Anion tuning of chiral bis(urea) low molecular weight gels†
Gareth O.
Lloyd
a,
Marc-Oliver M.
Piepenbrock
b,
Jonathan A.
Foster
b,
Nigel
Clarke
c and
Jonathan W.
Steed
*b
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK
bDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK. E-mail: jon.steed@durham.ac.uk
cThe Department of Physics and Astronomy, University of Sheffield, Hicks Building, Hounsfield Road, Sheffield, S3 7RH, UK
First published on 7th November 2011
Abstract
A series of chiral bis(urea) compounds based on oligomethylene spacers with S-phenylethyl end groups have been investigated as low molecular weight gelators. The series shows gelation of a variety of liquids and their structural, morphological and rheological properties are reported. The bis(urea) compounds also act as supramolecular hosts for anions such as chloride and acetate and the weakening of the gels by competitive anion complexation on this series of archetypical, low molecular weight gels is reported. Modification of gel properties using gel mixtures is also described and the gelation process studied using a fluorescent napthyl analogue.
Introduction
There is currently great interest in low molecular weight gelators (LMWGs), both in terms of the fundamental relationship between molecular structure and the resulting gel characteristics, and in a range of actual and emerging application areas.1–10 Applications that have been successfully implemented are in separation,11 drug delivery,12–14 and the templating of inorganic and polymer materials.15,16 Some emerging applications17 include dynamic gels,18,19 biological applications20 especially using gels as cell growth media21,22 and using gels as a medium to control crystal growth.23 The tuning of the gelation characteristics of LMWGs using a variety of methods such as thermal treating,24 shearing (e.g. thixotropy),25–30 photochemical transformations,31–33 changes in pH,34–37 host–guest chemistry38–40 and use of chemical stimuli such as complexation with metal cations, anions and bioactive compounds,41–44 have been reported and have a number of interesting technological possibilities.45 Chemical tuning of gels by addition of ions or small molecules is of particular interest as this represents a way of introducing useful functionality and modifying the properties of these soft materials. Tuning of gel properties by non-covalent binding of anions is gathering considerable momentum.42,43,46 The majority of the reported examples of anion tuning are based on breaking the gel down to a sol by adding anions on a competitive basis. Conversely, there are examples of systems in which anion binding actually triggers gelation47 and systems in which the anions have a complex effect on the gel behaviour.48 We, and others, have also presented examples of anion tuning of metal-containing LMWGs.44,48,49 Recently, several groups have started to target the possibility of tuning the properties and characteristics of low molecular weight salt gelators by varying the identity of the anion component of the salt.50,51 We now present a full study of the gelation ability of a series of chiral bis(urea) compounds,52,53 the relationship between structure and gel characteristics, and the influence of anion binding on the bulk materials properties of the gels, including their rheology.54,55Experimental section
Synthesis
Chemicals for the synthesis of all compounds were purchased from Sigma-Aldrich and were used without further purification.In a typical synthesis a solution of (S)-(-)-α-methylbenzyl isocyanate (0.90 g, 6.1 mmol) in dry CHCl3 (100 mL) was added dropwise to a stirred solution of the appropriate α-ω-diaminoalkane (3.0 mmol) under reflux. After complete addition the reaction was allowed to reflux under an inert N2 atmosphere for up to 24h. The resulting precipitate formed was collected by filtration and washed with CHCl3 (3 × 10 mL). The resulting white powders were obtained in good yields (75–82%).
1-[(1S)-1-Phenylethyl]-3-[2-({[(1S)-1-phenylethyl]carbamoyl} amino)ethyl] urea (12). (S)-1-phenylethyl isocyanate (2.1 g, 13.6 mmol) was added to 150 mL dry CHCl3 in a three necked round bottom flask fitted with a condenser and flowing N2. To this solution was added ethylene diamine (0.408 g, 0.454 mL, 6.8 mmol) dropwise. The reaction mixture was refluxed for 24 h under N2. A white precipitate formed and was filtered off using a Büchner funnel. Yield: 1.96 g; 81%; 5.5 mmol. 1H NMR (400 MHz, d-DMSO, δ (ppm), J/Hz) 1.30 (6H, d, J = 7.1, –CH3) 2.99 (4H, m, –CH2–) 4.73 (2H, pseudo-quin, –CH–) 5.86 (2H, br. m, –NH–CH2) 6.42 (2H, d, J = 8.1, –CH–NH–) 7.29 (10H, m, Ar. C–H). 13C{1H} NMR: (400 MHz, d-DMSO, δ (ppm)) 14.05; 23.30; 48.56; 125.75; 126.35; 128.13; 145.76; 157.37. ES+MS m/z = 355.3 [44%] C20H26N4O2 + H+, 377.2 [100%] C20H26N4O2 + Na+. Anal. Calc. C 67.78% H 7.38% N 15.81%. Found C 66.94% H 7.32% N 15.57%. m.p. 216 ± 1 °C. FT-IR: (v, cm−1) 3321 (m) 2970 (w) 2930 (w) 2866 (w) 1620 (s) 1571 (s) 1526 (m) 1496 (w) 1442 (m) 1363 (w) 1259 (s) 1234 (m) 913 (w) 750 (m) 696 (m) 646 (m) 562 (w).
Crystal data for 12: C20H26N4O2, M = 354.45, colourless needle, 0.40 × 0.11 × 0.10 mm3, monoclinic, space group P21 (No. 4), a = 12.1914(14), b = 4.6342(6), c = 17.341(2) Å, β = 107.849(5)°, V = 932.57(19) Å3, Z = 2, Dc = 1.262 g cm−3, F000 = 380, Smart-6 K, Mo-Kα radiation, λ = 0.71073 Å, T = 120(2)K, 2θmax = 56.5°, 7921 reflections collected, 4407 unique (Rint = 0.2294). Final GooF = 0.896, R1 = 0.0972, wR2 = 0.2219, R indices based on 2170 reflections with I > 2σ(I) (refinement on F2), 235 parameters, 1 restraint. Lp and absorption corrections applied, μ = 0.084 mm−1.
1-[(1S)-1-Phenylethyl]-3-[2-({[(1S)-1-phenylethyl]carbamoyl} amino)propyl] urea (13). Prepared as 12 using 1,3-diaminopropane. 1H NMR: (400 MHz, d-DMSO, δ (ppm), J/Hz) 1.35 (6H, d, J = 7.0, –CH3), 1.45 (2H, sept, J = 6.6, –CH2–), 3.00 (4H, m, NH–CH2), 4.77 (2H, dq, J = 8.2 & 7.0, CMeH), 5.86 (2H, t, J = 5.8, NH–CH2), 6.39 (2H, d, J = 8.2, NH–CH), 7.25–7.36 (10H, m, ArH). Anal. Calc. C 68.45%, H 7.66%, N 15.20%. Found C 68.33% H 7.69% N 15.13%. ES+MS m/z = 369 [34%] C21H28N4O2 + H+, 391 (100%) C21H28N4O2 + Na+.
1-[(1S)-1-Phenylethyl]-3-[2-({[(1S)-1-phenylethyl]carbamoyl} amino)butyl] urea (14). Prepared as 12 using 1,4-diaminobutane. 1H NMR (400 MHz, d-DMSO, δ (ppm), J/Hz) 1.28–1.32 (4H, m, –CH2-), 1.29 (6H, d, J = 7.0, CH2), 2.94 (4H, m, NH–CH2-), 4.72 (2H, dq, J = 8.2 & 7.1, CMeH), 5.77 (2H, t, J = 5.6, NH–CH2), 6.24 (2H, d, J = 8.2, NH–CH), 7.18–7.32 (10H, m, ArH). Anal. Calc. C 69.08%, H 7.91%, N 14.65%. Found C 68.83% H 7.92% N 14.55%. ES+MS m/z = 383 [6%] C22H30N4O2 + H+, 405 (100%) C22H30N4O2 + Na+.
Crystal data for 14 (form I): C22H30N4O2, M = 382.50, colourless needle, 0.35 × 0.21 × 0.16 mm3, monoclinic, space group C2 (No. 5), a = 35.285(15), b = 4.651(2), c = 12.784(5) Å, β = 97.518(7)°, V = 2079.7(15) Å3, Z = 4, Dc = 1.222 g cm−3, F000 = 824, Smart-6 K, Mo-Kα radiation, λ = 0.71073 Å, T = 120(2)K, 2θmax = 56.4°, 12725 reflections collected, 5076 unique (Rint = 0.0619). Final GooF = 1.152, R1 = 0.0761, wR2 = 0.1907, R indices based on 4339 reflections with I > 2σ(I) (refinement on F2), 253 parameters, 1 restraint. Lp and absorption corrections applied, μ = 0.080 mm−1.
Crystal data for 14 (form II): C22H30N4O2, M = 382.50, colourless needle, 0.21 × 0.07 × 0.05 mm3, monoclinic, space group P21 (No. 4), a = 4.5914(1), b = 19.6481(4), c = 11.2393(3) Å, β = 90.8720(10)°, V = 1013.81(4) Å3, Z = 2, Dc = 1.253 g cm−3, F000 = 412, Smart-6 K, Mo-Kα radiation, λ = 0.71073 Å, T = 120(2)K, 2θmax = 61.0°, 18937 reflections collected, 3189 unique (Rint = 0.0399). Final GooF = 1.104, R1 = 0.0376, wR2 = 0.0886, R indices based on 2763 reflections with I > 2σ(I) (refinement on F2), 253 parameters, 1 restraint. Lp and absorption corrections applied, μ = 0.082 mm−1.
1-[(1S)-1-Phenylethyl]-3-[2-({[(1S)-1-phenylethyl]carbamoyl} amino)pentyl] urea (15). Prepared as 12 using 1,5-diaminopentane. 1H NMR: (400 MHz, d-DMSO, δ (ppm), J/Hz) 1.19 (2H, m, –CH2–), 1.29 (6H, d, J = 7.0, CH3), 1.30–1.36 (4H, m, –CH2–), 2.94 (4H, m, NH–CH2–), 4.72 (2H, dq, J = 8.1 & 7.0, CMeH), 5.75 (2H, t, J = 5.6, NH–CH2), 6.23 (2H, d, J = 8.1, NH–CH), 7.17–7.32 (10H, m, ArH). Anal. Calc. C 69.67%, H 8.13%, N 14.13%. Found C 69.33% H 8.10% N 14.14%. ES+MS m/z = 397 [44%] C23H32N4O2 + H+, 419 (94%) C23H32N4O2 + Na+, 793 (25%) 2C23H32N4O2 + Na+, 815 (100%) 2C23H32N4O2 + Na+.
Crystal data for 15: C23H32N4O2, M = 396.53, colourless needle, 0.49 × 0.24 × 0.10 mm3, monoclinic, space group P21 (No. 4), a = 12.317(2), b = 4.6108(8), c = 19.488(3) Å,β = 107.519(4)°, V = 1055.4(3) Å3, Z = 2, Dc = 1.248 g cm−3, F000 = 428, Smart-6 K, Mo-Kα radiation,λ = 0.71073 Å, T = 120(2)K, 2θmax = 55.1°, 7923 reflections collected, 4625 unique (Rint = 0.0499). Final GooF = 1.018, R1 = 0.0624, wR2 = 0.1354, R indices based on 3061 reflections with I > 2σ(I) (refinement on F2), 262 parameters, 1 restraint. Lp and absorption corrections applied, μ = 0.081 mm−1.
1-[(1S)-1-Phenylethyl]-3-[2-({[(1S)-1-phenylethyl]carbamoyl} amino)hexyl] urea (16). Prepared as 12 using 1,6-diaminohexane. 1H NMR: (400 MHz, d-DMSO, δ (ppm), J/Hz) 1.18–1.20 (4H, m, –CH2–), 1.29 (6H, d, J = 7.0, CH3), 1.30–1.36 (4H, m, –CH2–), 2.94 (4H, m, NH–CH2–), 4.72 (2H, dq, J = 8.1 & 7.0, CMeH), 5.75 (2H, t, J = 5.6, NH–CH2), 6.23 (2H, d, J = 8.1, NH–CH), 7.18–7.32 (10H, m, ArH). Anal. Calc. C 70.21%, H 8.35%, N 13.65%. Found C 70.04% H 8.34% N 13.47%. ES+MS m/z = 411 [60%] C24H34N4O2 + H+, 433 (98%) C24H34N4O2 + Na+, 821 (32%) 2C24H34N4O2 + Na+, 843 (100%) 2C24H34N4O2 + Na+.
1-[(1S)-1-Phenylethyl]-3-[2-({[(1S)-1-phenylethyl]carbamoyl} amino)heptyl] urea (17). Prepared as 12 using 1,7-diaminoheptane. 1H NMR: (400 MHz, d-DMSO, δ (ppm), J/Hz) 1.20–1.24 (6H, m, –CH2–), 1.29 (6H, d, J = 7.0, CH3), 1.30–1.36 (4H, m, –CH2–), 2.94 (4H, m, NH–CH2–), 4.71 (2H, dq, J = 8.1 & 7.0, CMeH), 5.74 (2H, t, J = 5.6, NH–CH2), 6.23 (2H, d, J = 8.1, NH–CH), 7.18–7.32 (10H, m, ArH). Anal. Calc. C 70.72%, H 8.55%, N 13.20%. Found C 70.48% H 8.55% N 13.19%. ES+MS m/z = 425 [45%] C25H36N4O2 + H+, 447 (100%) C25H36N4O2 + Na+.
Crystal data for 17: C25H36N4O2, M = 424.58, colourless needle, 0.50 × 0.23 × 0.08 mm3, monoclinic, space group P21 (No. 4), a = 12.275(4), b = 4.6302(15), c = 20.791(7) Å,β = 102.108(9)°, V = 1155.4(7) Å3, Z = 2, Dc = 1.220 g cm−3, F000 = 460, Smart-6 K, Mo-Kα radiation,λ = 0.71073 Å, T = 120(2)K, 2θmax = 54.0°, 12691 reflections collected, 4874 unique (Rint = 0.1213). Final GooF = 0.988, R1 = 0.0592, wR2 = 0.1468, R indices based on 4077 reflections with I > 2σ(I) (refinement on F2), 280 parameters, 1 restraint. Lp and absorption corrections applied,μ = 0.078 mm−1.
1-[(1S)-1-Phenylethyl]-3-[2-({[(1S)-1-phenylethyl]carbamoyl} amino)octyl] urea (18). Prepared as 12 using 1,8-diaminooctane. 1H NMR: (400 MHz, d-DMSO, δ (ppm), J/Hz) 1.26–1.30 (8H, m, –CH2–), 1.35 (6H, d, J = 7.0, CH3), 1.36–1.41 (4H, m, –CH2–), 3.01 (4H, m, NH–CH2–), 4.78 (2H, dq, J = 8.1 & 7.0, CMeH), 5.80 (2H, t, J = 5.4, NH–CH2), 6.29 (2H, d, J = 8.1, NH–CH), 7.24–7.39 (10H, m, ArH). Anal. Calc. C 71.20%, H 8.73%, N 12.77%. Found C 71.03% H 8.73% N 12.54%. ES+MS m/z = 439 [72%] C26H38N4O2 + H+, 461 (100%) C26H38N4O2 + Na+.
1-[(1R)-1-(1-Naphthyl)ethyl]-3-[2-({[(1R)-1-(1-naphthyl)ethyl] carbamoyl}amino)ethyl]urea (2). (R)-1-(1-naphthyl) isocyanate (1 g, 5.08 mmol) was added to 150 mL dry CHCl3 in a three necked round bottom flask fitted with a condenser and flowing N2. To this solution was added ethylene diamine (0.152 g, 0.169 mL, 2.54 mmol) dropwise. The reaction mixture was refluxed for 24 h under N2. The product formed as a white precipitate and was isolated by filtration using a Büchner funnel. Yield: 0.95 g; 82%; 2.1 mmol. 1H NMR: (400 MHz, d-DMSO, δ (ppm), J/Hz) 1.44 (6H, d, J = 6.8, –CH3) 3.02 (4H, m, –CH2–) 5.54 (2H, pseudo-quin, –CH–) 5.87 (2H, br s, –NH–CH2) 6.57 (2H, d, J = 8.1, –CH–NH–) 7.44–7.56 (8H, m, J = 7.3; 7.6; 8.1, Ar. C–H) 7.79 (2H, d, J = 7.6, Ar. C–H) 7.92 (2H, d, J = 7.3, Ar. C–H) 8.12 (2H, d, J = 8.1, Ar. C–H). 13C{1H} NMR: (400 MHz, d-DMSO, δ (ppm)) 14.05; 22.45; 44.64; 121.98; 123.23; 125.42; 125.46; 126.00; 126.97; 128.54; 130.31; 133.37; 141.31; 157.30. Anal. calc. C 73.98% H 6.65% N 12.33%. Found C 73.57% H 6.64% N 12.14%. M.p. 243 ± 1 °C. ES+MS (m/z) 455 [78%] C28H30N4O2 + H+, 477 (100%) C28H30N4O2 + Na+, 931 (52%) 2C28H30N4O2 + Na+. FT–IR: (v, cm−1) 3315 (m) 2972.4 (w) 2938 (w) 2868 (w) 1614 (m) 1569 (s) 1509 (m) 1450 (m) 1371 (w) 1301 (w) 1261 (m) 1232 (m) 1167 (w) 1108 (m) 1088 (m) 790 (m) 770 (s) 631 (m).
Rheometry
Rheology experiments were performed using a TA Instruments Advanced Rheometer 2000. A cylindrical couette geometry was used for experiments run on gel samples. All experiments were run at 20 °C. Frequency sweep measurements were performed over a range of 1 to 100 Hz with a constant osc. torque value of 100 Nm. Stress sweep measurements were performed over an osc. torque range of 1 Nm to 20000 Nm with a constant frequency of 1 Hz. Time or temperature sweep measurements were performed at a constant osc. torque value of 100 Nm and constant frequency value of 1 Hz. Details of anion tuning rheological experiments are given in the ESI.†NMR titrations
1H NMR titration experiments were carried out at room temperature using either a Varian Inova-500 spectrometer operating at 500 MHz or Varian Mercury-400 spectrometer operating at 400 MHz. All chemical shifts are reported in ppm relative to the residual protic solvent as an internal reference. A solution of the host species of known concentration, typically 0.02 M, was made up in an NMR tube using the appropriate deuterated solvent (0.5 ml). Solutions of the anions, as TBA+ salts, were made up in volumetric flasks (2 ml) with a concentration five times greater than that of the host. The guest solution was typically added in 10 μl aliquots, representing 0.1 equivalents of the guest with respect to the host. Larger aliquots were used in some cases where no inflection of the trace was evident. Spectra were recorded after each addition and the trace followed. Results were analysed using the curve-fitting program HypNMR,56,57 simultaneously fitting as many peaks as could be accurately followed throughout the experiment. Details of titration experiments and Job plot's are given in the ESI.†Crystallography
Single crystal diffraction data was collected on a Bruker SMART 6000 CCD. Powder samples were dried and processed through a 100 μm microsieve and run on silicon zero background sample holders using a Bruker D5000 diffractometer and copper radiation.Fluorescence spectroscopy
Samples were scanned in 1 ml glass vials using a Jobin-Yvon Horiba Fluorolog 3-222 Tau-3 Spectrofluorimeter with a front facing illumination method and were corrected for the spectral response of the machine.Microscopy
SEM samples were partially dried under room temperature and atmosphere, and then dried under high vacuum on a piece of silicon wafer. They were then coated with a thin layer of platinum of 3–5 nm using a 308UHR Ultra High Resolution Coating System (Biological & Biomedical Sciences, Durham). Samples were imaged using a S-5200 UHR FE–SEM Hitachi Scientific instrument (Biological & Biomedical Sciences, Durham). TEM samples were deposited onto a holey carbon grid and were immediately examined in a JEOL 2100F FEG TEM (Physics, Durham) operating at 200kV.Results and discussion
Synthesis and gel formation
The seven compounds of type 1n (where n = 2–8) are readily synthesised by the coupling of (S)-(−)-α-methylbenzyl isocyanate with the appropriate α,ω-diaminoalkane to give the desired chiral bis(urea) products. Compound 2 is prepared by reaction of ethylene diamine with (R)-1-(1-naphthyl)ethyl isocyanate. The products precipitated from the reaction solution and were isolated in good yields (75–82%; see experimental section for details). The phenylethyl and napthylethyl substituents were deliberately introduced in order to limit crystallinity by limiting space group choice to Sohnke groups and provide a sterically cumbersome moiety, hence promoting gelation.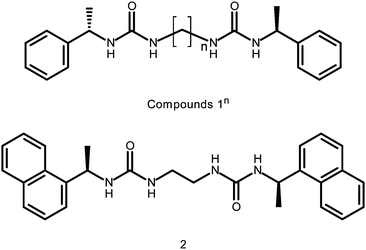
All compounds were tested for gelation ability by dissolving 1% by weight in 1 ml of a variety of solvents, namely MeCN, CHCl3, MeOH, EtOH, DMF, DMSO, toluene (Tol), ethylAcetate (EtOAc), (CH3)2CO, THF and solvent mixtures with water or Tol as the predominant solvent. The mixtures were initially heated to dissolve the gelator, followed by cooling of the solution to room temperature (Fig. S1, ESI†). Gelation was assessed by means of an “inversion test”. Table 1 summarises the overall gelation results for the nine compounds. Compounds 1n (where n = even) and 2 are efficient gelators of a wide variety of solvents, especially less polar solvents and solvent mixtures. The compounds do not gel pure, polar solvents namely water, DMF, DMSO, MeOH and EtOH. The series 1n (where n = odd) do not form gels under any circumstances except 13 which forms a weak gel in CHCl3 after sonication, with poor reproducibility (Fig. 1). This kind of alternating odd/even effect has been observed in related systems.58–63 Sonication10 generally improves the homogeneity of the gels, and in the case of 18, gels were only formed when sonication was used. All gels were found to be thermally reversible and in the case of compound 2 gels in DMSO:H2O mixtures, the gels are thixotropic.
| Compound | Solvents gelled | Appearancea |
|---|---|---|
a TG = transparent gel; OG = Opaque gel.
b See Table S1 for more details.
c See main text for method of preparation.
d Range of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2–1:1.
e gel + crystals mixed phases for all ratios.
f Range of 4:1–2:3.
g Requires sonication for gelation to occur. :2–1:1.
e gel + crystals mixed phases for all ratios.
f Range of 4:1–2:3.
g Requires sonication for gelation to occur.
|
||
| 12 b | CHCl3/MeCN/DMSO:H2O/EtOH:H2O/MeOH:H2O/Ethyl Acetate/THF/Acetone/Tol:DMSOc | TG to OG |
| 14 | CHCl3/MeCN/DMSO:H2Od/MeOH:H2Oe | TG |
| 16 | CHCl3/MeCN/DMSO:H2Of/MeOH: H2Oe | TG |
| 18 | CHCl3/MeCNg/DMSO:H2Of/MeOH: H2Oe | TG |
| 2 b | CHCl3/MeCN/DMSO:H2O/EtOH:H2O/MeOH:H2O/Ethyl Acetate/THF/Acetone/Tol:DMSO | TG to OG |
| 12, 14, 16, 18 and 2 | Tol, Hexane, Cyclohexane, MeOH, EtOH, DMSO, H2O, DMF | Precipitates or insoluble or stays in solution |
 | ||
| Fig. 1 Alternation of gel (even n) and sol (odd n) formation in CHCl3 by compounds 1(n = 2–8). All vials are at 1% by weight in 2 ml of solvent. | ||
To form gels of hydrophobic solvents, such as toluene, where compounds 1n (n = even) are not soluble, the gelator was dissolved in a minimum volume of DMSO, MeCN, EtOH or MeOH and added to a larger volume of the hydrophobic solvent resulting in gel formation. For example, thermoreversible gels of 12 with toluene as the major solvent component, can be formed at a solvent ratio of 9.5:0.5 Tol to DMSO at 0.05% gelator by weight, making 12 a supergelator. Hydrogels could also be made by a similar method by mixing solutions of gelator in a polar solvent with water to produce hydrogels with a high percentage of H2O or DMSO (90 to 95% of the solvent, Fig. S2–S3, ESI†). Given the ability of a concentrated solution of 12 in DMSO to gel both water and non-polar solvents like toluene, we attempted to selectively gel one of these solvents from a mixture of the two with a view to developing liquid–liquid separations applications such as oil spill remediation.64–68 A few drops of a concentrated solution of 12 in DMSO were added to a stirred or still mixture of H2O and toluene (with ratios from 8:2 to 2:8). The toluene is gelled exclusively upon addition of 12 in all cases when the total weight percentage of gelator added is less than 2%. If excess 12 (above 2% by weight, the saturation point in Tol) is added the compound begins to gel the water as well. Subsequent heating of this solution with water and the gelled toluene does not result in dissolution of the gel, and results in an apparent stiffening of the toluene gel (the gels are firmer under a spatula). In addition, the effect of small amounts of water on gel behaviour of organic solvent was investigated. In the case of MeCN and CHCl3 the gels of these solvents were found to be stronger (as measured by the magnitude of their storage modulus, G′, by stress sweep rheometry, e.g. 400 Pa in the “wet” solvent compared to 4000 Pa in “dry” solvent) when the solvent was dried over molecular sieves to remove water compared to when the solvents were not dried. This effect is more pronounced in the case of THF gels; addition of one or two drops of water results in the complete dissolution of the gels.
Crystalline structures of 1n
Gelators 12, 14, 15 and 17 were characterised by single crystal X-ray crystallography. Compound 14 exists in two polymorphic forms (I and II) obtained from aqueous DMSO and aqueous methanol, respectively. The crystals of 12 were also obtained from aqueous DMSO (3:7 v/v), while the 15 and 17 samples were obtained from aqueous MeOH upon cooling of hot concentrated solutions. The formation of the urea tape hydrogen bonding motif is common to all the structures.
Compound 12 crystallises in the Sohnke monoclinic space group P21, with the whole molecule as the asymmetric unit (Fig. S4, ESI†). The most obvious feature of the structure is the R12(6) urea tape motif26,69 formed by both urea substituents on each molecule forming typical and symmetric urea hydrogen bonds (Fig. 2 and S4, ESI†). The arrangement of the hydrogen bonding of the two urea tapes is in an antiparallel chain. There is a gauche arrangement of the phenylethyl groups (Fig. S4, ESI†). For this very short alkylene chain the two terminal C–N–C–C torsion angles are ca. 80° rather than the 180° expected for an all-trans oligomethylene chain, resulting in a ‘kink’ in the chain. The crystal morphology is a long flat needle. This high aspect ratio is related to the fibre morphology seen in the gel state imaged by TEM and SEM (vide infra). Face indexing the crystals showed that the fastest growing face is (010). The urea-tape packing direction is [010] indicating that the urea–urea packing drives the faster growth of the (010) face and hence determines the needle morphology of the crystal. The flat longer face of the crystal is (100) and the thin face is the (001).
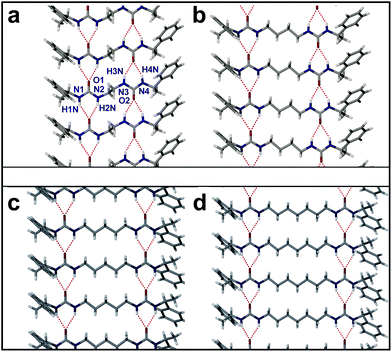 | ||
| Fig. 2 Crystal packing diagrams showing the urea tape motif of (a) 12, (b) 14 (form I), (c) 15, (d) 17. The urea tape motifs are anti-parallel and the phenylethyl groups are in a gauche arrangement for 12 and 14. The urea tape motifs are parallel and the phenylethyl groups are in an anti arrangement for 15 and 17. | ||
The 14 (Form I), 15 and 17 crystal structures were also determined and showed similar characteristics to that of the 12 structure. In all cases the most obvious feature of the crystal packing is the R12(6) urea tape motif formed by both urea substituents on each molecule (Fig. 2). The structures of the 14 (I), 15 and 17 materials display the expected all-trans conformations of the alkylenic chains and hence the two independent urea tapes are antiparallel for 14 (I), whereas they are all co-aligned in the 15 and 17 case giving an overall polar as well as chiral structure, Fig. 2. The orientation of the bulky phenylethyl groups is strongly correlated to the urea orientation because of the need to minimise steric bulk round the urea carbonyl group which both minimises intramolecular steric interactions, and makes the oxygen atom more available as an intermolecular hydrogen bond acceptor for the two NH donors.70,71 This conformational consideration results in a gauche arrangement of the phenylethyl groups in 12 and 14 (I) and an anti arrangement for 15 and 17. The gauche substituents could result in a more awkward shape and hence less efficient crystal packing compared to the compounds with anti substituents, however, there is little difference between these four compounds' densities, for example. In 14 (I) and 17 a lower density is found (1.22 g cm−3) compared to 12 and 15 cases (1.26 g cm−3 and 1.25 g cm−3, respectively). In the 14 (I), case the packing may be considered less efficient since it is based on rotational rather than screw symmetry (C2 instead of P21).72 However, there is no obvious, consistent difference between the packing of gelators 12,4 and non-gelators 15,7 except for the mutual orientation of the two hydrogen bonded tapes.
When compound 14 was crystallised from a MeOH:H2O mixture a conformational polymorph was obtained (Fig. S5, ESI†).73,74 Crystallisation of 14 was performed by taking a room temperature concentrated solution of 14 in MeOH and adding drops of water until crystallisation began. This polymorph (form II) crystallises in space group P21. The density of form II is higher at 1.25 g cm−3 than that of form I. The R12(6) urea tape motif, formed by both urea substituents, is present once again and the two urea tapes are in an antiparallel arrangement as for 12 and form I of 14. The all-trans conformation of the alkylenic chain, as seen in form I, is also present in form II. The significant change in the molecular structure of 14 in form II compared with form I is the angle of the urea groups in relation to the all-trans oligomethylene chain. In form I, the backbone is essentially flat with C–N–C–C torsion angles close to 180° (171.6° and 177.2°; Fig. 3a). In form II, the C–N–C–C torsion angles are 95.3° and −78.3° resulting in a ‘kinked’ molecule, similar to that seen in the structure of 12 (Fig. S4b, ESI†). The phenylethyl groups remain in a gauche arrangement. The overall packing in form II is of a herringbone-like motif in contrast to the interlocked motif of form I and of 12 (Fig. 3b).
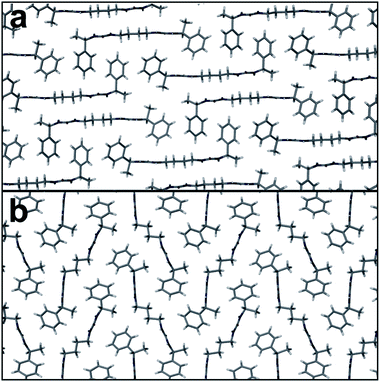 | ||
| Fig. 3 Packing arrangements of 14 in (a) form I and (b) form II. Both structures show similar antiparallel urea tape hydrogen bonding, which runs into the plane of the figure. | ||
The form II structure of 14 is denser than form I and we conclude that it is not poor crystallinity or ‘bad 3D packing’ that gives gelation, but it is the antiparallel urea hydrogen bonded chains in bis(ureas) that is linked to gelation while the parallel, polar arrangement results in crystalline materials. The antiparallel mode has been postulated in gels of a number of cyclic bis(ureas).26 In tris(urea)s (which mimic bis(urea)s in their crystal packing) the known gelator crystal structure is antiparallel, whereas the known crystal structure of the tris(urea) analogue of 1 has a parallel motif, however, this crystal form is isolated in the presence of Cl− which may act to switch the antiparallel urea chains in the gel to parallel chains in the crystal form.75,76
In the cases of 12 and 14 examination of the xerogels by powder X-ray diffraction (PXRD) (Fig. 4 and S6, ESI†) showed that the single crystal structure is also the structure of the dried gel fibres. The xerogels studies in the case of 12 were obtained from MeCN, CHCl3 and DMSO:H2O, while for 14 gels were obtained from CHCl3 and match the calculated PXRD pattern of the form II crystal structure. Correlation of single crystal structure and xerogel structure has been observed in some cases before, but is relatively unusual and suggests that the single crystal X-ray data represents a good structural model for the gel fibers themselves.8,77,78 The PXRD patterns of precipitates from synthesis of 16 show good crystallinity whereas the PXRD patterns of xerogels are very poorly crystalline, and it is not clear if the precipitates and the xerogels are the same phase (Fig. S7, ESI†). The PXRD patterns of the as-synthesised form and xerogels of 18 show mostly amorphous character (Fig. S7, ESI†).
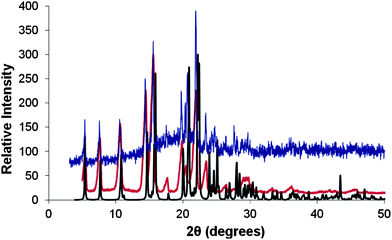 | ||
| Fig. 4 PXRD patterns of the xerogel of compound 12 formed from the drying of the CHCl3 gel (Blue line), drying of MeCN gel (Red line) and the simulated PXRD of the single crystal data of compound 12 (Black line). There is a systematic drift to lower 2θ values of the powder sample peaks compared to the simulated due to the single crystal structure being obtained at 120 K compared to the powder samples which were obtained are room temperature. Intensities have been normalised to the same scale. | ||
Gel characteristics
Gel morphologies of both gels and xerogels of 12,4,6,8 were studied by scanning electron microscopy (SEM), transmission electron microscopy (TEM) and cryo-SEM. For SEM imaging, samples of gels of 12,4,6,8 were dried under vacuum and then coated with a thin layer of platinum metal. A characteristic thread-like morphology was found for the compounds 14, 16 and 18 (Fig. 5), with helicity apparent in the fibres formed by compounds 14 attributed to the molecular chirality of the gelator.79–83 A rod-shaped morphology is evident in the SEM micrographs of the gels formed by 12 in solvents other than water, while a different fibre morphology is observed for the hydrogels (Fig. 5a and S8, ESI†). We postulate that the morphology difference is due to the way in which the gels dry under dynamic vacuum. It is likely that this rod-shaped morphology is formed by recrystallised micro-crystalline material that does not truly represent the gel structure. | ||
| Fig. 5 (a) SEM image of the xerogel of 12 gel from MeCN (solvent) showing the rod-shaped nature of the dried gel sample due to crystallisation during drying. (b) SEM image of the xerogel of 14 gel from MeCN showing the thread-like nature of the gel fibres and the helical twist induced by the chiral gelator. (c) SEM image of the xerogel of a MeCN gel of 16 showing the crystalline fibrous nature of the gel. (d) SEM image of the xerogel of a MeCN gel of 18. | ||
To address this issue the 12 in CHCl3 gels were also characterised using TEM. The preparation technique results in a fresh sample being exposed abruptly to high vacuum resulting in an altered drying process. The placement of thin layers of gel on the carbon mesh support and the imaging results revealed a more truly representative image of the gel. As shown in Fig. 6a the TEM images show a much more gel-like fibrous morphology.78,84–88 We attempted to obtain an electron diffraction pattern from the larger fibres but this proved impossible due to the breakdown of the fibres under the electron beam, although a very short-lived but good quality initial diffraction image was obtained, indicating the fibrous strands are crystalline in nature. The morphology for the 12 gels found using TEM was confirmed by cryo-SEM (Fig. 6b).
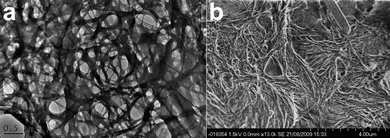 | ||
| Fig. 6 (a) TEM image of compound 12 gel formed in a CHCl3 at 0.5% by weight. (b) Cryo-SEM image of a CHCl3 gel of 12 showing the fibrous gelatinous morphology as seen in the TEM image, which is in strong contrast to the dried crystalline SEM samples seen in Fig. 5 and S8, ESI†. To the top right of the image can be seen the edge of crystalline structure similar in morphology to the dried gel. | ||
With this technique the gels are frozen in liquid nitrogen and imaged as the frozen material. Initial imaging first showed no obvious gel features but by gradually heating the sample (under dynamic vacuum) the frozen solvent is slowly sublimed from the surface. This reveals a gel fibre network, confirming the fibrous morphology seen in the TEM images. This technique of removing the solvent under high vacuum and low temperature results in less surface tension effects from evaporation, resulting in less sampling-induced perturbation of the gel morphologies.
The behaviour of fibres of the gels when exposed to mechanical stress was investigated using temperature sweep, stress sweep and frequency sweep rheometry in a variety of solvents at a number of difference concentrations.84,89–93 Frequency sweep rheometry with a small amplitude stress, revealed the solid-like nature of the gels at 20 °C with the storage modulus, G′, being typically an order of magnitude greater than the loss modulus, G′′ (Fig. S9, ESI†).94 This viscoelastic behaviour is associated with classical gels, and therefore supports the notion that the cooling of these samples from a clear solution to a solid-like material results in a true gel state.
The non-linear rheological response was investigated using stress sweep experiments, during which an oscillatory torque was imposed with a fixed frequency (1 Hz) over a range of shear stress amplitudes. The gels showed a typical G′ value, essentially constant below the critical value of oscillatory torque (“yield stress”). At this yield stress point, the sample starts to flow (Fig. S10 and S11, ESI†).84,93
The effect of the concentration on the gel strength was also investigated using rheology. Plotting G′ against concentration of 12 reveals concentration dependence similar to that seen often for the dependence of Tgel with concentration (Fig. S12, ESI†).93 There have been theoretical models put forward for the mechanical properties of gels.95 Two examples that are pertinent to LMWGs are the colloidal gel description96 and the cellular solid model.90,97,98 Attempting to fit either of the two models to the data shown in Fig. 5 gives no obvious match. However, there is a saturation point beyond which increasing concentration does not result in increased gel strength at approximately 0.45% by weight. As neither model takes this into account, the use of data points below this weight percentage threshold should provide a set of data that can be modelled.89,99 The cellular solid model is predicted to have a relationship of G′ ∞ [conc]n, where n can vary between 1 and 2.5, which is in good agreement with our observations, where as for the colloidal gel description states that n should be much greater than 2.5. The cellular solid model describes an open-cell cellular material which consists of load bearing struts interconnected via crosslinks or junction points which deform by bending. This description of the gel structure matches well with the electron microscopy determined morphologies of the gels thus suggesting the cellular solid model is a good theoretical description for the gels presented here.
The powder diffraction patterns of dried gel samples for compounds 12 and 14 reveal that the dried crystalline xerogel materials obtained from gels of CHCl3 and MeCN (EM images Fig. 5 and 6) have the same structures as the single crystal forms (14 being the same as the form II polymorph) (Fig. S6, ESI†). From this crystallographic data, and taking the assumption that the structure of the crystals is the same as the fibres seen for the gel, we can take the density of the struts (ρs) joining junctions of a gel to be 1.262 g cm−3 and 1.25 g cm−3 for 12 and 14, respectively. For a wide range of materials with a range of cellular concentrations, the formula G′ ≈ 3Es(c/ρs)2/8 holds true,100 where Es is the Young's modulus of the struts and c is the concentration of the compound making up those struts. The Young's moduli of the individual fibres for compounds 12 and 14, determined using the data collected from the rheological study of 12 and 14 in MeCN, can be calculated to be Es ∼ 1.0 GPa (±0.1) and Es ∼ 1.7 GPa (±0.3), respectively. These values are comparable to that calculated for other LMWGs and values associated with the macroscopic mechanical behaviour of semi-crystalline polymers.75
Anion tuning of gel rheological characteristics
Tetrabutyl ammonium (TBA+) salts of various anions were added to gels of 12,4,6,8 either as solids or in solution, and the resulting mixtures were visually inspected for gel dissolution. Between 0.1 and 2 equivalents of anion relative to gelator were added to preformed 1 ml samples of gels of 0.1 to 2% by weight, either a solid or as a solution of dissolved salt in 0.1 ml of the same gel solvent. Adding the salts to hot dissolved gels gave identical results. In principle anion binding by the gelators should result in competition between gelator–gelator hydrogen bonding of the urea tape type, and urea-anion interactions. In the case of gels in MeCN, CH3Cl, ethyl acetate, THF, acetone and Tol:MeCN the gel broke down to give a solution when at least one equivalent of F−, Cl−, Br−, NO3− or MeCO2− was added, either as a solid or concentrated solution. Weakly coordinating anions BF4− and PF6− had no effect indicating that neither the TBA+ cation nor anions of low hydrogen bond basicity have any noticeable effect on the gel. Hydrogels of all of the gelators are not affected by the addition of the anions, either in the form of TBA+ salts or Na+ salts. The solution anion binding affinity of representative anions by gelators were determined using NMR spectroscopic titration in solution at 50 °C, above Tgel, and are shown in Table 2. The gelator-anion stoichiometry was confirmed as 1:1 by Job plot analysis for all compounds (Fig. S13–S17, ESI†). In each case of anion binding by 12 it proved necessary to include a competing dimerisation process within the model with log β20 = 3.78(1). Gelator dimerisation in solution is an obvious precursor step to gel formation. For the longer homologues (which are significantly more soluble) significant dimerization was not observed at NMR concentrations. In general, MeCO2− is bound strongest by all the compounds, followed by Cl− then NO3−. For Br− and BF4− the binding proved to be too weak to determine binding constants.
| Anion | 12 | 14 | 16 | 18 | 2 |
|---|---|---|---|---|---|
| MeCO2− | 4.26(1) | 3.10(3) | 3.35(2) | 3.18(2) | 4.30(1) |
| Cl− | 3.52(2) | 2.57(1) | 2.61(1) | 2.57(1) | — |
| NO3− | 2.93(6) | — | — | — | — |
Adding anions to gels based on 12, 14, 16 or 18 should significantly reduce the rheological strength of the gels according to the degree of interaction between the gelator and anion. Rheology was used as a tool to investigate the behaviour of the gels prior to complete dissolution by adding small amounts of anion to the gels, both in the case of preformed gels or hot solutions of the dissolved gels. In general, it was found that the cumulative addition of the anions led to a decrease in the gel elastic modulus and yield stress for all gelators 12, 14, 16 and 18 depending on the identity of the anion and the concentration added (Fig. S18–S20, ESI†). It was found that the most consistent and reliable results were obtained by adding anions to the hot solution prior to gel formation. Addition of anions to the upper surface of preformed gels resulted in more uneven disruption of the gel, with the upper surface of the gels breaking down while the lower surface was unaffected. Therefore the following results are all based on the addition of anions to the gels prior to gelation. Stress sweep measurements before and after addition of anions as TBA+ salts to samples of gels of 12 in MeCN showed that while BF4− had no effect on the observed storage and loss moduli (G′ and G′′), the other anions dramatically compromised the gel strength and greatly decreased the values of the moduli. Fig. 7 (Fig. S18–S20, ESI†) shows that addition of small amounts of anions (0.1 equivalents with respect to the gelator concentration) which results in the reduction in the G′ by up to two orders of magnitude depending on the identity of the anion, indeed addition of 0.1 equivalents of MeCO2− or F− completely disrupts gel formation. In addition to the decrease in the G′, the anions cause a decrease in the “yield stress” of the gel (which is an indicator of the breaking strength of the fibres and their degree of connectivity) and the Tgel (which is an indication of the amount of gelator in the solid gel phase in relation to the solution phase),93 also indicating a weakening of the gel structure (Fig. S21–S22, ESI†).
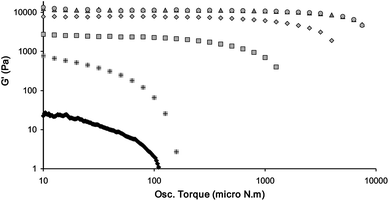 | ||
| Fig. 7 Influence of anions (0.1 equivalents of anion added as their TBA+ salts) on the storage modulus (G′) at a frequency of 1 Hz and a temperature of 20 °C, as a function of osc. torque of the 0.3% by weight gel of compound 12 in MeCN. The anions added are BF4− (△); Br− (◇); NO3− (□); Cl− (+) and MeCO2− (Black ◇). The pure gel is represented as brown ○. F− is not shown as it forms a liquid. Both axes are log scale. | ||
In addition to weakening the gels, time sweep rheometry experiments reveal that the gels took longer to form if anions were added. The trend of the anion binding affinities in the pre-gel solution mirrors the inhibitory effect anions have on the gel formation, the gel rheology and the gel structure after forming. Thus MeCO2−, the strongest bound anion has by far the most significant influence on G′, G′′, Tgel and yield stress. This leads to the effect that the gel structure of gels of 12 is completely broken and dissolution of 12 occurs when as little as 0.05 equivalents of MeCO2− are added (similarly low quantities are required for gels of 14,6,8). Similarly, Cl− has a significantly greater effect than NO3− on the gel rheological properties. Therefore the order of anion binding in solution and the order of reduction in G′, G′′, Tgel and “yield stress” by addition to the gels of 1n (n = even) are the same and is MeCO2− ∼ F− > Cl− > NO3− > Br− > BF4−.
The gelator 14 also shows the property of being tuneable through the addition of anions (Fig. S19, ESI†). MeCO2− does not have as great as effect on the gel strength of 14 as on gels of 12, with G′ nearly halving when 0.1 equivalents is added to the 14 gel, where as in the case of the 12 gels, full disruption of gel is noted. This behaviour maps the trend seen in the binding strength of the two gelators for MeCO2− in solution, the stronger binding 12 being more easily disrupted than the weaker binding 14. In the case of gels of 16 in CHCl3, when TBA+ salts of the anions Cl−, Br−, BF4− and MeCO2− were added at sub-stoichiometric amounts, a similar trend to that seen in gels of 12 in the disruption of gel formation was noted (Fig. S18, ESI†). This indicates that the solvent plays little role in the disruption if the gelator–anion complex is soluble in that solvent and the solvent does not compete effectively in the complex formation. The study of gelator 18 revealed that the gels are not as affected by the addition of anions as in the cases of gelators 12, 14 and 16 (Fig. S20, ESI†). Both “yield stress” and G′ decrease by a quarter compared to the values for the pure 18 gels. As the gelators 12, 14, 16 and 18 all show a decrease in rheological strength as shown in the decrease in the parameters loss moduli, yield stress and Tgel, for a fixed concentration of gelator, such as decrease suggest a lower interconnectivity of the individual threads, supporting our hypothesis that the anions disrupt the gelation process.98
Gelation and anion tuning of 2
Compound 2 was designed as a fluorescent analogue to compound 12. The gelation behaviour of 2 is almost identical to that of 12, except that 2 gels at slightly lower concentrations than 12. In contrast to 12, compound 2 showed no significant drying effects and the SEM images reveal that 2 forms fibrous strands (Fig. 8) but the fibres are crystalline upon drying (Fig. S23, ESI†). This difference can be seen by the naked eye as gels of 12 are generally opaque and appear fibrous and crystalline under the optical microscope while gels of 2 are more transparent. Compound 2 shows helicity in the fibres, which was not evident in gels of compound 12 but present in gels of 14 (Fig. 5). The rheology of 2 is also similar to that of 12 (Fig. S24–S26, ESI†). | ||
| Fig. 8 Helical fibres found within gels of compound 2. (a) 0.1% by weight CHCl3 gel and (b) 0.06% by weight MeCN gel. The helicity is in an opposite sense in the two samples suggesting that the direction of twist is not determined by the absolute configuration of 2. | ||
Compound 2 readily formed gels in DMSO:H2O mixtures that exhibit thixotropic properties. Thixotropic gels are gels that begin to flow upon application of a shearing force, but upon removal of the shearing force the gel properties return. This behaviour contrasts to conventional gels which continue to flow once sheared. This “gel repair” property has been seen to occur in some gels but is still a relatively uncommon phenomenon.25,29,30,91,92,101–104 The process was investigated using stress sweep rheometry. Following gelation within the rheometer concentric cylinder couette, a stress sweep was performed resulting in the breaking of the gel. The rheological properties of the broken gel were then monitored over time, with constant low shear and frequency values (Fig. 9). The gel underwent shearing at time point 58 min, as shown in Fig. 9, upon which shearing force was removed and the sample monitored for gel formation. The gel could be seen to redevelop its solid-like characteristics with the gradual increase in G′ while the G′′ value remained practically the same eventually leading to a gel with a G′ measurement of an approximate magnitude of order greater value than G′′ but which was weaker (55% of the original G′) than the original gel formed by cooling.
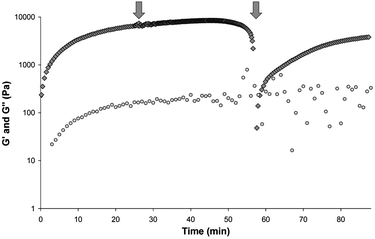 | ||
| Fig. 9 A gel of 2 at 0.6% by weight in a DMSO:H2O 5:5 solvent mixture is shown to reform after being sheared to breaking point. The gel is allowed to set for 25 min upon which a stress sweep is performed from a range of 10 micro Nm to 15000 micro Nm (Time 25.2 min to 57.4 min). The gel starts to flow at around 10000 micro N.m. Upon removal of this shear force the measurements are continued at a constant low frequency and stress value (i.e a time sweep is performed). The gel reforms over time, the G′ value reaching ∼ 55% of its original value. G′ shown as grey filled ◇ and G′′ as grey filled ○. G′ and G′′ axis values shown in log scale. | ||
A SEM study of the gel fibre morphology gave good insight into how this particular gel system might be undergoing the shearing breakdown of the gel and thixotropic “self-repair”. SEM imaging of a pristine sample revealed a well connected fibrous network with fibres that have a uniform thickness along their length (Fig. 8). Inspection of images of samples that had been sheared and have “self-repaired”, Fig. 10, revealed that the fibres have ends that are braided but still connect the fibres together into a network. It appears that although the fibres are broken by the shearing process, the fibres are able to reconnect as a result of the dynamic nature of the supramolecular gel reconnecting the spherulites. It is possible that the presence of π-stacking interactions in 2 aid in this process.
 | ||
| Fig. 10 SEM images of the dried thixotropic gel formed by 2 in a DMSO:H2O mixture after shearing and “self-repair”. (a) the ends of fibres that join the fibres together are thinned. (b) Close up image of the areas that join the gel fibres of the thixotropic gel together. | ||
It was anticipated that the gelation of 2 would result in a change in fluorescence of the naphthyl group105 allowing insight into the mode of aggregation of the gel. Fig. 11 shows the fluorescent emission spectra obtained when a hot solution of 2 is cooled below Tgel. The structured emission band (λ = 327, 335 and 353 nm) increases in intensity with a slight red shift (in the 335 nm peak) and loss of the fine structure.
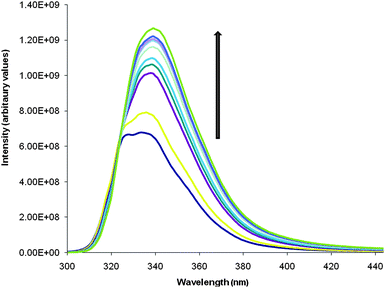 | ||
| Fig. 11 Fluorescence spectra of a gel of 2 at 0.06% by weight in DMSO:H2O. The dark blue spectrum (lowest intensity spectrum) is of the hot solution. The general increase of the intensity is seen as the temperature decreases and as a result the gel forms. As well as this there is a small red shift in the band. | ||
The aggregation of 2 within the gel fibres results in a restriction in the motion of the molecules and therefore reduces the non-radiative dynamic decay pathways of the naphthyl excited states resulting in the increase in the intensity (also known as the phenomenon of aggregation-induced enhanced emission; AIEE).85,106–114 The slight red shift can be attributed to the stacking of the naphthyl groups together, through π–π interactions, within the solid state of the gel (a hydrophobic environment).115,116 This red shift in the band is characteristic of J-aggregation of the naphthyl groups.106,108,117–119 In addition to this experiment, where a hot solution is cooled and gelation occurs fairly quickly, a second experiment was performed on a gel solution that was at the CGC level (0.04% by weight in DMSO:H2O (7:3)). Upon cooling there is much less change in the appearance of the emission profile due to a lack of gel formation at this low concentration. Upon shaking the solution to initiate nucleation, and, therefore gelation, the spectra show gelation occurring with occurrence of the red shift and intensity increase (Fig. S27, ESI†). This experiment highlights the importance of kinetic nucleation factors for the initiation of the growth of the gel fibres and the consequent gelation of the solution.
The anion tuning of the gels of 2 was also investigated using rheological measurements. As with compounds 1 these gels show a gel to sol transformation upon addition of excess anion in the form of a TBA+ salt, even though 2 is less soluble in most solvents when compared to compounds 1. A measurement of the binding constants of 2 for MeCO2− at 50 °C in MeCN using NMR spectroscopic titration suggested a model in which dimerization of 2 with a self-association constant of log β20 = 2.92(10), is in competition with the formation of both 1:1 and 1:2 guest–host complexes; log β11 = 4.30(1); and log β21 = 7.63(3). The value of β11 is comparable to 12.54 However, the dimerisation constant, β20, is smaller. The formation of a two host to one guest complex also contrasts with the behaviour of 12. Acetate binding results in substantial weakening of the gel which is apparent from the rheology of gels of 2 in the presence of increasing concentrations of TBA+ MeCO2− (Fig. 12, S28 and S29, ESI†). Compound 12 forms gels in MeCN that can be fully “dissolved” with addition of as little as 0.1 equivalents of TBA+ MeCO2−.
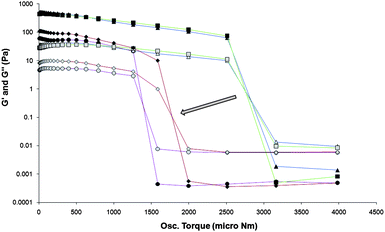 | ||
| Fig. 12 Rheological study of the tuning of gels of 2 using the addition of anion in the form of TBA+ MeCO2−. Stress sweep measurements are performed on gels of 2 at 0.1% by weight in MeCN with varied amounts of TBA+ MeCO2− added. G′ represented as dark filled symbols and G′′ as light filled symbols. △ symbols (blue guide line) are for the pure gel, □ symbols (green guide line) are for 0.05 equivalents of TBA+ MeCO2− added, ◇ (red guide line) symbols are for 0.2 equivalents of TBA+ MeCO2− added and ○ symbols (purple guide line) are for 0.5 equivalents of TBA+ MeCO2− added. The “yield stress”, as represented by the swapping of the G′ being larger than G′′, decreases with increasing equivalents of anion added (shown with an arrow). “Yield stress” values can be estimated to be where the eye-guiding colour lines intersect. G′ and G′′ values are shown on a log scale. | ||
In comparison, 2 forms a gel in MeCN that requires a much larger concentration of anion to dissolve; one equivalent of TBA+ MeCO2− is required to fully dissolve the gel. The rheological characterisation of the tuning of 2 by addition of TBA+ MeCO2− reveals how the strengths of the gels, as represented by G′ values measured over the setting time of the gel, are decreased with increasing amounts of TBA+ MeCO2− added (Fig. S28, ESI†). The frequency sweep characterisations of the gels show how they are true gels. This is shown by the G′ value being an order of magnitude larger than G′′ and by the fact that over the frequency range measured the G′ and G′′ values are constant (Fig. S29, ESI†). The weakening of the gels by the addition of the TBA+ MeCO2− is also represented by the decrease in the “yield stress” of the gels as can be seen in Fig. 12. This indicates that the binding strength between the host gel forming compound and the anion is not the only determining point in allowing the breakdown of these gels. The solubility of both the gel compound and the gel compound and anion complex, and the way in which the equilibrium between the solvated components and solid gel matrix is affected by these solubilities, plays a vital role in the anion tuning of the gel. The anion-induced weakening of gels of 2 was also followed using fluorescence spectroscopy. TBA+ MeCO2− was layered on to the top of a 0.1% by weight gel of 2 in MeCN at room temperature as a concentrated solution in a small volume of MeCN and the vial placed in the spectrometer. The fluorescence emission was then followed as the gel dissolved (Fig. S30, ESI†). A small blue shift and decrease in the intensity of the band centred at 338 nm is observed, and represents the opposite of the changes in the emission profile during gel formation.
Mixed gels
Mixing of related gelators allows access to a wide range of potential composite gels.120–125 In the present case, mixtures of the gelators resulted in materials with both different rheological characteristics and morphologies. The mixing of gelators 12 and 2 led to a gel with a similar appearance to gels formed by 12, i.e. an opaque, stiff (rheologically similar) gel. However, the SEM images of dried, 0.5% by weight (1:1 molar ratio of the two gelators) CHCl3 gels, revealed notable changes in the morphology. The morphology of the xerogels of 12 in gels of CHCl3 are more crystalline, while xerogels of 2 in CHCl3 are fibrous and helical (Fig. 5 and 8, respectively). The mixed gel proved different in two ways (Fig. 13a–d). Firstly, instead of the crystallisation observed for 12, the gel fibres are more stable and sections of the SEM images of the xerogels contain what appears to be a physical mixture of the fibres of both 12 and 2, with the larger fibres (similar to those seen in the cryoSEM images) likely to be that of 12 and the finer fibres that of 2 (Fig. 13c–d). Interspersed amongst these physical mixtures are areas of discrete helical structures which represent a new morphology for these gelators. These regions likely represent a mixed gel (Fig. 13a–b). Mixing the gelators was also found to be an effective way of tuning of rheological characteristics of the gels. The comparison of the rheological characteristics of each of the gelator compounds and their mixtures gives insight into the molecular bases of the tuning. The order of rheological strength of the gelators is 12 ≪ 16 > 18 > 14 (Fig. 14). The gels of mixed gelators in a 1:1 mixture of 14 with 16 and 14 with 18 (adding up to 1 wt% of total gelator in CHCl3) shows how the mixtures generate gels with intermediate strength to the pure gelators. This means if a gel is needed with a specific weight percent and gel rheological strength, it can be made by mixing the gelators in the correct stoichiometry. In addition to the rheological changes, SEM images indicated that there is a change in morphology as well from mixing of the gelators. Although the fibrous morphology is seen again there is a stark contrast in the thickness and size of the fibres (Fig. 13e–h)
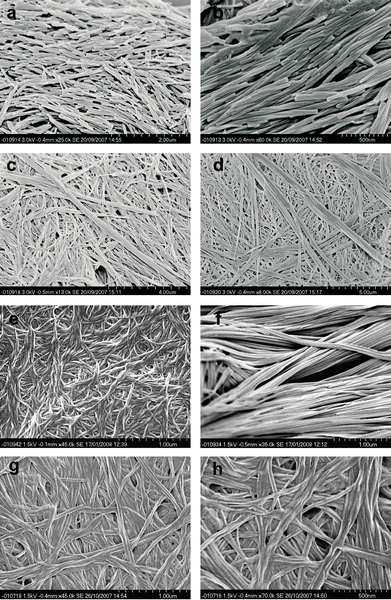 | ||
| Fig. 13 SEM images of mixed 12 and 2 gels. (a) and (b) Helical structures found in a xerogel from a 1:1 mixed gel of 12 and 2 in MeCN at 0.5% by weight. (c) and (d) Possible physical mixtures of gel fibres found from the same mixed gel. SEM images of (e) 14 gel morphology, (f) 16 gel morphology, (g) and (h) gel of 1:1 mixture of 14 and 16. All gels of 14 and 16 are at 1% by weight in total and are gels of CHCl3. | ||
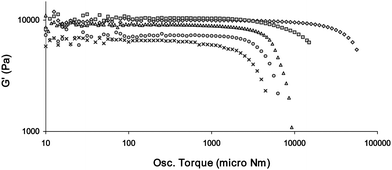 | ||
| Fig. 14 Stress sweep rheology experiments showing the tuning of the rheological strength of gels made of mixed gelators (all are 1% by weight in total and are gels of CHCl3). The gelators are, in order of strongest to weakest, 16 (◇); 14 and 16 (1:1) (□); 18 (△); 14 and 18 (1:1) (○); and 14 (X). G′ and Osc. Torque values are shown on a log scales. | ||
Conclusions
We have shown how the combination of urea groups and chiral functionality within a low molecular weight compound can be used to form simple but versatile gelators. This versatility includes a range of solvents that can be gelled, an array of morphologies, and varied rheological characteristics (including the fact that most of the gelators can be considered as supergelators)126 and some degree in the variation in the functional groups. There is a distinct alternation effect with gels only forming when there is an even number of methylene groups in the spacer and hence an anti-parallel arrangement of urea carbonyl groups.58–63 The rheological characteristics of the gels can be tuned in a chemically specific way by taking advantage of the solution anion binding ability of bis(urea)s. Anions that are weakly bound have much less effect than those which interact more strongly. This work points to a novel synergy between solution based thermodynamic affinity measurements and gel properties. To date, the majority of studies in anion tuning of gels have shown how anions can be used to impart gel to sol or sol to gel transitions, gel to crystal transitions or tune physical properties like colour or fluorescence.46 The gel materials reported here show the anion based gel to sol transition that is expressed in tuneable rheological characteristics that arise from anion-induced dissolution of the gelators and disruption of fibre formation during gelation. We hope that this understanding of the anion-based tuning of LMWGs will allow researchers to exploit the anion tuning of gel characteristics in a range of notable applications.4,23Acknowledgements
We thank Dr Bhudika G. Mendis for assistance with the TEM instrumentation and Dr Adam N. Swinburne for his help in obtaining the fluorescence experiments and Mrs A. C. Richardson for help in collecting the SEM images. We are grateful to the ESPRC funding. GOL also thanks the Commonwealth Scholarship Commission and the Herchel Smith Fellowship Fund (Cambridge) for funding.Notes and references
- D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12, 1237–1247 CrossRef CAS.
- M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615–3631 CrossRef CAS.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3159 CrossRef.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS.
- F. Zhao, M. L. Ma and B. Xu, Chem. Soc. Rev., 2009, 38, 883–891 RSC.
- P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699–2715 RSC.
- M. Suzuki and K. Hanabusa, Chem. Soc. Rev., 2009, 38, 967–975 RSC.
- G. Cravotto and P. Cintas, Chem. Soc. Rev., 2009, 38, 2684–2697 RSC.
- E. Krieg, H. Weissman, E. Shirman, E. Shimoni and B. Rybtchinski, Nat. Nanotechnol., 2011, 6, 141–146 CrossRef CAS.
- P. D. Thornton, R. J. Mart, S. J. Webb and R. V. Ulijn, Soft Matter, 2008, 4, 821–827 RSC.
- S. Sutton, N. L. Campbell, A. I. Cooper, M. Kirkland, W. J. Frith and D. J. Adams, Langmuir, 2009, 25, 10285–10291 CrossRef CAS.
- K. J. C. van Bommel, M. C. A. Stuart, B. L. Feringa and J. van Esch, Org. Biomol. Chem., 2005, 3, 2917–2920 RSC.
- B. T. Li, L. M. Tang, L. Qiang and K. Chen, Soft Matter, 2011, 7, 963–969 RSC.
- Q. Wei and S. L. James, Chem. Commun., 2005, 1555–1556 RSC.
- D. J. Adams, Macromol. Biosci., 11, pp. 160–173 Search PubMed.
- J. W. Sadownik and R. V. Ulijn, Chem. Commun., 2010, 46, 3481–3483 RSC.
- J. Boekhoven, A. M. Brizard, K. N. K. Kowlgi, G. J. M. Koper, R. Eelkema and J. H. van Esch, Angew. Chem., Int. Ed., 2010, 49, 4825–4828 CAS.
- S. Kiyonaka, K. Sada, I. Yoshimura, S. Shinkai, N. Kato and I. Hamachi, Nat. Mater., 2004, 3, 58–64 CrossRef CAS.
- E. F. Banwell, E. S. Abelardo, D. J. Adams, M. A. Birchall, A. Corrigan, A. M. Donald, M. Kirkland, L. C. Serpell, M. F. Butler and D. N. Woolfson, Nat. Mater., 2009, 8, 596–600 CrossRef CAS.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. E. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- J. A. Foster, M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke, J. A. K. Howard and J. W. Steed, Nature Chem., 2010, 2, 1037–1043 CrossRef CAS.
- A. Pal, S. Shrivastava and J. Dey, Chem. Commun., 2009, 6997–6999 RSC.
- V. Percec, M. Peterca, M. E. Yurchenko, J. G. Rudick and P. A. Heiney, Chem.–Eur. J., 2008, 14, 909–918 CrossRef CAS.
- J. van Esch, F. Schoonbeek, M. de Loos, H. Kooijman, A. L. Spek, R. M. Kellogg and B. L. Feringa, Chem.–Eur. J., 1999, 5, 937–950 CrossRef CAS.
- P. Mukhopadhyay, N. Fujita, A. Takada, T. Kishida, M. Shirakawa and S. Shinkai, Angew. Chem., Int. Ed., 2010, 49, 6338–6342 CrossRef CAS.
- T. D. Hamilton, D. K. Bucar, J. Baltrusaitis, D. R. Flanagan, Y. J. Li, S. Ghorai, A. V. Tivanski and L. R. MacGillivray, J. Am. Chem. Soc., 2011, 133, 3365–3371 CrossRef CAS.
- W. G. Weng, A. M. Jamieson and S. J. Rowan, Tetrahedron, 2007, 63, 7419–7431 CrossRef CAS.
- M. Lescanne, P. Grondin, A. d'Aleo, F. Fages, J. L. Pozzo, O. M. Monval, P. Reinheimer and A. Colin, Langmuir, 2004, 20, 3032–3041 CrossRef CAS.
- S. Wang, W. Shen, Y. L. Feng and H. Tian, Chem. Commun., 2006, 1497–1499 RSC.
- Z. J. Qiu, H. T. Yu, J. B. Li, Y. Wang and Y. Zhang, Chem. Commun., 2009, 3342–3344 RSC.
- V. Haridas, Y. K. Sharma, R. Creasey, S. Sahu, C. T. Gibson and N. H. Voelcker, New J. Chem., 2011, 35, 303–309 RSC.
- A. Shome, S. Debnath and P. K. Das, Langmuir, 2008, 24, 4280–4288 CrossRef CAS.
- J. J. Panda, A. Mishra, A. Basu and V. S. Chauhan, Biomacromolecules, 2008, 9, 2244–2250 CrossRef CAS.
- H. Komatsu, S. Matsumoto, S. Tamaru, K. Kaneko, M. Ikeda and I. Hamachi, J. Am. Chem. Soc., 2009, 131, 5580–5585 CrossRef CAS.
- Z. M. Yang, H. W. Gu, Y. Zhang, L. Wang and B. Xu, Chem. Commun., 2004, 208–209 RSC.
- F. Rodriguez-Llansola, J. F. Miravet and B. Escuder, Chem. Commun., 2011, 47, 4706–4708 RSC.
- J. A. Sáez, B. Escuder and J. F. Miravet, Chem. Commun., 2010, 46, 7996–7998 RSC.
- J. A. Foster and J. W. Steed, Angew. Chem., Int. Ed., 2010, 49, 6178–6724 CrossRef CAS.
- F. Fages, Angew. Chem., Int. Ed., 2006, 45, 1680–1682 CrossRef CAS.
- H. Maeda, Chem.–Eur. J., 2008, 14, 11274–11282 CrossRef.
- M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS.
- P. Byrne, G. O. Lloyd, L. Applegarth, K. M. Anderson, N. Clarke and J. W. Steed, New J. Chem., 2010, 34, 2261–2274 RSC.
- B. Escuder, F. Rodriguez-Llansola and J. F. Miravet, New J. Chem., 2010, 34, 1044–1054 RSC.
- G. O. Lloyd and J. W. Steed, Nature Chem., 2009, 1, 437–442 CrossRef CAS.
- T. Becker, C. Y. Goh, F. Jones, M. J. McIldowie, M. Mocerino and M. I. Ogden, Chem. Commun., 2008, 3900–3902 RSC.
- M.-O. M. Piepenbrock, N. Clarke and J. W. Steed, Langmuir, 2009, 25, 8451–8456 CrossRef CAS.
- A. Kishimura, T. Yamashita and T. Aida, J. Am. Chem. Soc., 2005, 127, 179–183 CrossRef CAS.
- L. Sambri, F. Cucinotta, G. De Paoli, S. Stagni and L. De Cola, New J. Chem., 2010, 34, 2093–2096 RSC.
- G. O. Lloyd and J. W. Steed, Soft Matter, 2011, 7, 75–85 RSC.
- J. van Esch, S. DeFeyter, R. M. Kellogg, F. DeSchryver and B. L. Feringa, Chem.–Eur. J., 1997, 3, 1238–1243 CrossRef CAS.
- J. van Esch, R. M. Kellogg and B. L. Feringa, Tetrahedron Lett., 1997, 38, 281–284 CrossRef CAS.
- M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Commun., 2008, 2644–2646 RSC.
- M. O. M. Piepenbrock, N. Clarke, J. A. Foster and J. W. Steed, Chem. Commun., 2011, 47, 2095–2097 RSC.
- C. Frassineti, S. Ghelli, P. Gans, A. Sabatini, M. S. Moruzzi and A. Vacca, Anal. Biochem., 1995, 231, 374–382 CrossRef CAS.
- P. Gans, University of Leeds, Leeds, 2006.
- N. Fujita, Y. Sakamoto, M. Shirakawa, M. Ojima, A. Fujii, M. Ozaki and S. Shinkai, J. Am. Chem. Soc., 2007, 129, 4134–4135 CrossRef CAS.
- K. Aoki, M. Kudo and N. Tamaoki, Org. Lett., 2004, 6, 4009–4012 CrossRef CAS.
- M. Suzuki, M. Nanbu, M. Yumoto, H. Shirai and K. Hanabusa, New J. Chem., 2005, 29, 1439–1444 RSC.
- T. Ishi-i, R. Iguchi, E. Snip, M. Ikeda and S. Shinkai, Langmuir, 2001, 17, 5825–5833 CrossRef CAS.
- T. Sumiyoshi, K. Nishimura, M. Nakano, T. Handa, Y. Miwa and K. Tomioka, J. Am. Chem. Soc., 2003, 125, 12137–12142 CrossRef CAS.
- J. H. Jung and S. Shinkai, J. Chem. Soc., Perkin Trans. 2, 2000, 2393–2398 RSC.
- S. Bhattacharya and Y. Krishnan-Ghosh, Chem. Commun., 2001, 185–186 RSC.
- M. Suzuki, T. Sato, H. Shirai and K. Hanabusa, New J. Chem., 2006, 30, 1184–1191 RSC.
- J. X. Peng, K. Q. Liu, X. F. Liu, H. Y. Xia, J. Liu and Y. Fang, New J. Chem., 2008, 32, 2218–2224 RSC.
- S. Debnath, A. Shome, S. Dutta and P. K. Das, Chem.–Eur. J., 2008, 14, 6870–6881 CrossRef CAS.
- D. Bardelang, F. Camerel, J. C. Margeson, D. M. Leek, M. Schmutz, M. B. Zaman, K. Yu, D. V. Soldatov, R. Ziessel, C. I. Ratcliffe and J. A. Ripmeester, J. Am. Chem. Soc., 2008, 130, 3313–3315 CrossRef CAS.
- P. Byrne, D. R. Turner, G. O. Lloyd, N. Clarke and J. W. Steed, Cryst. Growth Des., 2008, 8, 3335–3344 CrossRef CAS.
- L. S. Reddy, S. Basavoju, V. R. Vangala and A. Nangia, Cryst. Growth Des., 2006, 6, 161–173 CrossRef CAS.
- R. W. Hoffmann, Chem. Rev., 1989, 89, 1841–1860 CrossRef CAS.
- C. P. Brock and J. D. Dunitz, Chem. Mater., 1994, 6, 1118–1127 CrossRef CAS.
- A. Nangia, Acc. Chem. Res., 2008, 41, 595–604 CrossRef CAS.
- J. Bernstein, Chem. Commun., 2005, 5007–5012 RSC.
- C. E. Stanley, N. Clarke, K. M. Anderson, J. P. Lenthall and J. W. Steed, Chem. Commun., 2006, 3199–3201 RSC.
- J. H. van Esch and B. L. Feringa, Angew. Chem., Int. Ed., 2000, 39, 2263–2266 CrossRef.
- D. R. Trivedi, A. Ballabh, P. Dastidar and B. Ganguly, Chem.–Eur. J., 2004, 10, 5311–5322 CrossRef CAS.
- D. J. Adams, K. Morris, L. Chen, L. C. Serpell, J. Bacsa and G. M. Day, Soft Matter, 2010, 6, 4144–4156 RSC.
- A. Friggeri, C. van der pol, K. J. C. van Bommel, A. Heeres, M. C. A. Stuart, B. L. Feringa and J. van Esch, Chem.–Eur. J., 2005, 11, 5353–5361 CrossRef CAS.
- X. J. Wu, S. J. Ji, Y. Li, B. Z. Li, X. L. Zhu, K. Hanabusa and Y. G. Yang, J. Am. Chem. Soc., 2009, 131, 5986–5993 CrossRef CAS.
- S. Cicchi, G. Ghini, L. Lascialfari, A. Brandi, F. Betti, D. Berti, P. Baglioni, L. Di Bari, G. Pescitelli, M. Mannini and A. Caneschi, Soft Matter, 2010, 6, 1655–1661 RSC.
- C. C. Lee, C. Grenier, E. W. Meijer and A. Schenning, Chem. Soc. Rev., 2009, 38, 671–683 RSC.
- D. K. Smith, Chem. Soc. Rev., 2009, 38, 684–694 RSC.
- F. M. Menger and K. L. Caran, J. Am. Chem. Soc., 2000, 122, 11679–11691 CrossRef.
- A. Saha, S. Manna and A. K. Nandi, Langmuir, 2007, 23, 13126–13135 CrossRef CAS.
- B. Yuan, J. L. Li, X. Y. Liu, Y. Q. Ma and H. Y. Xu, Chem. Commun., 2011, 47, 2793–2795 RSC.
- D. Wang and J. C. Hao, Langmuir, 2011, 27, 1713–1717 CrossRef CAS.
- X. Y. Liu, in Low Molecular Mass Gelators: Design, Self-Assembly, Function, Springer-Verlag Berlin, Berlin, 2005, pp. 1–37 Search PubMed.
- J. L. Li, X. Y. Liu, R. Y. Wang and J. Y. Xiong, J. Phys. Chem. B, 2005, 109, 24231–24235 CrossRef CAS.
- P. Terech, D. Pasquier, V. Bordas and C. Rossat, Langmuir, 2000, 16, 4485–4494 CrossRef CAS.
- J. Brinksma, B. L. Feringa, R. M. Kellogg, R. Vreeker and J. van Esch, Langmuir, 2000, 16, 9249–9255 CrossRef CAS.
- P. Terech and S. Friol, Tetrahedron, 2007, 63, 7366–7374 CrossRef CAS.
- P. Terech, C. Rossat and F. Volino, J. Colloid Interface Sci., 2000, 227, 363–370 CrossRef CAS.
- R. G. Larson, The Structure and Rheology of Complex Fluids, Oxford University Press, Oxford, 1999 Search PubMed.
- N. M. Sangeetha, S. Bhat, A. R. Choudhury, U. Maitra and P. Terech, J. Phys. Chem. B, 2004, 108, 16056–16063 CrossRef CAS.
- W. H. Shih, W. Y. Shih, S. I. Kim, J. Liu and I. A. Aksay, Phys. Rev. A: At., Mol., Opt. Phys., 1990, 42, 4772–4779 CrossRef CAS.
- L. J. Gibson and M. F. Ashby, Proc. R. Soc. London, Ser. A, 1982, 382, 43–59 CrossRef CAS.
- G. A. Buxton and N. Clarke, Phys. Rev. Lett., 2007, 98, 23–26.
- A. R. Hirst, I. A. Coates, T. R. Boucheteau, J. F. Miravet, B. Escuder, V. Castelletto, I. W. Hamley and D. K. Smith, J. Am. Chem. Soc., 2008, 130, 9113–9121 CrossRef CAS.
- L. J. Gibson and M. F. Ashby, Cellular Solids: Structures and Properties, Cambridge University Press, Cambridge, 1997 Search PubMed.
- M. Shirakawa, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2005, 127, 4164–4165 CrossRef CAS.
- X. Haung, S. R. Raghavan, P. Terech and R. G. Weiss, J. Am. Chem. Soc., 2006, 128, 15341–15352 CrossRef CAS.
- J. L. Li and Z. Y. Liu, Adv. Funct. Mater., 2010, 20, 3196–3216 CrossRef CAS.
- C. Yan, A. Altenbas, T. Yucel, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, Soft Matter, 2010, 6, 5143–5156 RSC.
- Z. M. Yang, G. L. Liang and B. Xu, Chem. Commun., 2006, 738–740 RSC.
- J. Seo, J. W. Chung, E. H. Jo and S. Y. Park, Chem. Commun., 2008, 2794–2796 RSC.
- J. W. Chen, C. C. W. Law, J. W. Y. Lam, Y. P. Dong, S. M. F. Lo, I. D. Williams, D. B. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535–1546 CrossRef CAS.
- J. W. Chung, B. K. An and S. Y. Park, Chem. Mater., 2008, 20, 6750–6755 CrossRef CAS.
- J. P. Desvergne, A. G. L. Olive, N. M. Sangeetha, J. Reichwagen, H. Hopf and A. Del Guerzo, Pure Appl. Chem., 2006, 78, 2333–2339 CrossRef CAS.
- L. L. Zhu, X. Ma, F. Y. Ji, Q. C. Wang and H. Tian, Chem.–Eur. J., 2007, 13, 9216–9222 CrossRef CAS.
- B. K. An, S. K. Kwon, S. D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410–14415 CrossRef CAS.
- A. Y. Y. Tam, K. M. C. Wong and V. W. W. Yam, J. Am. Chem. Soc., 2009, 131, 6253–6260 CrossRef CAS.
- W. L. Leong, S. K. Batabyal, S. Kasapis and J. J. Vittal, Chem.–Eur. J., 2008, 14, 8822–8829 CrossRef CAS.
- B. K. An, D. S. Lee, J. S. Lee, Y. S. Park, H. S. Song and S. Y. Park, J. Am. Chem. Soc., 2004, 126, 10232–10233 CrossRef CAS.
- M. Asai, K. Sugiyasu, N. Fujita and S. Shinkai, Chem. Lett., 2004, 33, 120–121 CrossRef CAS.
- T. H. Kim, J. Seo, S. J. Lee, S. S. Lee, J. Kim and J. H. Jung, Chem. Mater., 2007, 19, 5815–5817 CrossRef CAS.
- A. Saha, S. Manna and A. K. Nandi, Chem. Commun., 2008, 3732–3734 RSC.
- Q. N. Pham, N. Brosse, C. Frochot, D. Dumas, A. Hocquet and B. Jamart-Gregoire, New J. Chem., 2008, 32, 1131–1139 RSC.
- F. Wurthner, C. Bauer, V. Stepanenko and S. Yagai, Adv. Mater., 2008, 20, 1695–1698 CrossRef.
- J. R. Moffat and D. K. Smith, Chem. Commun., 2009, 316–318 RSC.
- Z. Dzolic, K. Wolsperger and M. Zinic, New J. Chem., 2006, 30, 1411–1419 RSC.
- D. G. Velázquez and R. Luque, Chem.–Eur. J., 2011, 17, 3847–3849 CrossRef CAS.
- M. M. Smith and D. K. Smith, Soft Matter, 2011, 7, 4856–4860 RSC.
- S. Khanna, M. K. Khan and P. Sundararajan, Langmuir, 2009, 25, 13183–13193 CrossRef CAS.
- R. K. Das, R. Kandanelli, J. Linnanto, K. Bose and U. Maitra, Langmuir, 2010, 26, 16141–16149 CrossRef CAS.
- M. Zinic, F. Vogtle and F. Fages, in Low Molecular Mass Gelators: Design, Self-Assembly, Function, Springer-Verlag Berlin, Berlin, 2005, pp. 39–76 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data in the form of CIF files. Job plots of binding of anions. Rheological characterisation of all gelators and further rheology on anion tuning of the gels are presented. Additional fluorescence studies of the anion tuning of gelator 2 are also given. CCDC reference numbers 681966, 681967, 837366–837368. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sm06448g |
| This journal is © The Royal Society of Chemistry 2012 |