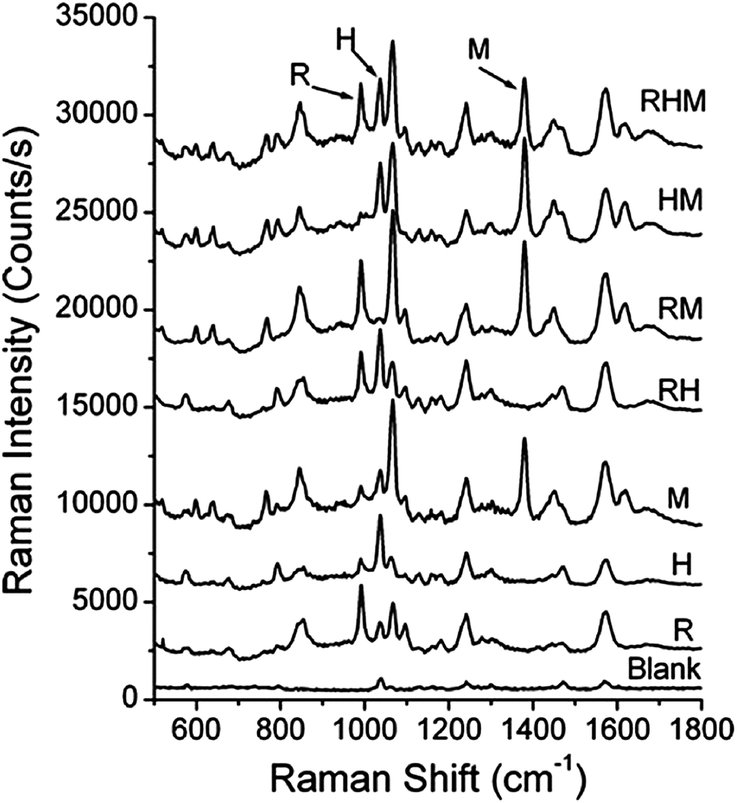 | ||
| Fig. 3 Simultaneous detection of three analytes: rabbit IgG (labeled as R), human IgG (labeled as H), as mouse IgG (labeled at M). The reporter molecules are 3-methoxybenzenethiol, 2-methoxybenzenethiol, 4-nitrobenzenethiol, respectively (reprinted (adapted) with permission from ref. 13, Copyright 2009 American Chemical Society). | ||
Small molecules can also be detected directly (i.e. without the use of ERLs). Advantages of direct detection include reduced assay times and improved selectivity in complex matrices vs. indirect detection methods. Previously, label-free SERS biosensors for the detection of protein in a complex sample matrix (i.e. milk) was achieved using silver dendrites.11,12 First, ovalbumin was extracted from milk using an immunomagnetic separation. After extraction and drying, molecule identification was achieved using principle component analysis to exploit differences between the background and analyte. Higher sensitivity was observed for the dried sample vs. solution-phase measurements in 0–5 μg mL−1 ovalbumin concentrations. As the ovalbumin concentration increased further, SERS intensities saturated and provided no quantitative information regarding ovalbumin concentration. All in all, the combination of immunomagnetic separation and SERS detection resulted in detection limits <1 μg mL−1 ovalbumin in less than 20 minutes (total assay time) in complex matrices making the technology extremely useful in the milk processing industry where rapid and accurate detection of contaminate proteins is required.
3. Artificial recognition elements
Previously, plastic antibodies provided alternative and more environmentally stable recognition elements than traditional antibodies.27 Here, a general overview of plastic antibody synthesis, limitations, and recent advances in plastic antibody technology is presented.MIPs are formed by polymerizing functional and cross linking monomers around a template molecule.60,68 Initially, functional monomers bind with template molecules forming template–monomer complexes. Upon polymerization, the template–monomer complexes are held in place by the highly crossed-linked polymer.28,60 Ideally, when template molecules are removed, molecular-specific cavities remain in the MIP that are shape and size-specific for the templated molecule.28,60,69 A schematic representation of MIP formation is shown in Fig. 4.60
 | ||
| Fig. 4 Scheme of molecular imprinting (reproduced with permission from ref. 60). | ||
Molecular imprinted polymer based biosensors are becoming increasingly popular because of increased stability, ease of use, and low cost vs. natural recognition elements.28,55 Previously, a MIP-based flow-through fluorosensor for digoxin detection in serum samples was compared to a traditional immunosensor.37 Briefly, a digoxin templated MIP was polymerized, ground, extracted, and integrated into a flow-through injection system where the fluorescence intensity of FITC-digoxin was monitored to determine digoxin binding. For comparison, a traditional immunosensor was constructed by functionalizing glass beads with anti-digoxin. Normalized fluorescence signals for both the MIP fluorosensor and immunosensor were monitored as a function of digoxin concentration. The MIP fluorosensor outperformed the traditional immunosensor with decreased detection limits (1.7 × 10−5vs. 1.2 × 10−3 mg L−1), improved precision (n = 6) (2 vs. 7% RSD), as well as an increased shelf life (18 vs. 3 months).37 The MIP fluorosensor illustrates some advantages of using molecular imprinted polymers in place of naturally occurring antibodies.
Two major challenges associated with MIP-based sensors are the incomplete removal of template during extraction and non-specific binding.27,28,55 Incomplete extraction can lead to template bleeding in subsequent assays resulting in an over-estimation of target biomolecule concentrations.36 Template bleeding can be reduced by using a structurally similar or “dummy” template molecule with minimal loss of signal from target molecules. In addition to template bleeding, MIPs are often plagued by non-specific binding responses.42 Tov et al. proposed incorporating a second polymerization step into the MIP synthesis to reduce the number of non-specific binding sites. The secondary polymerization step used monomers designed to react with only the polymer chains which do not actively bind to the template thereby effectively blocking the non-specific binding sites. Using this approach, selectivity of the target analyte lysozyme to the recognition sites increased by an order of magnitude. Additional modifications of MIP synthesis either through dummy templates or additional polymerization steps will likely lead to more selective and sensitive recognition capabilities in sensor applications.
Recognition of small molecules using MIPs can also be improved by mechanical grinding and sieving to improve polymer particle uniformity. Traditionally, MIPs are synthesized in bulk, ground, and then sieved before use which leads to heterogeneous structures with a distribution of recognition sites.49 Alternatively, purification of MIP particles is possible through the use of affinity column purification.49,70 Hoshino and coworkers synthesized fluorescently labeled MIP particles templated with melittin. Next, the materials were passed through a melittin functionalized agarose bead column where retention of the MIP particles was dictated by the degree of template affinity for the agarose column.49 After passing through the column, the eluted MIP particles were analyzed using fluorescence spectroscopy. Approximately 40% of the particles eluted from the column indicating no interaction with melittin. Washing the affinity column using 1 °C water facilitated the removal of the plastic antibodies from the column and revealed a Kd = 0.66–2.3 nM which is comparable to typical antibody–antigen interactions. All in all, affinity purification yielded MIP nanoparticles with a more narrow affinity distribution than unpurified polymer particles. Affinity purification can be applied to many different MIP–template systems and will likely become an integral step in plastic antibody template synthesis for biosensor applications.
Industrial applications of MIPs require an automated, direct, reproducible, and large scaled synthesis of high affinity plastic antibodies.33,59 Piletsky et al. proposed an automated MIP synthesis on an immobilized solid-phase template for the production of reproducible, high affinity MIP particles.58,59 In the automated synthesis, MIP particles were polymerized around solid-phase immobilized templates where unreacted monomers and low binding affinity particles were easily removed with washing. Three unique MIP syntheses which took place over five days were found to exhibit similar binding properties (Kd = 6.3 × 10−8 ± 1.7 × 10−9 M).58 In addition to producing high-affinity MIP particles, solid-phase synthesis methods readily allowed for further surface modification with a variety of ligands for use in biosensing applications.59 These high affinity MIP particles were functionalized with labels for fluorescence studies, electrochemical labels for cyclic voltammetry detection, or thiol groups for immobilization onto gold surfaces. The ease of surface modification on high-affinity MIP particles further improved biosensor integration for industrial applications.
4. Incorporating plastic antibodies into nanosensors towards real applications
In recent years, the incorporation of nanomaterials into MIPs produced sensitive and selective biosensors. There are three methods to incorporate nanomaterials into MIP sensors: suspending nanomaterials into the MIP matrix, polymerizing nanoparticles into the MIP matrix, and polymerizing the MIP onto a nanomaterial surface. In addition to the method of nanomaterial incorporation into the MIP sensor, fundamental nanomaterial properties such as nanomaterial shape, size, surface chemistry, and stability in the MIP matrix must also be considered. Specifically, noble metal nanoparticle and MIP networks yielded selective detection of biomolecules,71–75 drugs,69,76–81 environmental contaminates,82,83 and explosives.84 Herein, noble metal nanomaterials and their incorporation into MIP electrochemical, fluorescence, SERS, and SPR sensors will be described.4.1 Fluorescence MIP sensors
Detection of biomolecules using fluorescence MIP sensors allows for rapid and selective solution-phase measurements.72–75,85 Introduction of noble metal nanoparticles facilitates large changes (increases or decreases depending on the mechanism) in fluorescence signals yielding highly sensitive and selective biosensors.72–75 For instance, Gültekin and coworkers polymerized a MIP layer onto organically modified gold nanoparticles for the sensitive and selective detection of dipicolinic acid, a main component of Bacillus spores where some of its species are attributed to bioterrorism (Bacillus anthracis) and food poisoning (Bacillus cerus).73This was achieved in a multi-step process. First, gold nanoparticles were functionalized with methacryloylamidocysteine. Next, a dipicolinic acid MIP was polymerized around the gold nanoparticles producing a MIP nanoshell. After the removal of the template molecules, target dipicolinic acid was added, and the fluorescence intensity decreased significantly via photoluminescence quenching from the gold nanoparticle through specific interactions with dipicolinic acid in the MIP binding sites.73,74 Fluorescence emission spectra at 600 nm were collected after incubating the MIP nanoshell in 0–1 × 10−4 M dipicolinic acid solutions which resulted in a detection limit of 0.1 μM dipicolinic acid (spore concentration = 3.2 × 104 CFU mL−1). The MIP nanoshells were then used to quantify dipicolinic acid in Bacillus cerus spore samples. This integration of MIPs and the photoluminescence properties of gold nanoparticles yielded promising results toward the rapid and sensitive detection of Bacillus cerus spores for food safety applications.
Since this work, development of fluorescence MIP sensor applications continued.72,75 Fluorescence MIP nanobiosensor responses can be magnified by incorporating silver nanomaterials.72,75 For example, silver coated gold nanoclusters were functionalized with methacryloylamidocysteine and coated with a dipicolinic acid MIP.72 After the dipicolinic acid MIP cluster and non-imprinted nanoclusters were polymerized and prepared for sensor use, dipicolinic acid (concentration = 10−7–10−4 M) solutions were allowed to incubate with the samples. As expected, the fluorescence intensity decreased systematically for the MIP nanocluster but did not change for the non-imprinted nanocluster. The detection limit decreased by 1.5× for the gold/silver MIP nanocluster vs. the gold MIP nanoshell sensors.72,73 While incorporation of noble metal nanoparticles into fluorescence MIP biosensors yielded selective biosensor responses, sub-μM detection limits are typically required for trace biomolecule detection but were unrealized using this device.
Recently, Ton and et al. demonstrated sub-μM detection limits of the fluorescent probe dansyl-L-phenylalanine with gold nanoparticles incorporated into a MIP functionalized optical fiber.85 First, a molecular imprinted polymer was polymerized onto an optical fiber tip with or without gold nanoparticles. Next, the fibers were incubated in dansyl-L-phenylalanine solutions (concentration = 0–10 μM) for 45 minutes, and the fluorescence response was measured (Fig. 5A). Integrating gold nanoparticles into the MIP optical fiber sensor decreased the limit of detection vs. controls by a factor of 50 (1 μM–20 nM). To decrease the incubation time required for sensing by decreasing the analyte diffusion distance, optical fibers with a thin (0.7 μm) MIP layer were fabricated. Binding of 10 μM dansyl-L-phenylalanine was observed within 2 minutes and fluorescence intensity saturated within 10 minutes (Fig. 5B). In addition, the sensor was rinsed with solvent and subsequently used to detect 2.5 μM dansyl-L-phenylalanine. The reversibility of the sensor response indicated close to real-time detection of fluorescent molecules. While incorporation of noble metal nanoparticles into the fluorescence MIP optical fiber yielded sub-μM detection limits, a fluorescent functional monomer was required limiting the application of this optical fiber MIP biosensor.
 | ||
| Fig. 5 (A) Fluorescence response of the MIP sensor tip without gold (“MIP no Au”, λEM = 495 nm) and the composite gold-MIP tip (“MIP Au”, λEM = 489 nm) after incubation with increasing concentrations of dansyl-L-phenylalanine (dansyl-L-Phe). Integration time: 100 ms. (B) Kinetics of the fluorescence response of the thin-film MIP tip incubated with dansyl-L-phenylalanine. Inset: Confocal fluorescence image of the thin-film MIP tip after incubation. Experiments were performed in triplicate; error bars represent standard deviations. The blank signal was recorded before each set of experiments (adapted with permission from ref. 85, Copyright 2013, Wiley). | ||
4.2 Electrochemical MIP sensors
In an effort to improve detection limits without the use of labeled monomers, MIPs can be combined with the simplicity, high sensitivity, and low-cost of electrochemical methods.80 Incorporation of noble metal nanomaterials into electrochemical MIP sensors can increase the number of binding sites as well as the probability of electron transfer events between the electrode surface and analyte.81 For instance, there are three methods to modify electrode surfaces with gold nanomaterials: direct deposition of gold nanoparticles on the electrode,86 electrode surface functionalization with ligands,71,80,81 and nanoparticles grown directly onto the electrode.83 Previously, gold coated silica MIP (Au@SiO2) nanoparticles were synthesized and deposited onto a gold electrode surface for the electrochemical detection of dopamine. Differential pulse voltammetry was used to detect 4.8 × 10−8 to 5.0 × 10−5 M dopamine (Fig. 6A).86 A systematic increase in the dopamine oxidation peak current was observed which resulted in a linear response and dopamine detection limits of 2.0 × 10−8 M (Fig. 6B). Importantly, incubation of the Au@SiO2 nanoparticle modified electrode in epinephrine, norepinephrine, ascorbic acid, and uric acid solutions exhibited smaller current response vs. dopamine which indicated high specificity of the MIP to dopamine. | ||
| Fig. 6 (A) Differential pulse voltammograms of increasing dopamine concentration in 0.2 PBS (pH = 7.0). Dopamine concentrations were (a) 0.05, (b) 0.1, (c) 0.3, (d) 0.5, (e) 1, (f) 2.5, (g) 4.8, (h) 10, (i) 17.9, (j) 24.6, (k) 32.4, and (l) 50 μM (from top to bottom), respectively. (B) The response curve for dopamine. The inset reveals low dopamine concentration responses (reprinted from ref. 86, with permission from Elsevier). | ||
While this study demonstrated the simplicity and specificity of MIPs, the performance of electrochemical MIP sensors depends on the adhesion and stability of the sensing layer to the electrode surface.71 In an effort to overcome these sensor limitations, two devices were fabricated. First, the electrode surface was functionalized with a thiol anchoring agent for gold nanoparticle attachment and end groups which are then available for attachment to the dopamine MIP sensing layer. As a control, electrochemical sensors without a gold nanoparticle layer were also fabricated. After incubating both devices in 1 mM dopamine, the current was measured using square wave voltammetry. Current was enhanced by ∼120% when gold nanoparticles were incorporated into the MIP sensing layer vs. the control device by increasing electrical conduction between dopamine and the electrode surface. The gold nanoparticle MIP electrochemical sensor also exhibited improved dopamine detection limits (0.35 nM) vs. sensors without anchoring agents (20 nM).71,86 The anchored gold nanoparticle MIP electrochemical sensors further illustrated the advantages of integrating gold nanoparticles into MIP sensors for small molecule detection.
While MIP based sensors exhibit many sensing advantages, practical challenges in the wide-spread use of these remain. For instance, attaching gold nanoparticles to electrode surfaces with subsequent MIP polymerization requires multiple fabrication steps which can be time consuming.71,86 To address this limitation, Li and coworkers proposed a one-step growth and electrodeposition of gold nanoparticles to an electrode procedure using a silica imprinted hydrogel templated with p-nitrophenol.83 The one-step synthesis yielded an imprinted silica gel with well-dispersed gold nanoparticles onto a gold coated electrode. Cyclic voltammetry promoted template extraction by scanning the potential from +0.6 and −1 V (three times) effectively disrupting the interactions between p-nitrophenol and the binding sites. Next, the current at +0.12 V was measured using differential pulse voltammetry for sensors incubated in p-nitrophenol (concentration = 3.0 × 10−8–3.5 × 10−4 M). Plotting the voltammetric magnitude vs. p-nitrophenol concentration yielded a linear response with a detection limit of 5.0 × 10−9 M. In addition, the molecular imprinted silica gel exhibited a 6-fold enhancement vs. non-imprinted controls. This one-step electrochemical nanoparticle MIP sensor approach could be applied to many different MIP-template systems which would reduce biosensor fabrication requirements.
4.3 SPR MIP sensors
While electrochemical MIP sensors exhibit low detection limits (10−9) and selectivities, real-time detection is not always possible. Alternatively, SPR MIP sensors exhibit real-time sensing capabilities and have been used to rapidly and selectively detect biomolecules,87,88 drugs,89 explosives,90,91 and proteins.92,93 MIPs can be incorporated into SPR sensors by coating an SPR surface with MIP,89,92 embedding nanomaterials in a MIP matrix,87,88 or polymerizing nanoparticle crosslinked MIPs onto a SPR surface.90,91 For instance, a MIP SPR sensor successfully quantified lysozyme, a model protein, by first immobilizing lysozyme imprinted MIP particles to a gold surface and measuring the change in refractive index through changes in the SPR response in 21–1400 nM lysozyme.92 The lysozyme MIP SPR sensor was shown to selectively detect lysozyme in a complex protein mixture in 45 minutes which is ∼5× faster than quartz crystal microbalance measurements.The detection limits of the MIP SPR sensors can be further improved by crosslinking Au nanoparticles around a template to form a gold nanoparticle MIP which is then immobilized on a SPR surface.90,91 Specifically, gold nanoparticles are first functionalized with thioaniline electropolymerizable units and are then electropolymerized on a thioaniline-monolayer-modified Au electrode in the presence of hexahydro-1,3,5-trinitro-1,3,5-triazine to form a bisaniline-crosslinked gold nanoparticle SPR sensor. Upon template binding, the π–donor bisaniline units associate with the π–acceptor nitro groups of the hexahydro-1,3,5-trinitro-1,3,5-triazine. The charge–transfer complexes in the nanoparticle matrix resulted in an amplified shift in the SPR spectrum. As expected, an immediate and systematic shift in the SPR curve occurred after treating the SPR sensor with 0–10 μM hexahydro-1,3,5-trinitro-1,3,5-triazine resulted in a detection limit of 4 nM.91 Additionally, the use of a more soluble, “dummy” template (Kemp's Acid) decreased the detection limits of hexahydro-1,3,5-trinitro-1,3,5-triazine to 12 fM as a result of the increased number of templates sites in the MIP matrix.
Riskin and coworkers further investigated the role of the “dummy” template structure in the bisaniline-crosslinked gold nanoparticle SPR sensor sensitivity by varying the template carboxylic acid for the detection of the explosives pentaerythritol tetranitrate, nitroglycerin, and ethylene glycol dinitrate.90 For example; when citrate is employed as the template, the sensitivity is 200 fM for the structurally similar pentaerythritol tetranitrate. Nitroglycerin and ethylene glycol dinitrate, however, exhibited sensitivities of 100 pM, and 400 nM, respectively, in the citrate template MIP. The differences in sensitivity indicated that the structure of the template molecule and resulting binding site is vital for selective detection of explosives. The decreased detection limits will likely lead to increased applications of gold nanoparticle MIP SPR sensors for explosives detection.
4.4 SERS MIP sensors
While SPR sensors exhibit low detection limits (10−15), the sensors were plagued with non-specific molecule responses. To combat this detection limitation, SERS can be utilized for molecular identification by combining MIP sensors with narrow and unique Raman vibrational bands.69,76–79,82,84 Previously, MIPs were polymerized directly onto patterned SERS-active grids for the detection of propranolol.69,77,78 Droplets of MIP monomer were deposited onto SERS-active patterned grids using a nano fountain pen and polymerized. This resulted in MIP droplets with diameters of 6–12 μm.69,77 SERS spectra were collected from the MIP droplet after polymerization, extraction, and rebinding of propranolol. The presence of the propranolol vibrational bands revealed identification of propranolol;69,77 however, the thickness of the MIP droplet varied in each single droplet as well as from droplet to droplet limiting the quantitative detection of propranolol.69,77,78To facilitate quantitative detection using SERS MIP sensors, 2,4,6-trinitrotoluene imprinted xerogel films were fabricated on SERS substrates.83,84 First, a MIP xerogel film was cast over the entire SERS substrate using spin coating, and template molecules were extracted. After extraction was verified, SERS spectra were then measured from solutions containing 0–0.6 mM 2,4,6-trinitrotoluene. Plotting the SERS intensity of the asymmetric nitrate stretch at 1352 cm−1 yielded a linear response over the concentration range and a detection limit of 3 × 10−6 M. In contrast to both fluorescence and electrochemical detection methods, unique vibrational bands of 2,4,6-trinitrotoluene allowed for both qualitative and quantitative detection even in the presence of structurally similar molecules. Unfortunately, the 2,4,6-trinitrotoluene MIP SERS sensor sensitivity was not adequate for sub-μM detection of trace explosives.
The small signals observed in the previous example were limited by either (1) the number of molecules near the SERS substrate and/or (2) the distance dependence of the SERS effect.76,84 Clearly, polymerizing a thin MIP layer directly onto a nanoparticle surface can increase the number of template binding sites close to the metal surface thereby increasing biosensor sensitivity.76,82 Bompart et al. fabricated a MIP SERS sensor by functionalizing gold nanoparticles to carboxylate polymer particles and subsequently coated the composite with propranolol MIP shells (Fig. 7A).76 SERS spectra of the MIP composite (Fig. 7B-2) and a control (Fig. 7B-1) were collected after incubating the device in a 10−5 M propranolol solution. The presence of characteristic propranolol bands at 483, 733, 1380, and 1563 cm−1 indicated the presence of propranolol in the MIP composite. After the MIP composite was washed with (9 methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetic acid), the propranolol vibrational bands were no longer observed (Fig. 7B-3). In contrast, propranolol vibrational bands were observed for the non-imprinted polymer composite incubated in 10−5 M propranolol thereby indicating non-specific binding (Fig. 7B-4). All in all, the MIP composite successfully demonstrated a lower detection limit (10−7 M) vs. the non-imprinted polymer composite (10−5 M) and MIP particles (10−4 M). Additionally, the MIP composite facilitates successful quantification of propranolol in a complex biological matrix. The decreased detection limits using MIP composite SERS sensors will likely lead to more selective and sensitive recognition capabilities for biosensor applications.
:1 acetic acid), the propranolol vibrational bands were no longer observed (Fig. 7B-3). In contrast, propranolol vibrational bands were observed for the non-imprinted polymer composite incubated in 10−5 M propranolol thereby indicating non-specific binding (Fig. 7B-4). All in all, the MIP composite successfully demonstrated a lower detection limit (10−7 M) vs. the non-imprinted polymer composite (10−5 M) and MIP particles (10−4 M). Additionally, the MIP composite facilitates successful quantification of propranolol in a complex biological matrix. The decreased detection limits using MIP composite SERS sensors will likely lead to more selective and sensitive recognition capabilities for biosensor applications.
 | ||
| Fig. 7 (A) Optical microscopy image (100× magnification) of composite MIP particles during SERS measurements (defocused laser spot). (B) SERS spectra of propranolol: (1) on aggregated gold colloids; (2) single composite MIP particle incubated in 10−5 M propranolol; (3) single composite MIP particle after washing with 9 methanol:1 acetic acid, followed by methanol to remove the propranolol; and (4) single composite non-imprinted polymer particle incubated in 10−5 propranolol (adapted with permission from ref. 76, Copyright 2010, Wiley). | ||
5. Conclusions and future outlook
In summary, biosensors facilitate quantification of a variety of biomolecules for clinical, industrial, and safety applications. Antibodies, aptamers, and DNA are widely used as recognition elements in affinity biosensors. The addition of noble metal nanomaterials to these devices improves signal transduction by allowing for numerous “enhanced” detection methods resulting in decreased assay times and detection limits vs. traditional immunoassays. Furthermore, artificial recognition elements such as plastic antibodies were shown to mimic traditional receptors and were more environmentally stable, easier to synthesize, and cheaper than traditional antibodies. The incorporation of a purification step after MIP synthesis using affinity purification yielded high affinity MIP particles with a more narrow affinity distribution than unpurified MIPs for a particular molecule but resulted in a low yield of high affinity MIP particles. Finally, automated immobilized solid-phase template methods yielded reproducibly synthesized, high affinity MIP particles for industrial applications.Further improvements in MIP sensor sensitivity were achieved with the addition of noble metal nanomaterials into electrochemical, fluorescence, SERS, and SPR detection platforms. Nanomaterial shape, metal, and surface functionalization were found to greatly impact signal enhancements in MIP biosensors. Additionally, decreasing the MIP sensing layer thickness significantly decreased the assay time required for sensitive detection. Specifically, fM detection limits with real-time detection were observed using a gold surface functionalized with a cross-linked nanoparticle MIP SPR sensor; however, the SPR MIP sensor was plagued with issues of non-specific binding. To overcome inherent non-specific binding limitations of MIPs, molecular identification was achieved using SERS MIP sensors using both SERS-active substrates and solution-phase gold nanoparticles. Importantly, signal enhancements from nanomaterial incorporation required short distances between the nanomaterials and template sites which can ultimately limit quantitative detection. Tuning the polymer matrix allows for the sensitive and selective detection of a seemingly endless variety of analytes in complex, real-world applications. As such, MIPs could be used to overcome challenges faced by biosensors. These challenges include:
• Biosensors must be environmentally stable and exhibit a long shelf life. Plastic antibodies are inherently stable in a variety of environments and as a result, increased shelf life vs. naturally occurring recognition elements is observed.
• Recognition elements used in biosensors require specific responses for target analytes in complex matrices (i.e. blood, mucus, and plasma). Without purification, the binding affinities of plastic antibodies will not equal or exceed binding affinities of antibodies, aptamers, or DNA. Further improvements in MIP specificity are required; however, plastic antibodies could be used as a molecule specific filter allowing for complex matrix simplification.
• Real-time responses are vital for point-of-care applications. Currently, real-time detection using MIPs is limited by traditional biosensors designs that rely on analytes diffusing into binding sites for detection. The use of flow-through devices are expected to allow for real-time detection by promoting biomolecule diffusion into the binding site.
• Low detection limits (sub-nM) are needed for trace analyte detection. Limits of detection could be improved by incorporating noble metal nanomaterials with MIPs possibly allowing for trace biomolecule detection.
The incorporation of noble metal nanomaterials into MIP biosensors will continue to improve analyte sensitivity and as a result, applicability of nanomaterial biosensor detection methods. Regardless of the signal transduction method or nanomaterial used; plastic antibody design, incorporation, and synthesis will continue to drive real-world applications of MIP sensors for biological and environmental sample analysis.
Acknowledgements
This work was funded by the National Science Foundation, (CHE-1150135).Notes and references
- F. Scheller, F. Schubert, D. Pfeiffer, R. Hintsche, I. Dransfeld, R. Rennenberg, U. Wollenberger, K. Riedel, M. Pavlova, M. Kuhn, H. G. Muller, P. M. Tan, W. Hoffman and W. Moritz, Analyst, 1989, 114, 653–662 RSC.
- S. M. Borisov and O. S. Wolfbeis, Chem. Rev., 2008, 108, 423–461 CrossRef CAS PubMed.
- P. Vadgama and P. W. Crump, Analyst, 1992, 117, 1657–1670 RSC.
- H. S. Song and T. H. Park, Biotechnol. J., 2011, 6, 1310–1316 CrossRef PubMed.
- Immunoassay, ed. E. P. Diamandis and T. K. Christopoulos, Academic Press, San Diego, CA, 1996 Search PubMed.
- P. Englebienne, Analyst, 1998, 123, 1599–1603 RSC.
- K. Aslan, C. C. Luhrs and V. H. Peréz-Luna, J. Phys. Chem. B, 2004, 108, 15631–15639 CrossRef CAS.
- V. Pavlov, Y. Xiao, B. Shlyahovsky and I. Willner, J. Am. Chem. Soc., 2004, 126, 11768–11769 CrossRef CAS PubMed.
- N. T. K. Thanh and Z. Rosenzweig, Anal. Chem., 2002, 74, 1624–1628 CrossRef CAS.
- V. K. Gasparyan, Colloids Surf., B, 2010, 80, 180–183 CrossRef CAS PubMed.
- L. He, T. Rodda, C. L. Haynes, T. Deschaines, T. Strother, F. Diez-Gonzalez and T. P. Labuza, Anal. Chem., 2011, 83, 1510–1513 CrossRef CAS PubMed.
- L. He, C. L. Haynes, F. Diez-Gonzalez and T. P. Labuza, J. Raman Spectrosc., 2011, 42, 1428–1434 CrossRef CAS.
- G. Wang, H. Y. Park, R. J. Lipert and M. D. Porter, Anal. Chem., 2009, 81, 9643–9650 CrossRef CAS PubMed.
- J. Driskell, K. M. Kwarta, R. J. Lipert and M. D. Porter, Anal. Chem., 2005, 77, 6147–6154 CrossRef CAS PubMed.
- N. Guarrotxena and G. C. Bazan, Chem. Commun., 2011, 47, 8784–8786 RSC.
- D. S. Grubisha, R. J. Lipert, H. Y. Park, J. Driskell and M. D. Porter, Anal. Chem., 2003, 75, 5936–5943 CrossRef CAS PubMed.
- S. Szunerits and R. Boukherroub, Chem. Commun., 2012, 48, 8999–9010 RSC.
- K. M. Mayer and J. H. Hafner, Chem. Rev., 2011, 111, 3828–3857 CrossRef CAS PubMed.
- A. J. Haes, L. Chang, W. L. Klein and R. P. Van Duyne, J. Am. Chem. Soc., 2005, 127, 2264–2271 CrossRef CAS PubMed.
- W. P. Hall, S. N. Ngatia and R. P. Van Duyne, J. Phys. Chem. C, 2011, 115, 1410–1414 CAS.
- X. Wang, Y. Yu, Y. Chen, L. Li, F. Liu and N. Li, Anal. Bioanal. Chem., 2011, 400, 2085–2091 CrossRef CAS PubMed.
- A. J. Haes and R. P. Van Duyne, J. Am. Chem. Soc., 2002, 124, 10596–10604 CrossRef CAS PubMed.
- H. R. Sim, A. W. Wark and H. J. Lee, Analyst, 2010, 135, 2528–2532 RSC.
- H. Bai, R. Wang, B. Hargis, H. Lu and Y. Li, Sensors, 2012, 12, 12506–12518 CrossRef CAS PubMed.
- S. Balamurugan, K. M. Mayer, S. Lee, S. A. Soper, J. H. Hafner and D. A. Spivak, J. Mol. Recognit., 2013, 26, 402–407 CrossRef CAS PubMed.
- K. C. Bantz, A. F. Meyer, N. J. Wittenberg, H. Im, O. Kurtulus, S. H. Lee, N. C. Lindquist, S. H. Oh and C. L. Haynes, Phys. Chem. Chem. Phys., 2011, 13, 11551–11567 RSC.
- W. J. Cheong, S. H. Yang and F. Ali, J. Sep. Sci., 2013, 36, 609–628 CrossRef CAS PubMed.
- K. Haupt and K. Mosbach, Chem. Rev., 2000, 100, 2495–2504 CrossRef CAS PubMed.
- S. A. Piletsky, H. Matuschewski, U. Schedler, A. Wilpert, E. V. Piletska, T. A. Thiele and M. Ulbricht, Macromolecules, 2000, 33, 3092–3098 CrossRef CAS.
- C. Xie, B. Liu, Z. Wang, D. Gao, G. Guan and Z. Zhang, Anal. Chem., 2008, 80, 437–443 CrossRef CAS PubMed.
- S. A. Piletsky, E. V. Piletskaya, T. A. Sergeyeva, T. L. Panasyuk and A. V. El'skaya, Sens. Actuators, B, 1999, 60, 216–220 CrossRef CAS.
- G. Wulff, Chem. Rev., 2002, 102, 1–27 CrossRef CAS PubMed.
- A. Nematollahzadeh, A. Shojaei, M. J. Abdekhodaie and B. Sellergren, J. Colloid Interface Sci., 2013, 404, 117–126 CrossRef CAS PubMed.
- U. G. Spizzirri and N. A. Peppas, Chem. Mater., 2005, 17, 6719–6727 CrossRef CAS.
- M. Lasákova, D. Thiébaut, P. Jandera and V. Pichon, J. Sep. Sci., 2009, 32, 1036–1042 CrossRef PubMed.
- B. T. S. Bui, F. Merlier and K. Haupt, Anal. Chem., 2010, 82, 4420–4427 CrossRef CAS PubMed.
- G. P. González, P. F. Hernando and J. S. D. Alegría, Anal. Bioanal. Chem., 2009, 394, 963–970 CrossRef PubMed.
- Y. Yonamine, Y. Hoshino and K. J. Shea, Biomacromolecules, 2012, 13, 2952–2957 CrossRef CAS PubMed.
- M. Moschallski, A. Evers, T. Brandstetter and J. Rühe, Anal. Chim. Acta, 2013, 781, 72–79 CrossRef CAS PubMed.
- F. X. Gao, X. T. Ma, X. W. He, W. Y. Li and Y. K. Zhang, Colloids Surf., A, 2013, 433, 191–199 CrossRef CAS PubMed.
- X. Zhang, X. Du, X. Huang and Z. Lv, J. Am. Chem. Soc., 2013, 135, 9248–9251 CrossRef CAS PubMed.
- O. Y. Tov, S. Luvitch and H. Bianco-Peled, J. Sep. Sci., 2010, 33, 1673–1681 CrossRef CAS PubMed.
- Z. Zeng, Y. Hoshino, A. Rodriguez, H. Yoo and K. J. Shea, ACS Nano, 2010, 4, 199–204 CrossRef CAS PubMed.
- A. Bossi, F. Bonini, A. P. F. Turner and S. A. Piletsky, Biosens. Bioelectron., 2007, 22, 1131–1137 CrossRef CAS PubMed.
- F. T. C. Moreira, S. Sharma, R. A. F. Dutra, J. P. C. Noronha, A. E. G. Cass and M. G. F. Sales, Biosens. Bioelectron., 2013, 45, 237–244 CrossRef CAS PubMed.
- S. Ambrosini, S. Beyazit, K. Haupt and B. T. S. Bui, Chem. Commun., 2013, 49, 6746–6748 RSC.
- Y. Hoshino, H. Koide, T. Urakami, H. Kanazawa, T. Kodama, N. Oku and K. J. Shea, J. Am. Chem. Soc., 2010, 132, 6844 Search PubMed.
- Y. Hoshino, T. Kodama, Y. Okahata and K. J. Shea, J. Am. Chem. Soc., 2008, 130, 15242–15243 CrossRef CAS PubMed.
- Y. Hoshino, W. W. Haberaecker III, T. Kodama, Z. Zeng, Y. Okahata and K. J. Shea, J. Am. Chem. Soc., 2010, 132, 13648–13650 CrossRef CAS PubMed.
- Y. Hoshino and K. J. Shea, J. Mater. Chem., 2011, 21, 3517–3521 RSC.
- M. Boopathi, M. V. S. Suryanarayana, A. K. Nigam, P. Pandey, K. Ganesan, B. Singh and K. Sekhar, Biosens. Bioelectron., 2006, 21, 2339–2344 CrossRef CAS PubMed.
- S. Piszkiewicz, E. A. Kirkbride, N. Doreng-Stearns, B. R. Henderson, M. A. Lenker, E. Tang, L. H. Kawashiri, C. S. Nichols, S. C. Moore and S. G. Sogo, Chem. Commun., 2013, 49, 5954–5956 RSC.
- B. T. S. Bui and K. Haupt, Anal. Bioanal. Chem., 2010, 398, 2481–2492 CrossRef CAS PubMed.
- W. J. Cheong, F. Ali, J. H. Choi, J. O. Lee and K. Y. Sung, Talanta, 2013, 106, 45–59 CrossRef CAS PubMed.
- Y. Fuchs, O. Soppera and K. Haupt, Anal. Chim. Acta, 2012, 717, 7–20 CrossRef CAS PubMed.
- R. Suedee, Int. J. Pharm. Sci. Rev. Res., 2013, 20, 235–268 CAS.
- K. Haupt, Nat. Mater., 2010, 9, 612–614 CrossRef CAS PubMed.
- A. Poma, A. Guerreiro, M. J. Whitcombe, E. V. Piletska, A. P. F. Turner and S. A. Piletsky, Adv. Funct. Mater., 2013, 23, 2821–2827 CrossRef CAS.
- E. Moczko, A. Poma, A. Guerreiro, I. P. d. V. Sansalvador, S. Caygill, F. Canfarotta, M. J. Whitcombe and S. A. Piletsky, Nanoscale, 2013, 5, 3733–3741 RSC.
- G. Vasapollo, R. Del Sole, L. Mergola, M. R. Lazzoi, A. Scardino, S. Scorrano and G. Mele, Int. J. Mol. Sci., 2011, 12, 5908–5945 CrossRef CAS PubMed.
- A. G. Mayes and M. J. Whitcombe, Adv. Drug Delivery Rev., 2005, 57, 1742–1778 CrossRef CAS PubMed.
- N. R. Jana and J. Y. Ying, Adv. Mater., 2008, 20, 430–434 CrossRef CAS.
- C. L. Haynes, A. J. Haes, A. D. McFarland and R. P. Van Duyne, in Topics in Fluorescence, ed. J. R. Lakowicz, Plenum Press, New York, 2003, Vol. 8 Search PubMed.
- C. L. Haynes, A. D. McFarland and R. P. Van Duyne, Anal. Chem., 2005, 77, 338A–346A CrossRef CAS.
- A. A. Volkert, V. Subramaniam, M. R. Ivanov, A. M. Goodman and A. J. Haes, ACS Nano, 2011, 5, 4570–4580 CrossRef CAS PubMed.
- G. Mie, Ann. Phys., 1908, 25, 377–445 CrossRef CAS.
- A. A. Volkert, V. Subramaniam and A. J. Haes, Chem. Commun., 2011, 47, 478–480 RSC.
- C. C. Hwang and W. C. Lee, J. Chromatogr., A, 2002, 962, 69–78 CrossRef CAS.
- K. Kantarovich, I. Tsarfati, L. A. Gheber, K. Haupt and I. Bar, Anal. Chem., 2009, 81, 5686–5690 CrossRef CAS PubMed.
- A. R. Guerreiro, I. Chianella, E. Piletska, M. J. Whitcombe and S. A. Piletsky, Biosens. Bioelectron., 2009, 24, 2740–2743 CrossRef CAS PubMed.
- S. Gam-Derouich, S. Mahouche-Chergui, S. Truong, D. B. Hassen-Chehimi and M. M. Chehimi, Polymer, 2011, 52, 4463–4470 CrossRef CAS PubMed.
- A. Gültekin, S. E. Diltemiz, A. Ersöz, N. Y. Sariözlü, A. Denizli and R. Say, Talanta, 2009, 78, 1332–1338 CrossRef PubMed.
- A. Gültekin, A. Ersöz, D. Hür, Y. N. Sariozlu, A. Denizli and R. Say, Appl. Surf. Sci., 2009, 256, 142–148 CrossRef PubMed.
- A. Gültekin, A. Ersöz, A. Denizli and R. Say, Sens. Actuators, B, 2012, 162, 153–158 CrossRef PubMed.
- A. Gültekin, A. Ersöz, A. Denizli and R. Say, Talanta, 2012, 93, 364–370 CrossRef PubMed.
- M. Bompart, Y. De Wilde and K. Haupt, Adv. Mater., 2010, 22, 2343–2348 CrossRef CAS PubMed.
- K. Kantarovich, I. Tsarfati, L. A. Gheber, K. Haupt and I. Bar, Biosens. Bioelectron., 2010, 26, 809–814 CrossRef CAS PubMed.
- K. Kantarovich, I. Tsarfati-BarAd, L. A. Gheber, K. Haupt and I. Bar, Plasmonics, 2013, 8, 3–12 CrossRef CAS.
- S. Kostrewa, M. Emgenbroich, D. Klockow and G. Wulff, Macromol. Chem. Phys., 2003, 204, 481–487 CrossRef CAS.
- X. Kan, T. Liu, C. Li, H. Zhou, Z. Xing and A. Zhu, J. Solid State Electrochem., 2012, 16, 3207–3213 CrossRef CAS PubMed.
- H. Wang, H. Zhao, X. Quan and S. Chen, Electroanalysis, 2011, 23, 1863–1869 CrossRef CAS.
- J. Q. Xue, D. W. Li, L. L. Qu and Y. T. Long, Anal. Chim. Acta, 2013, 777, 57–62 CrossRef CAS PubMed.
- S. Li, D. Du, J. Huang, H. Tu, Y. Yang and A. Zhang, Analyst, 2013, 138, 2761–2768 RSC.
- E. L. Holthoff, D. N. Stratis-Cullum and M. E. Hankus, Sensors, 2011, 11, 2700–2714 CrossRef CAS PubMed.
- X. A. Ton, B. T. S. Bui, M. Resmini, P. Bonomi, I. Dika, O. Soppera and K. Haupt, Angew. Chem., 2013, 52, 8317–8321 CAS.
- D. Yu, Y. Zeng, Y. Qi, T. Zhou and G. Shi, Biosens. Bioelectron., 2012, 38, 270–277 CrossRef CAS PubMed.
- J. Matsui, K. Akamatsu, S. Nishiguchi, D. Miyoshi, H. Nawafune, K. Tamaki and N. Sugimoto, Anal. Chem., 2004, 76, 1310–1315 CrossRef CAS PubMed.
- J. Matsui, K. Akamatsu, N. Hara, D. Miyoshi, H. Nawafune, K. Tamaki and N. Sugimoto, Anal. Chem., 2005, 77, 4282–4285 CrossRef CAS PubMed.
- M. Kara, L. Uzun, S. Kolayli and A. Denizli, J. Appl. Polym. Sci., 2013, 2273–2279 CrossRef CAS.
- M. Riskin, Y. Ben-Amram, R. Tel-Vered, V. Chegel, J. Almog and I. Willner, Anal. Chem., 2011, 83, 3082–3088 CrossRef CAS PubMed.
- M. Riskin, R. Tel-Vered and I. Willner, Adv. Mater., 2010, 22, 1387–1391 CrossRef CAS PubMed.
- G. Sener, L. Uzun, R. Say and A. Denizli, Sens. Actuators, B, 2011, 160, 791–799 CrossRef CAS PubMed.
- S. Lépinay, K. Kham, M. C. Millot and B. Carbonnier, Chem. Pap., 2012, 66, 340–351 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |