Screening of a selection of commercially available homogeneous Ru-catalysts in valuable olefin metathesis transformations†
Frédéric
Caijo
*a,
Fabien
Tripoteau
a,
Aurélien
Bellec
a,
Christophe
Crévisy
b,
Olivier
Baslé
b,
Marc
Mauduit
*ab and
Oliver
Briel
*c
aOMEGA CAT SYSTEM SARL, ENSCR, Avenue du Général Leclerc, CS 50837, 35708 Rennes Cedex 7, France. E-mail: f.caijo@omcat-system.com; Fax: +33 (0)299 388 516; Tel: +33 (0)299 388 516
bEcole Nationale Supérieure de Chimie de Rennes, CNRS, UMR 6226, Avenue du Général Leclerc, CS 50837, 35708 Rennes Cedex 7, France. E-mail: marc.mauduit@ensc-rennes.fr; Fax: +33 (0)223 238 108; Tel: +33 (0)223 238 112
cPrecious Metals Chemistry, UMICORE AG&Co. KG, Rodenbacher Chaussee 4, 63457 Hanau-Wolfgang, Germany. E-mail: oliver.briel@eu.umicore.com; Fax: +49 6181 592970; Tel: +49 6181 593894
First published on 19th September 2012
Abstract
A library of thirteen different commercially available Ru-based catalysts was evaluated in valuable metathesis reactions for the production of fragrance and bioactive molecule precursors. Rigorous library screening clearly illustrated the different catalytic behaviour of the catalyst selection and highlighted its significant advantage to provide efficiency in specific metathesis applications. Interestingly, this strategy offered substantial improvement over the state of the art, with the efficient synthesis of the macrocyclic Exaltolide 2 at low catalyst loading and dilution conditions.
Introduction
Initially used in polymer chemistry in the early 1960s, the olefin metathesis has considerably expanded its sphere of application and subsequently influenced the organic synthesis area over the past two decades. In fact, this metal-catalyzed methodology has become a powerful and effective synthetic tool for the construction of Csp2–Csp2 bonds in an eco-friendly manner.1 Olefin metathesis shows interesting properties such as exceptional tolerance to various functional groups, remarkable chemo-, regio-, and stereo-selectivity along with accessibility to a wide range of ring size products. These features allowed for the drastic simplification of the retrosynthetic design of numerous natural-product syntheses.2During the early 2000s in response to the increasing popularity gained through academic research, pharmaceutical companies have considered the vast potential of olefin metathesis and evaluated the production of biologically active ingredients. Cilepruvir3 (from Boehringer Ingelheim) and TMC4354 (from Janssen Pharmaceutical), two potent antiviral hepatitis C drugs, and SB-4627955 (from GlaxoSmithKline), a potent Cathepsin K inhibitor, are perfect examples of how ring-closing metathesis (RCM) on a large-scale can lead to the desired macrocyclic fragment (multi-kg).
The gold rush for olefin metathesis during the last decade has a direct correlation to important breakthroughs gained in the development of more efficient ruthenium-based catalysts (for instance, high TON and TOF,6 thermally stable,7 recoverable).8 In fact, the large number of catalysts currently available constantly overcomes limitations, leading to the production of more and more original building blocks when successful.9 Nevertheless, when the metathesis step fails or provides low yields, despite intensive screening of experimental conditions, the appropriate choice of catalyst must be questioned.10 Among all catalysts reported in the literature, only twenty-five of them are currently commercially available. This is primarily due to their difficult accessibility and/or a complicated Intellectual Property (IP) situation. Despite this limitation, the selection of available catalysts is representative enough to ensure valid, efficient catalyst screening.
Implementation of the commonly accepted statement: “the more reactive the catalyst is, the better efficiency will be achieved” may lead to disappointing results. In fact, high catalyst activity is often associated to poor stability and/or production of large amounts of undesired by-products, such as self-metathesis products. Therefore, a systematic and rigorous screening of the commercially available catalyst library should be completed to achieve optimal yield and selectivity in the presence of low catalyst loading.
In this study, we envisioned the screening of thirteen well-defined and commercially available Ru-based pre-catalysts at fixed and low (1 mol%) loading (Fig. 1). These complexes were selected taking into account their different catalytic behaviours: (i) standard precatalysts11 such as Grubbs II (G II), Hoveyda–Grubbs II (HG II), M2 or M20; (ii) fast-initiation pre-catalysts12 such as M51, M71SIMes, M71SIPr, M73SIMes, M73SIPr, M831, M832 or M853; (iii) thermally stable pre-catalysts13 such as M22. Three olefin metathesis transformations (two RCM and one cross-metathesis (CM)) were selected for their straightforward ability to provide useful precursors of fragrances (δ-decalactone 1 and Exaltolide 2) and of a natural bioactive molecule (Aplysamine 6 3) (Fig. 2). At first, these synthetic targets seem to involve truly simple metathesis reactions. Nevertheless, they require solving important challenges in the limitation of by-products formation, resulting from the undesired self-metathesis. A careful catalyst screening under optimal conditions should prevent this recurrent hurdle from occurring during the metathesis reaction.
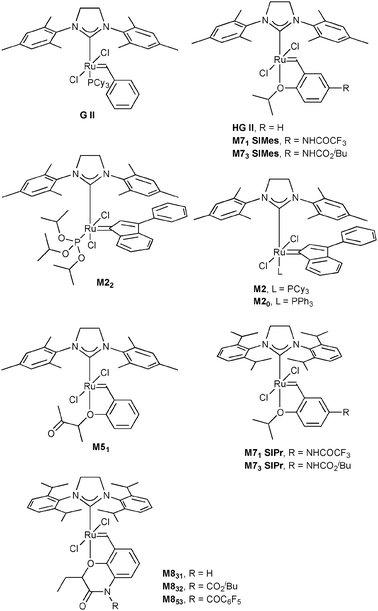 | ||
| Fig. 1 Selection of commercially available Ru-based catalysts. | ||
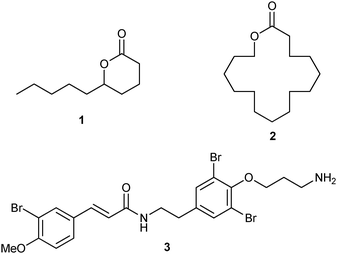 | ||
| Fig. 2 Targeted compounds. | ||
Results and discussion
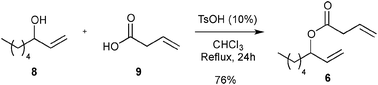
First, the selected commercially available Ru-catalysts of the library were evaluated and classified according to their reactivity in performing the RCM of a benchmark substrate, methylallyl-allyl diethylmalonate 4, under standard reaction conditions [e.g. 1 mol% of catalyst, CD2Cl2 (0.1 M), 30 °C].14 As depicted in Fig. 3, the kinetic profiles of selected Ru-catalysts revealed major differences (see ESI†). The most active catalyst, (M853), achieved complete conversion to the desired product within 5 minutes, while the “thermally latent” catalysts, (M2 and M22),15 revealed low or no activity under these standard reaction conditions. Importantly, popular catalysts G II or HG II presented moderate activities under the selected reaction conditions. Therefore, based on 1H NMR conversion after 10 min the resulting reactivity classification is presented below.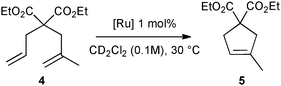
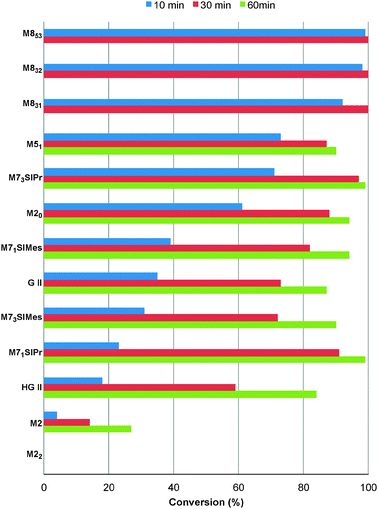
With the reactivity features in hand, metathesis catalysts were evaluated and screened in the RCM of ester 6, which yields lactone 7,16 a direct precursor of the well-known fragrance δ-decalactone 1 (Table 1). This lactone is usually used in foods and beverages for its peach-like odour and cream flavour. Enantiopure (R)-1 can be isolated by extraction from a natural product, however the extraction process remains fairly expensive.17 The biotechnological strategy, on the other hand, represents a cost effective alternative for the production of decalactone fragrances, unfortunately it leads mostly to the five-membered γ-decalactone (by bioconversion of the ricinoleic fatty acid).18 Therefore, the synthesis of the six-membered lactone 1 by the RCM pathway can also be considered as a promising chemical alternative for large-scale production.19 Interestingly, the involved oct-1-en-3-ylbut-3-enoate 6 is easily synthesized on a multi-gram scale and good isolated yield (76%) through the reaction of cheap 3-octenol 8 (also called the mushroom alcohol) and 3-butenoic acid 9 in the presence of p-toluene-sulfonic acid (Fig. 4).20 It is worth noting that enantioenriched mushroom alcohols (>99.9% ee, (R) or (S)) are also available and are efficiently produced via kinetic resolution involving lipases.21 For this study, however, only the racemic ester 6 was evaluated.
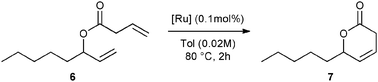
| Entry | [Ru] | 7/6 ratioa | Yieldb (%) |
|---|---|---|---|
| a Determined by 1H NMR spectroscopy. b Isolated yield after silica gel chromatography. c nd: not determined. | |||
| 1 | M853 | 75/25 | 55 |
| 2 | HG II | 79/21 | 56 |
| 3 | M22 | 100/0 | 77 |
| 4 | M73SIPr | 84/16 | 75 |
| 5 | M831 | 88/12 | 75 |
| 6 | M2 | 88/12 | 68 |
| 7 | M832 | 85/15 | 68 |
| 8 | M20 | 81/19 | 56 |
| 9 | M71SIPr | 100/0 | 87 |
| 10 | M51 | 57/43 | 54 |
| 11 | G II | 47/53 | ndc |
| 12 | M73SIMes | 45/55 | ndc |
| 13 | M71SIMes | 24/76 | ndc |
 | ||
| Fig. 4 Synthesis of 6. | ||
 | ||
| Fig. 3 RCM kinetics of methylallyl-allyl diethylmalonate 4. | ||
Ring-closing metathesis to obtain cyclic unsaturated esters constitutes a challenging process. In fact, the metathesis product is often so sensitive to reaction conditions that, in most cases, it undergoes formation of undesired by-products under prolonged reaction time.22 Therefore, the isolated yield of the desired product should be highly related to activity, selectivity, and stability of the metathesis catalyst. Initial screening of the reaction conditions revealed that the M853 catalyst (0.1 mol%), previously described as the most active for methylallyl-allyl diethylmalonate cyclization, offered an incomplete conversion along with a moderate (55%) isolated yield after 2 h at 80 °C (Table 1, entry 1). It is also important to note that the HG II catalyst displayed a similar behaviour under the described conditions (entry 2). Further screening of the catalyst library led to large improvements in the efficiency of the reaction. In fact, the M71SIPr catalyst reached complete conversion within 2 h along with high (87%) isolated yield (entry 9). Interestingly, total disappearance of the starting material was also achieved with the thermally latent M22 catalyst activated at 80 °C, but with altered 77% isolated yield (entry 3).
Hydrogenation of 7 was performed under classical hydrogenation conditions yielding 89% of isolated lactone 1 (Fig. 5).
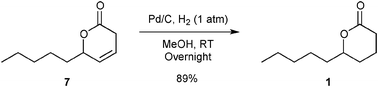 | ||
| Fig. 5 Hydrogenation of 7. | ||
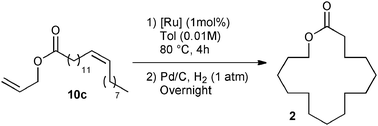
Exaltolide 2 belongs to the musk-ketone family, and used as a base note in perfumery.23 Interestingly, the ring closing metathesis of esters 10 should yield macrolactone 11, a precursor of the desired fragrance (see Table 2). Macrocyclization of lactones was intensely studied in the past decade and remains one of the most challenging synthetic targets in RCM.24 A representative example of this challenge was the macrocyclization to form the 14-membered tridecanolactone, which required highly diluted reaction conditions (up to 0.003 M) to prevent undesired cross-metathesis and large amounts of dimer (and/or oligomers) by-products.25 Ring-closing metathesis to form the 16-membered macrolactone 11 was previously described in the literature.26 High dilution reaction conditions were also necessary when terminal olefins were introduced into the catalytic cyclization process (ester 10a).26b An interesting strategy to promote the RCM while decreasing self-metathesis involved the use of a trisubstituted alkene instead of a terminal olefin. Despite the improved selectivity, the dilution of the media remained high (0.008 M).25 In recent years, the amount of organic solvent used has caused great concern. The search for efficient macrocyclization, limiting large waste production, constitutes a field of research, which attracted interest of both academia and industry.
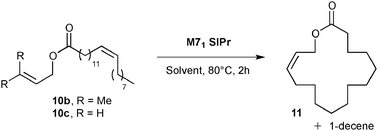
Preliminary results in the RCM of cyclic esters prompted us to examine our catalyst library in the challenging esters 10 macrocyclization to form the unsaturated precursor of exaltolide. Starting from commercially available bio-resourced erucic acid 12b, extracted from vegetable oils, esters 10b–c were synthesized and evaluated in the metathesis process using the M71SIPr catalyst. First, we did not observe any reaction with the sterically-demanding ester 10b bearing tri- and disubstituted alkenes (Table 2, entry 1). Therefore, we focused our attention on the less congested and more reactive diene-ester 10c (Fig. 6).
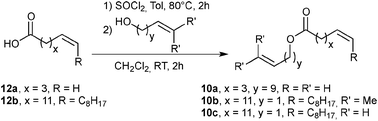 | ||
| Fig. 6 Esterification of 12a and erucic acid.27 | ||
Preliminary studies on dilution demonstrated that RCM of 10c was efficiently performed (45% of isolated yield) at a quite high concentration (0.01 M of toluene) using a low loading of catalyst (1 mol% of M71SIPr) (Table 2, entry 4). The yield dramatically decreased beyond this concentration (entry 5) and no desired product was formed under neat conditions (entry 6). No influence of the solvent was observed when replacing toluene by ethyl acetate (entry 7).
The commercially available catalyst library was subsequently evaluated in the RCM of ester 10c under the optimum reaction conditions: 1 mol% of catalyst loading and 4 h of reaction at 80 °C in 0.01 M of toluene. Additionally, the unsaturated lactone 11 demonstrated instability under purification conditions. Therefore, the RCM product was fully hydrogenated to form Exaltolide 2 in a one-pot reaction. Upon the conclusion of the metathesis reaction, Pd/C was added and the reaction flask pressured with 1 atm hydrogen overnight at room temperature.28 This strategy proved to be beneficial, since the desired product 2 was obtained under the same reaction conditions but with a better isolated yield than its unsaturated precursor 11 (compare Table 2, entry 4 with Table 3, entry 1). The challenge of this process was demonstrated by the moderate isolated yield observed for a large majority of catalysts of the library, reaching at best a moderate 50% isolated yield over two steps (Table 3, entries 6–13).
| Entry | [Ru] | Yielda (%) |
|---|---|---|
| a Isolated yield after silica gel chromatography. | ||
| 1 | M71SIPr | 50 |
| 2 | M22 | 69 |
| 3 | M73SIMes | 66 |
| 4 | M831 | 63 |
| 5 | M73SIPr | 63 |
| 6 | G II | 50 |
| 7 | HG II | 44 |
| 8 | M832 | 43 |
| 9 | M853 | 42 |
| 10 | M20 | 41 |
| 11 | M2 | 37 |
| 12 | M51 | 36 |
| 13 | M71SIMes | 34 |
Good yields, around 65%, were obtained with M73SIPr, M831 and M73SIMes catalysts (entry 3–5). Interestingly, the NHC–phosphite M22 complex presented the highest efficiency and offered 69% isolated yield of the desired product 2 (entry 2). This result illustrates the benefit of our library screening, since the least active catalyst in methylallyl-allyl diethylmalonate cyclization appeared as the most efficient in the macrolactonization. It is also important to note that in terms of catalyst efficiency and dilution conditions, the latest result constitutes a substantial improvement over the state of the art.25,26
Finally, the library of commercially available pre-catalysts was evaluated in a cross-metathesis reaction to generate 19, a precursor of Aplysamine 6 (3). Aplysamine 6, a bromotyrosine derivative extracted from the Australian marine sponge Pseudoceratina sp. displays good activity as an inhibitor of isoprenylcysteine carboxyl methyltransferase (Icmt).29 In 2009, Ullah and Arafeh reported the first total synthesis of Aplysamine 6 involving amide bond formation as a key step.30 Nevertheless, cross-metathesis reaction had previously only been investigated in a simplified model with acrylic amide and styrene.31 Therefore, we considered an advanced metathesis strategy offering the amide 19, a direct precursor of Aplysamine 6, via cross-metathesis of easily accessible substrates. In fact, 14 and 18 required only one or two simple steps starting from commercially available 3-bromo-4-methoxybenzaldehyde 13 and tyramine hydrochloride 15 (Fig. 7).30
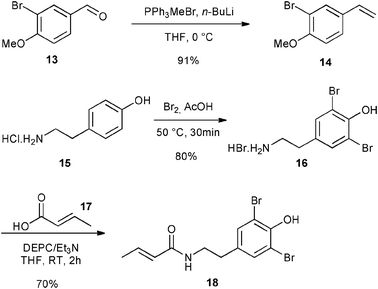 | ||
| Fig. 7 Synthesis of CM partners for synthesis of 19. | ||
Cross-metathesis reaction with deactivated acrylamides substrates suffers from poor reactivity and frequently requires high catalyst loading.32 The commercially available catalysts from our library were evaluated using 1 mol% of catalyst loading in 0.2 M of dichloromethane solution during 2 h at 40 °C. Under these standard conditions, M71SIPr, M73SIPr, M831, M51, M832, HG II and M853 catalysts showed good activity with a conversion into 19 between 88 and 76% (Table 4, entries 2–8). The cited catalysts also presented similar selectivities, affording analogous amounts of styrene self-metathesis by-products (ranging between 21 and 26% of initial styrene). The efficiency irremediably decreased with the other catalysts achieving less than 60% of conversion for most of them such as M73SIMes (entry 9), G II (entry 11) or M2 (entry 12). In the worst case, no reaction was observed with the M22 complex (entry 14). Finally, using the M71SIPr catalyst, product 19 could be isolated in 74% yield after chromatography (entry 2).
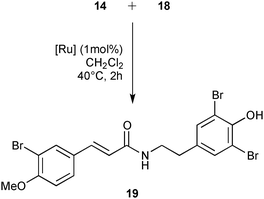
| Entry | [Ru] | 19 (%) |
|---|---|---|
| a An excess (1.5 equiv.) of 18 was used for this experiment, all other experiments were performed with an excess of 14 (2 equiv.). b Conversion determined by 1H NMR spectroscopy. c 19 was isolated in 74% yield after purification on silica gel. | ||
| 1a | M71SIPr | 24 |
| 2 | M71SIPr | 88c |
| 3 | M73SIPr | 85 |
| 4 | M831 | 84 |
| 5 | M51 | 82 |
| 6 | M832 | 80 |
| 7 | HG II | 79 |
| 8 | M853 | 76 |
| 9 | M73SIMes | 69 |
| 10 | M71SIMes | 68 |
| 11 | G II | 60 |
| 12 | M2 | 38 |
| 13 | M20 | 13 |
| 14 | M22 | 0 |
Conclusions
In conclusion, we have illustrated the fact that when developing new metathesis methodologies, the evaluation of a catalyst library can become very valuable. In fact, the multiplicity and plurality of parameters to consider when developing novel catalytic processes make the appropriate choice of catalyst difficult to predict. In the examples presented in this report, it was observed that an empiric preference exclusively based on catalyst activity may lead to unproductive results. Notably, a large screening of a commercially available catalyst library provided efficient syntheses of precursors of fragrances (δ-decalactone 1 and Exaltolide 2) and a natural bioactive molecule (Aplysamine 6 3).Experimental
General information
1H (400 MHz) and 13C (100 MHz) NMR spectra were recorded on a Bruker ARX400 spectrometer. Chemical shifts are reported in ppm with the solvent resonance as the internal standard (CDCl3: 1H δ 7.26 ppm, 13C δ 77.0 ppm, DMSO-d6: 1H δ 2.5 ppm, 13C δ 39.5 ppm). Data are reported as follows: chemical shift δ in ppm, multiplicity (s = singlet, d = doublet, t = triplet, q = quadruplet, m = multiplet), coupling constants (Hz) and integration. Anhydrous toluene and THF were obtained by passing through drying columns. DCM was distilled over CaH2. All commercial chemicals were used as received unless otherwise noted.General procedure for the synthesis of 6-pentyl-3,6-dihydro-2H-pyran-2-one 7
Ru-catalyst (0.1 mol%) was added to a solution of 6 (200 mg, 1.01 mmol) in toluene (51 mL). The reaction mixture was stirred for 2 h at 80 °C. After cooling down to room temperature, the solvent was evaporated and the residue was purified by silica gel chromatography (cyclohexane–EtOAc 100/0 to 90/10) to produce 7 as a yellow oil (see Table 1 for yields). 1H NMR (400 MHz, CDCl3): δ = 5.78–5.77 (m, 2H), 4.99–4.83 (m, 1H), 3.01–2.96 (m, 2H), 1.71–1.60 (m, 2H), 1.34–1.18 (m, 6H), 0.82 (t, J = 6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ = 169.2, 126.7, 121.4, 79.7, 35.7, 31.5, 29.9, 24.0, 22.5, 14.0.General procedure for the synthesis of Exaltolide 2
Ru-catalyst (1 mol%) was added to a solution of ester 10c (200 mg, 0.53 mmol) in toluene (53 mL). The reaction mixture was stirred for 4 h at 80 °C. After cooling down to room temperature, Pd/C (15 mg) was added and the reaction flask pressured with 1 atm hydrogen. After stirring overnight at room temperature, the reaction mixture was filtrated over a celite pad and the solvent was evaporated. The residue was purified by silica gel chromatography (cyclohexane–CH2Cl2 70/30) to produce 2 as a colourless oil (see Table 3 for yields). 1H NMR (400 MHz, CDCl3): δ = 4.13 (t, J = 5.8 Hz, 2H), 2.32 (t, J = 6.6 Hz, 2H), 1.68–1.59 (m, 4H), 1.42–1.25 (m, 20H); 13C NMR (100 MHz, CDCl3): δ = 174.0, 64.0, 34.4, 28.4, 27.8, 27.1, 27.1, 26.9, 26.7, 26.3, 26.0, 25.9, 25.8, 25.1, 24.9.General procedure for the synthesis of (E)-3-(3-bromo-4-methoxyphenyl)-N-(3,5-dibromo-4-hydroxyphenethyl)acrylamide 19
Ru-catalyst (1 mol%) was added to a solution of 14 (200 mg, 0.93 mmol) and 18 (170 mg, 0.47 mmol) in CH2Cl2 (2.3 mL). The reaction mixture was stirred for 2 h at 40 °C. The crude mixture was analysed by 1H NMR to determine the conversion (see Table 4). After cooling down to room temperature, Et2O (5 mL) was added and the solution was kept for 1 h at 5 °C to complete the precipitation. The solid was filtrated over a Büchner filter to remove amide by-products. Conversions were given by 1H NMR spectroscopy. When the M71SIPr catalyst was used, the desired product 19 was purified by silica gel chromatography (cyclohexane–EtOAc 60/40) and isolated in 74% (185 mg) as a white solid. 1H NMR (400 MHz, DMSO-d6): δ = 9.69 (s, 1H), 8.03 (dd, J = 5.7, 5.6 Hz, 1H), 7.78 (d, J = 2.1 Hz, 1H), 7.54 (dd, J = 8.6, 2.1 Hz, 1H), 7.40 (s, 2H), 7.32 (d, J = 15.8 Hz, 1H), 7.13 (d, J = 8.7 Hz, 1H), 6.51 (d, J = 15.8 Hz, 1H), 3.87 (s, 3H), 3.38 (t, J = 6.8 Hz, 2H), 2.68 (t, J = 6.8 Hz, 2H); 13C NMR (100 MHz, DMSO-d6): δ = 164.9, 156.1, 148.8, 136.8, 133.9, 132.3, 131.6, 131.5, 129.0, 128.4, 121.1, 112.8, 111.7 (2C), 111.0, 56.3, 39.8, 33.2.Acknowledgements
Funding for this project was partially provided by the European Community through the seventh framework program (CP-FP 211468-2 EUMET), the CNRS and the Ministère de la Recherche et de la Technologie. MM and FC thank Bretagne-Valorisation and Rennes Métropole for their financial support related to the development of metathesis catalysts.Notes and references
- For comprehensive reviews on olefin metathesis, see: (a) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18 CrossRef CAS; (b) R. H. Grubbs, in Handbook of metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 1–3 Search PubMed; (c) R. R. Schrock and A. H. Hoveyda, Angew. Chem., Int. Ed., 2003, 42, 4592 CrossRef CAS; (d) S. J. Connon and S. Blechert, Angew. Chem., Int. Ed., 2003, 42, 1900 CrossRef CAS; (e) D. Astruc, New J. Chem., 2005, 29, 42 RSC; (f) R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760 CrossRef CAS; (g) Y. Chauvin, Angew. Chem., Int. Ed., 2006, 45, 3741 CrossRef CAS; (h) R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 3748 CrossRef CAS; (i) P. H. Deshmukh and S. Blechert, Dalton Trans., 2007, 2479 RSC; (j) C. Samojlowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708 CrossRef CAS; (k) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746 CrossRef CAS; (l) S. Kotha and M. K. Dipak, Tetrahedron, 2012, 68, 397 CrossRef CAS.
- (a) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4490 CrossRef CAS; (b) A. Fürstner, Chem. Commun., 2011, 47, 6505 RSC.
- (a) T. Nicola, M. Brenner, K. Donsbach and P. Kreye, Org. Process Res. Dev., 2005, 9, 513 CrossRef CAS; (b) N. K. Yee, V. Farina, I. N. Houpis, N. Haddad, R. P. Frutos, F. Gallou, X.-J. Wang, X. Wei, R. D. Simpson, X. Feng, V. Fuchs, Y. Xu, J. Tan, L. Zhang, J. Xu, L. L. Smith-Keenan, J. Vitous, M. D. Ridges, E. M. Spinelli, M. Johnson, K. Donsbach, T. Nicola, M. Brenner, E. Winter, P. Kreye and W. Samstag, J. Org. Chem., 2006, 71, 7133 CrossRef CAS.
- O. Briel and C. S. J. Cazin, N-Heterocycle Carbene Complexes in Industrial Processes, in N-Heterocycle Carbenes in Transition Metal Catalysis and Organocatalysis, ed. C. S. J. Cazin, Springer, 2010, pp. 315–325 Search PubMed.
- (a) H. Wang, S. N. Goodman, Q. Dai, G. W. Stockdale and W. M. Clark, Jr, Org. Process Res. Dev., 2008, 12, 226 CrossRef CAS; (b) H. Wang, H. Matsuhashi, B. D. Doan, S. N. Goodman, X. Ouyang and W. M. Clark, Jr, Tetrahedron, 2009, 65, 6291 CrossRef CAS.
- For pre-catalysts efficient at low loading, see for example: (a) M. Gatti, L. Vieille-Petit, X. Luan, R. Mariz, E. Drinkel, A. Linden and R. Dorta, J. Am. Chem. Soc., 2009, 131, 9498 CrossRef CAS; (b) T. Vorfalt, S. Leuthäuβer and H. Plenio, Angew. Chem., Int. Ed., 2009, 48, 5191 CrossRef CAS; (c) K. M. Kuhn, T. M. Champagne, S. H. Hong, W.-H. Wei, A. Nickel, C. W. Lee, S. C. Virgil, R. H. Grubbs and R. L. Pederson, Org. Lett., 2010, 12, 984 CrossRef CAS; (d) V. Sashuk, L. H. Peeck and H. Plenio, Chem.–Eur. J., 2010, 16, 3983 CrossRef CAS; (e) X. Bantreil, R. A. M. Randall, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2010, 29, 3007 CrossRef CAS.
- For thermally stable catalytic systems, see for example: (a) L. H. Peeck and H. Plenio, Organometallics, 2010, 29, 2761 CrossRef CAS; (b) A. Kabro, T. Roisnel, C. Fischmeister and C. Bruneau, Chem.–Eur. J., 2010, 3, 12255 CrossRef; (c) O. Songis, A. M. Z. Slawin and C. S. J. Cazin, Chem. Commun., 2012, 48, 1266 RSC.
- Selected examples of recoverable homogeneous pre-catalysts: (a) J. S. Kingsbury, J. P. A. Harrity, P. J. Bonitatebus and A. H. Hoveyda, J. Am. Chem. Soc., 1999, 121, 791 CrossRef CAS; (b) C. Hongfa, J. Tian, H. S. Bazzi and D. E. Berbreiter, Org. Lett., 2007, 9, 3259 CrossRef CAS; (c) R. Gawin, A. Makal, K. Wozniak, M. Mauduit and K. Grela, Angew. Chem., Int. Ed., 2007, 46, 7206 CrossRef CAS; (d) H. Clavier, F. Caïjo, E. Borré, D. Rix, F. Boeda, S. P. Nolan and M. Mauduit, Eur. J. Org. Chem., 2009, 4254 CrossRef CAS.
- Olefin metathesis on challenging applications, see for example: (a) D. E. White, I. C. Stewart, R. H. Grubbs and B. M. Stoltz, J. Am. Chem. Soc., 2008, 130, 810 CrossRef CAS; (b) I. C. Stewart, T. Ung, A. A. Pletnev, J. M. Berlin, R. H. Grubbs and Y. Schrodi, Org. Lett., 2007, 9, 1589–1592 CrossRef CAS; (c) C. Samojlowicz, E. Borré, M. Mauduit and K. Grela, Adv. Synth. Catal., 2011, 353, 1993 CrossRef CAS.
- A. Fürstner, L. Ackermann, B. Gabor, R. Goddard, C. W. Lehmann, R. Mynott, F. Stelzer and O. Thiel, Chem.–Eur. J., 2001, 7, 3236 CrossRef.
- GII complex: (a) M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953 CrossRef CAS; (b) HG II complex: S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168 CrossRef CAS; (c) M2 complex: H. Clavier, C. A. Urbina-Blanco and S. P. Nolan, Organometallics, 2009, 28, 2848 CrossRef CAS; (d) M20 complex: C. A. Urbina-Blanco, S. Manzini, J. Pérez Gomez, A. Doppiu and S. P. Nolan, Chem. Commun., 2011, 47, 5022 RSC.
- (a) M51 complex: D. Arlt, M. Bieniek and R. Karch, PCT Int. Appl., WO2008034552, 2008 Search PubMed, M7 complexes: ; (b) M. Mauduit, H. Clavier and I. Laurent, PCT Int. Appl., WO2008065187, 2008 Search PubMed; (c) D. Rix, F. Caïjo, I. Laurent, F. Boeda, H. Clavier, S. P. Nolan and M. Mauduit, J. Org. Chem., 2008, 73, 4225 CrossRef CAS and see also ref. 8d; M8 complexes: ; (d) M. Mauduit and F. Caijo, PCT Int. Appl., WO2012013208, 2012 Search PubMed.
- M22 complex: X. Bantreil, T. E. Schmid, R. A. M. Randall, A. M. Z. Slawin and C. S. J. Cazin, Chem. Commun., 2010, 46, 7115 RSC.
- T. Ritter, A. Hejl, A. G. Wenzel, T. W. Funk and R. H. Grubbs, Organometallics, 2006, 25, 5740 CrossRef CAS.
- However, these latent pre-catalysts are really efficient at higher temperatures, see ref. 11c and 13.
- (a) S. Miyano, T. Hattori, T. Suzuki and M. Itai, JP2001151766, 2001; (b) T. Hattori, Y. Suzuki, O. Uesugi, S. Oi and S. Miyano, Chem. Commun., 2000, 73 RSC; (c) T. Hattori, Y. Suzuki, Y. Ito, D. Hotta and S. Miyano, Tetrahedron, 2002, 58, 5215 CrossRef CAS.
- M. Alchihab, J. Destain, M. Aguedo and P. Thonart, Biotechnol., Agron., Soc. Environ., 2010, 14, 681 Search PubMed.
- X.-D. Wang, G. Mauvais, R. Cachon, C. Divies and G. Feron, J. Biosci. Bioeng., 2000, 90, 338 CAS.
- Y. Zhao, L. Liang, W. Jia and Z. Wang, CN102010391, 2011.
- P. R. Andreana, J. S. McLellan, Y. Chen and P. G. Wang, Org. Lett., 2002, 4, 3875 CrossRef CAS.
- F. Felluga, C. Forzato, F. Ghelfi, P. Nitti, G. Pitacco, U. M. Pagnoni and F. Roncaglia, Tetrahedron: Asymmetry, 2007, 18, 527 CrossRef CAS.
- Catalyst degradation initiates side-reactions: M. Ulman and R. H. Grubbs, J. Org. Chem., 1999, 64, 7202 CrossRef CAS.
- A. S. Williams, Synthesis, 1999, 1707 CrossRef CAS.
- (a) A. Fürstner and K. Langemann, J. Am. Chem. Soc., 1997, 119, 9130 CrossRef; (b) V. P. Kamat, H. Hagiwara, T. Katsumi, T. Hoshi, T. Suzuki and M. Ando, Tetrahedron, 2000, 56, 4397 CrossRef CAS; (c) A. Michrowska, P. Wawrzyniak and K. Grela, Eur. J. Org. Chem., 2004, 2053 CrossRef CAS; (d) D. Burtscher and K. Grela, Angew. Chem., Int. Ed., 2009, 48, 442 CrossRef CAS.
- J. L. Christopher, W. Bielawski and R. H. Grubbs, J. Am. Chem. Soc., 2001, 123, 11312 CrossRef.
- (a) A. Fürstner and K. Langemann, Synthesis, 1997, 792 CrossRef; (b) H. Hagiwara, T. Nakamura, N. Okunaka, T. Hoshi and T. Suzuki, Helv. Chim. Acta, 2010, 93, 175 CrossRef CAS.
- For synthesis of 10a, see D. Villemin, Tetrahedron Lett., 1980, 21, 1715 CrossRef CAS ; for synthesis of 10b–c see ESI†.
- No trace of unsaturated compound was observed.
- M. S. Buchanan, A. R. Carroll, G. A. Fechner, A. Boyle, M. Simpson, R. Addepalli, V. M. Avery, J. N. A. Hooper, T. Cheung, H. Chen and R. J. Quinn, J. Nat. Prod., 2008, 71, 1066 CrossRef CAS.
- N. Ullah and K. M. Arafeh, Tetrahedron Lett., 2009, 50, 158 CrossRef CAS.
- W. Erb and E. Payet, Actual. Chim., 2010, 345, 33 CAS.
- T. L. Choi, A. K. Chatterjee and R. H. Grubbs, Angew. Chem., Int. Ed., 2001, 40, 1277 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20524f |
| This journal is © The Royal Society of Chemistry 2013 |