Exciton delocalization and dynamics in helical π-stacks of self-assembled perylene bisimides†
Jong Min
Lim
a,
Pyosang
Kim
a,
Min-Chul
Yoon
a,
Jooyoung
Sung
a,
Volker
Dehm
b,
Zhijian
Chen
bc,
Frank
Würthner
*b and
Dongho
Kim
*a
aDepartment of Chemistry, Yonsei University, Seoul, 120-749, Korea. E-mail: dongho@yonsei.ac.kr; Fax: +82-2-2123-2434; Tel: +82-2-2123-2436
bInstitut für Organische Chemie & Center for Nanosystems Chemistry, Universität Würzburg, Würzburg, 97074, Germany. E-mail: wuerthner@chemie.uni-wuerzburg.de; Fax: +49-931-31-847756; Tel: +49-931-31-85340
cSchool of Chemical Engineering and Technology, Tianjin University, Tianjin, 300072, China
First published on 5th October 2012
Abstract
Whilst the excitonic properties of J-aggregates have been investigated in great detail, those of H-aggregates have not been systematically investigated yet. In this regard, we have explored the exciton dynamics and excited-species formation processes in columnar H-aggregates of planar PBI dyes that are stacked in a helical fashion by various spectroscopic techniques such as time correlated single-photon counting and femtosecond pump–probe measurements with anisotropy changes. The outcome of this study is that photogenerated excitons in helically stacked PBI dyes experience complicated relaxation processes that involve excited-state interactions such as exciton delocalization and excimer formation. To scrutinize the exciton dynamics in the helically stacked aggregates, we have also included distorted bay-substituted PBI dyes as reference molecules that exhibit either no or only relatively small-sized dimeric aggregate structures. The comparative study revealed that the excited-state interactions in the large-sized helically stacked aggregates extend beyond two PBI units, leading to a final excimer (here, excimer means not only an “excited dimer” but an “excited multimer”) trap state within ∼50 ps. Although in competition with this relaxation path into the excimeric trap state, exciton diffusion has been revealed by exciton–exciton annihilation processes, occurring at high excitation power. Whilst the excimer formation process interrupts the direct observation of exciton diffusion in these columnar PBI aggregates, the exciton migration distance could be estimated by the incorporation of non-fluorescent PBI quencher molecules. From this analysis we can conclude that the exciton diffusion can reach a length of about 10 monomer units. Although this value appears to be shorter than those values observed for J-aggregates, this result shows that columnar PBI stacks might still be useful for optoelectronic applications if the relaxation process leading to excimer traps is prevented, e.g. by structural modifications of the molecules.
Introduction
Self-assemblies of π-conjugated molecular systems have drawn much attention due to their potential applications in photovoltaic and optoelectronic devices.1–5 These systems are promising materials for novel organic semiconductors, bridging the gap between single molecules and organic semiconductor films with high-charge carrier mobility.6 For instance, one-dimensional (1D) structures composed of single-stranded π-stacked monomers are of particular interest due to their potential advantages of superior charge transport along the long axis of aggregates, which is prerequisite for applications in nanoscale transistors, sensors, power sources, battery anodes and organic light-emitting displays.7–13 On the other hand, in contrast with charge transport phenomena, far less studies are available on exciton transport along 1D π-stacked aggregate structures, although this property is of equal importance. In this regard, it is necessary to elucidate the nature of molecular excitons generated in self-assembled molecular aggregates for their practical applications.8,11,12,14–19Among various π-conjugated molecules, self-assembled π-stacks composed of perylene tetracarboxylic acid bisimide (PBI) dyes with high chemical robustness and inherent high fluorescence quantum yield can be ideal candidates for the investigation of photodynamic processes in self-assembled molecular architectures.20,21 Since the single strand of PBI monomers can be easily fabricated by controlling the intermolecular forces such as hydrogen bonding and π–π stacking interactions, there have been various approaches to construct novel self-assembled supramolecular structures with characteristic photophysical properties.20–26 For instance, a PBI molecule which exhibits a distorted perylene core with a twist angle of 25–30° due to its four tert-butylphenoxy substituents, reveals well-ordered J-type PBI aggregates in nonpolar solvents such as methylcyclohexane (MCH).27 In this case, the hydrogen-bonding interaction induced by aminoethyl substituents at the imide positions of the PBI also supports the π-stacking arrangement along 1D helical stacks. The PBI aggregates reveal sharp J-type absorption and emission bands, indicating a strong excitonic coupling which is a prerequisite for facile exciton transport process in photonic and photovoltaic applications. Indeed, it was demonstrated that the J-type aggregation of PBI dyes is a promising artificial system for energy migration without exciton trapping.28
Unfortunately, however, in the absence of specific intermolecular interactions, such as hydrogen-bonding as mentioned above, most PBI molecules stack cofacially and form H-type aggregates, which can be analyzed by various geometric factors such as tilt angle to the long axis of aggregates, twist angle and distance between neighboring PBI molecules.29,30 Thus, in this work, we have explored the exciton relaxation processes and the role of the excited species in helically stacked PBI aggregates (Chart 1, PBI 1). The understanding of exciton dynamics that precedes the excimer formation process and the role of photoexcited species in helically stacked dye aggregates will provide important information for the utilization of self-assembled supramolecular complexes in tailored energy and charge transport devices.31 PBI 1 having a planar structure without bay substituents forms columnar aggregates with helical stacking in MCH (Chart 1, PBI 1).29,30 The columnar stacks of PBI 1 with a rotational displacement, in which the monomer units are separated by 3.5 Å, can be an appropriate system to substantiate the role of excited species in the exciton dynamics.9,29 Unlike typical excimers which constitute only π–π aggregates in their excited states, extended π-stacking already exists for PBI 1 in its ground state, and the excimeric species are associated with structural relaxation into dimer units with significantly smaller rotational displacement between the two PBIs.10 Owing to a mismatch between the Franck–Condon state and the excimer state, the excimer-type species also exhibit bathochromically displaced emission bands with rather long fluorescence lifetimes.29 Due to the structural reorganization in the excited state, the excimer state can participate in the exciton self-trapping process.29 Moreover, it is also expected that charge transfer (CT) may participate in the formation of the relaxed excited species.29–32 In other words, photoexcited species in helically stacked PBI aggregates can be of excimer or charge transfer (CT) state with geometric distributions among PBI monomers.
 | ||
| Chart 1 Molecular structures of PBI dyes employed in this study. PBIs 1 and 4 adopt helical aggregate structures in MCH solvent at high concentrations while PBIs 2 and 3 have dimer and monomer forms, respectively. PBI 4 with its electron-donating 3,4,5-tridodecyloxyphenyl substituents shows non-fluorescence behaviour. | ||
As reference molecules, we have adopted bay substituted PBIs to induce structural distortions in the PBI bay area, leading to an alteration in the π-stacked aggregate structures (Chart 1, PBIs 2 and 3).33 While PBI 1 forms helical aggregates with extended rod-like structures, the bay substituted PBIs 2 and 3 do not easily achieve aggregates larger than dimers due to their core distortion, with dihedral angles of 15 and 27°, respectively. Two naphthalene imide subunits of the PBI are twisted by steric hindrance at the bay region of the PBI which influences the intermolecular π–π interactions between the PBI dyes, leading to significantly smaller association constants, and as a consequence, the formation of only dimeric self-assembled structures in the accessible concentration range (up to ∼1 mM).33 Interestingly, despite the similarities in their steady-state spectra with broad absorption and excimeric emission features, PBIs 1 and 2 reveal completely different excited-state dynamics, especially exciton–exciton annihilation processes. This feature indicates that the exciton dynamics can occur in the whole assembled structures and the coherent exciton can exist beyond the segment of PBI dimers.
To support our arguments, a certain spatial limit in exciton diffusion process in the PBI helical aggregates has been estimated. The exciton diffusion length can be estimated from the analyses of transient absorption decay profiles and intrinsic lifetimes of the excitonic state.28 Unfortunately, however, in the helically stacked columnar aggregates, the aforementioned excimeric state can interrupt this approach with its exciton self-trapping process. Alternatively, we have explored the exciton dynamics in hetero-PBI aggregates which can provide a clue in exciton diffusion processes. In the formation of aggregates in which PBI 4 is introduced as an exciton quencher, due to its non-fluorescence nature, the exciton diffusion length can be scrutinized with a careful control of the ratio between PBIs 1 and 4.34
Overall, we have characterized the exciton relaxation dynamics in various self-assemblies of π-conjugated systems by changing their aggregate structures. By controlling the bay substituents, we have obtained large-sized aggregates, dimer and monomer structures in nonpolar MCH solvent for PBIs 1, 2 and 3, respectively, while monomeric forms of the PBI systems were achieved in typical organic solvents such as chloroform. Especially, we have focused on the excimer formation and exciton diffusion processes in the helically stacked columnar aggregate system. To derive information on the exciton dynamics among aggregate units having identical electronic structures, we examined the spectroscopic observables with excitation power density and anisotropy changes. Herein, based on the interpretations of exciton dynamics in the covalent polymers, we have examined the photodynamic nature of excitons generated in the π-stacked PBI aggregate systems.
Results
Steady-state absorption and fluorescence measurements
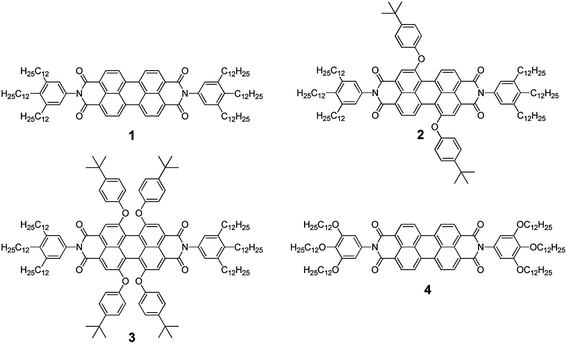
Fig. 1 shows the steady-state absorption spectra of helically stacked PBI 1 dissolved in MCH in the concentration range of 10−7 to 10−3 M. A highly diluted MCH solution (10−7 M) of PBI 1 revealed a monomeric absorption feature which is well recognized by the characteristic vibronic progression of the S0–S1 transition between 400 and 550 nm.29 These spectral features of PBI 1 in highly diluted MCH are also comparable to those of PBI 1 in chloroform (Fig. 1 and S1, ESI†). As the concentration of PBI 1 is increased in MCH, the absorption spectra become broad and less-structured with an appearance of an additional absorption band at longer wavelength region. Since the exciton coupling is generated by the interactions of molecular transition dipole moments in close proximity, the two representative types of aggregates reveal characteristic absorption behaviors.28,35 Generally, while H-type aggregates show a hypsochromic shift as compared with monomeric absorption spectra, the red-shifted absorption spectra of J-type aggregates exhibit sharp and intense transitions. These features indicate that the aggregate system of PBI 1 is neither exactly J- nor H-type. However, as suggested by Chen et al. based on the exciton model, the experimental data are consistent with aggregate structures of rotationally displaced PBI dyes in which the monomeric transition dipoles stack in a helical fashion.29 More recently, helical stacks of PBI 1 aggregates were also elucidated by quantum mechanical studies such as DFT calculations.10,36,37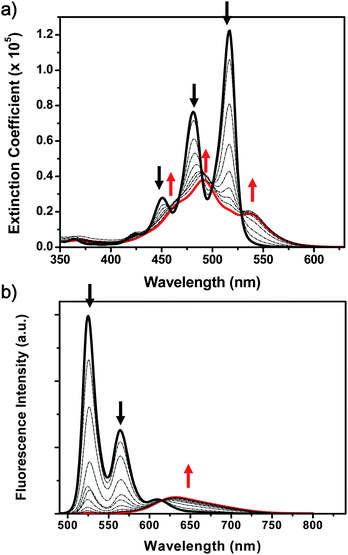 | ||
| Fig. 1 Concentration-dependent (a) UV-vis absorption and (b) emission spectra of PBI 1 in methylcyclohexane. The concentration was varied in the range from 10−7 (black bold) to 10−3 M (red bold). | ||
Gradual changes in the fluorescence spectra of PBI 1 were observed as the concentration was increased (Fig. 1). As the steady-state absorption features represent the electronic coupling among the PBI monomer units, the fluorescence emission arises from the relaxed excimer state.29 While monomeric emission bands in 500–650 nm disappeared, a broad emission band at ∼650 nm was newly observed as the concentration was increased. Time-resolved fluorescence measurements also revealed the lifetime changes as the concentration increased (Fig. S2, ESI†); fluorescent species with longer lifetime (>30 ns) became dominant. Interestingly, when we observed the fluorescence temporal profiles in a short time window, the rise profiles with a time constant of ∼200to 300 ps appeared as the concentration was increased in the spectral region of excimer fluorescence at 700 nm (Fig. S3, ESI†).
The distorted PBIs, bay-substituted PBIs 2 and 3, exhibit electronic interactions among the PBI core systems and electron donating bay-substituents.38 Compared to the planar PBI 1, the monomeric distorted PBIs show broader and red-shifted absorption spectra in chloroform (Fig. S1, ESI†). The distorted molecular structures of the PBIs and resulting electronic interactions also affect their self-assembled structures. When the PBIs (2 and 3) were dissolved in MCH, larger aggregates were not achieved. Formation of aggregates assembled by more than two or three monomers is not feasible due to the structural distortions of constituent monomers 2 and 3. The calculated formation constants of 2 and 3 also indicate that no larger aggregates can be expected under our experimental conditions (ca. 10−7 to 10−3 M).29,33 However, owing to the difference in the number of bay-substituents, PBIs 2 and 3 exhibit distinctly different aggregate features from each other. While the fluorescence emission of PBI 3 in MCH reveals vibronic structures comparable to its monomeric form in chloroform, PBI 2 reveals excimeric fluorescence features in MCH with reduced quantum yield (Fig. S1, ESI†). Because of their assembly features in MCH solvent as dimer (2) and monomer states (3), respectively, these distorted PBI dyes are appropriate reference systems for the helical aggregates of PBI 1. It should be noted that dimerized PBI 2 (10−3 M) in MCH does not show the rise profile at the region of excimer fluorescence.
Modification of the phenyl substituent at the terminal nitrogens of PBI is another method to control the self-assembly of PBI dyes.34 Owing to the strong electron-donating 3,4,5-tridodecyloxyphenyl substituents and resulting non-emissive charge separated excited state, the monomeric form of PBI 4 reveals non-fluorescent behavior and slightly more red-shifted absorption spectra as compared with PBI 1 (Fig. S1, ESI†).39,40 The aggregate form of PBI 4 in MCH solvent also reveals slightly red-shifted absorption spectra as compared with that of PBI 1. Thus, we expect that PBI 4 is supposed to act as an exciton quencher in the self-assembled hetero-PBI aggregates composed of PBIs 1 and 4 (vide infra).
Time-resolved anisotropy measurements
A highly concentrated solution PBI 1 in MCH (10−3 M) shows rise dynamics for the excimeric fluorescence emission without a corresponding decay profile at the spectral region of monomeric emission (Fig. S3, ESI†). Also it was reported for mixtures of monomeric and aggregated species that the steady-state fluorescence anisotropy values are determined as 0.11 for the monomer band and 0.003 for the aggregate band, respectively.29 These features indicate for the aggregated dye manifold that there exist complex dynamics during the relaxation processes from the nascent exciton to excimer state. To further elucidate the photophysical processes in these aggregates, we have monitored time-resolved anisotropy changes in time-resolved fluorescence and excited-state absorption spectra. Since the generated excitons can migrate in the helically stacked columnar aggregates where rotational displacements exist between the neighboring PBI dyes, the anisotropy dynamics can give a clue to their complex relaxation processes. The anisotropy change implies a dependence on the relative orientation between pump and probe polarization which is caused by reorientation of excitonically coupled transition dipole moments or by differently oriented transition dipole moments.28,41 In our case, the former can be related to the rotational diffusion motion of the PBI dye systems. While the monomeric PBI shows a rotational diffusion motion of ∼1.5 ns, the rotational diffusion time of concentrated PBI in MCH becomes much slower (Fig. S4 and S5, ESI†). Moreover, the initial fluorescence anisotropy value was reduced as the concentration was increased (Fig. 2a, Fig. S4, ESI† and ref. 29). In highly diluted condition (10−6 M) in MCH, the initial anisotropy value was estimated to be ∼0.33. This value gradually decreased as the aggregate structures were attained; the initial anisotropy value was estimated to be 0.1 at a PBI 1 concentration of 10−3 M. In contrast to the slower rotational diffusion motion, the fluorescence anisotropy of PBI 1 aggregates reveals fast decay profiles (Fig. 2). As the concentration of PBI 1 is increased in MCH, the relative intensity of the longer time component corresponding to the rotational diffusion motion is reduced. At highly concentrated condition (10−3 M), a fast decay component of ∼100 ps becomes dominant and the influence of rotational diffusion motion almost vanishes in the decay profile of the fluorescence anisotropy.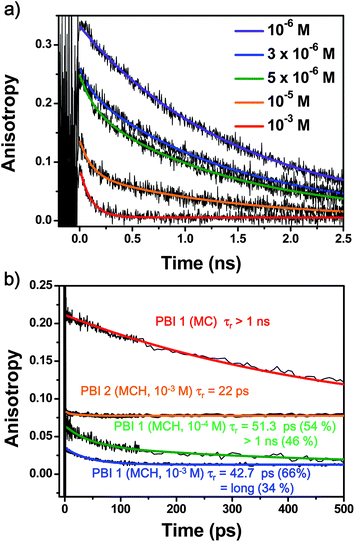 | ||
| Fig. 2 Concentration-dependent anisotropy changes of PBIs 1 and 2 in methylcyclohexane; (a) time-resolved fluorescence anisotropy decay of PBI 1 and (b) fs-transient absorption anisotropy decays of PBIs 1 and 2. The concentration of PBI 1 was 10−4 or 10−3 M and of PBI 2 was 10−3 M. The anisotropy decay of monomeric PBI 1 in dichloromethane (MC) is also included. | ||
Furthermore, we have examined the anisotropy decay profiles in the excited state of the PBI aggregates (10−4 to 10−3 M in MCH) and monomer form (in dichloromethane) by using femtosecond transient absorption anisotropy measurement. Upon photoexcitation of PBI aggregates at 550 nm, we monitored the anisotropy decay of excited-state absorption (ESA) signals at 620 nm to minimize the contribution by ground-state bleaching (GSB, at 500–600 nm) and stimulated emission (SE). As in the case of fluorescence anisotropy, the reduced initial values as well as the fast decay time constant dominantly describe the anisotropy dynamics of excited state absorption, indicating that ultrafast dipole interactions take place within the helically stacked PBI dyes in the exciton relaxation processes. Whereas there is no specific fast dynamics in the monomeric forms of PBI 1 (in dichloromethane), the aggregates (10−3 M) exhibit a decreased anisotropy value of 0.03 and relatively fast decay profiles with a time constant of 40–50 ps (Fig. 2), which might be attributed to the exciton formation process and their electronic interactions along the long axis of the π-stacks.
To clarify the origin of anisotropy dynamics in the helical stacks of PBI 1, we refer to the transient absorption anisotropy and fluorescence up-conversion temporal profiles of dimerized PBI 2. It should be noted that two independent spectroscopic tools, transient absorption anisotropy (Fig. 2b) and fluorescence up-conversion measurements (Fig. S6, ESI†), reveal similar decay profiles of dimerized PBI 2 with a time constant of ∼20 ps. Based on these results, we can interpret that the anisotropy decay profiles correspond to the excited dimer formation process. At high concentration (10−3 M), interestingly, the temporal profile of dimerized PBI 2 exhibits characteristic transient anisotropy features. The initial anisotropy value is estimated to be 0.08, which is lower than that of PBI 1 monomer, and higher than that of PBI 1 aggregates. After the decay process with a time constant of ∼20 ps, the anisotropy maintains nearly at a constant value. Considering the fact that the initial anisotropy value is ascribed to the interactions between PBIs, the excitonic interactions that reduce the anisotropy are not pronounced in the PBI 2 dimer system. This conjecture can be supported by the relatively high anisotropy intensity of the dimerized PBI 2. Thus, in both cases, large-sized columnar PBI 1 and small-sized PBI 2, we could expect that the reduced initial anisotropy values are affected by the exciton formation processes. In other words, our results suggest that the coherent exciton can exist beyond the segment of the PBI dimer.29,42–44 After the exciton formation process, excimer species such as excited dimer and excited multimers, seem to be produced for PBIs 2 and 1, respectively.
Femtosecond transient absorption measurements
To scrutinize the relaxation pathways from the nascent exciton state to the excimer states, we have utilized fs-transient absorption measurements by changing the excitation laser power. In the monomeric forms of PBI 1 in dichloromethane or chloroform, we could distinguish their excited state dynamics by their fast solvent relaxation process and S1 → S0 transition as well as residual species of the triplet excited state.22,23 Thus, we could assign the singlet excited state lifetime of the monomeric PBI 1 (Fig. S7, ESI†). Following the initial fast decay profiles of a few ps for solvent relaxation process, the transient species reveals its decay profile with a time constant of 1.4 ns (Fig. S7, ESI†). While the strong negative signals are originated from the GSB and SE processes, the ESA signals emerge as positive ones in the spectral region longer than 630 nm. These results are consistent with those in the time-resolved fluorescence measurements (Fig. S5, ESI†). We also observed the appearance of transient species that are sustained beyond our experimental time window (∼3 ns) that might be originated from the triplet excited state of the PBI molecule.22,23 Furthermore, there are no excitation power dependences on the transient absorption decay profiles in the monomeric forms.The transient species of PBI 1 aggregates (10−3 M in MCH) exhibit broad ESA and GSB signals in the entire spectral region (Fig. 3). Compared to the transient absorption spectra of PBI 1 monomers in chloroform (Fig. S7, ESI†), those of aggregates reveal broad GSB and ESA without SE signals. The transient species exhibit no spectral changes in our experimental time window (3 ns) and the temporal profile of ESA signals exhibits longer decay processes than that of the monomer.
 | ||
| Fig. 3 Femtosecond transient absorption spectra of (a) PBI 1 and (b) PBI 2 in MCH (inset: excitation power dependent signals in the initial time domain and thick arrows indicate the probe wavelengths of 490 and 700 nm for PBI 1 and PBI 2, respectively). The excitation power is controlled in the range of 0.8–4.6 mW. | ||
The monomeric transient species in chloroform exhibits similar temporal changes in the spectral shapes and dynamics to those of PBI 1, i.e. broad GSB and SE signals in the transient absorption spectra appeared, reflecting the characteristic red-shifted absorption and fluorescence emission in the steady-state spectra (Fig. S8, ESI†). Without any spectral shifts, the temporal profile of the transient species can be fitted to a single exponential decay with a time constant of 2 ns. Accordingly, PBI 2 aggregates in MCH reveal a longer decay time constant than PBI 1 monomers. However, bulky and highly distorted PBI 3 does not show any distinctive transient spectral differences between the two forms in chloroform and MCH except the spectral shift due to solvent polarities. In the steady state, we have already confirmed that PBI 3 reveals neither aggregate absorption nor excimeric fluorescence signals in the concentration range of ca. 10−7 to 10−3 M. The transient species in the two solvents, chloroform and MCH, reveal similar decay profiles. The non-fluorescence nature of PBI 4 substituted by the electron-donating trialkoxyphenyl group is also revealed in its excited state dynamics (Fig. S9, ESI†). Without prominent SE signals, the excited state absorption and ground-state bleaching signals of PBI 4 show a fast recovery process with a lifetime of ∼10 ps.
When multiple excitons are generated and more than two excitons are in close proximity, their exciton–exciton annihilation process allows an access to higher states that have fast relaxation channels.45 The inset of Fig. 3a reveals the transient absorption decay profiles of PBI 1 (10−3 M in MCH) in the initial time window (∼10 ps) under different excitation power. At high excitation energies (4.6 μJ), the transient absorption signal of PBI 1 shows a prominent fast decay profile with a time constant of 0.8 ps. This fast relaxation dynamics disappeared upon decreasing the excitation power which is a clear evidence for the exciton–exciton annihilation process in molecular aggregates. For the distorted PBIs, 2 and 3, there are no excitation power dependences on their transient absorption decay profiles (inset Fig. 3b). For the highly distorted PBI 3 this result is rather trivial because it is explained by the fact that no aggregates are present at the given concentration. On the other hand, PBI 2 forms dimer aggregates at high concentrations (10−3 M) but nevertheless does not show any specific excitation power dependency. Accordingly, despite the similarities in transient species (broad GSB and ESA signals without strong SE band and a long lifetime of a few nanoseconds), highly aggregated PBI 1 and dimerized PBI 2, show large differences in excitation power dependencies which is obviously related to their degree of aggregation: PBI 1 forms a rod-like large-sized columnar aggregate while PBI 2 does not form extended aggregates. This feature indicates that photogenerated excitons in extended aggregate systems are still mobile and can communicate with each other throughout the rod-like architecture until the excimer trap state is formed.
Formation of hetero-aggregates incorporating PBI exciton quencher PBI units
Generally, exciton luminescence decays have been analyzed to determine exciton transport phenomena.13,45–48 As a direct approach, one can observe the exciton transfer to the sites of energy or electron acceptors in the aggregate systems. When exciton acceptors are embedded into the aggregate system, they can act as exciton trap sites that regulate the exciton diffusion (or migration) processes.49–53 The insertion of quencher molecules (that exhibit lower energy than the donor molecules) into the molecular aggregates gives rise to a trapping of excitons, leading to a termination of the exciton migration, depending on the ratio between the luminescent energy donor and quenching acceptor moieties. In previous researches on squararine,54,55 and oligophenylenevinylene dyes,46–48 it was found that there are several factors which affect the trapping processes such as the relative concentration ratio of quencher and the overall aggregate structures. In our experiment, we have prepared hetero-PBI aggregates to investigate the exciton delocalization and diffusion preceding the excimer formation process within the confined length of aggregates by embedding exciton quenchers at certain positions along the long axis of the molecular aggregates (Fig. S9, ESI†). The spectroscopic analyses were made for the hetero-aggregate systems composed of PBI 1 and PBI 4 at different ratios where PBI 4 can act as an effective quencher due to its extremely short excited-state lifetime (Fig. S9, ESI†). The relative ratio between fluorescent PBI 1 and non-fluorescent PBI 4 was carefully controlled.We have observed fluorescence intensity change in the hetero-PBI aggregates depending on the concentration of PBI 4 (Fig. S11, ESI†). As the mole ratio of PBI 4 is increased, the excimeric fluorescence emission of the aggregates gradually decreased. To examine the trapping efficiency contributed by PBI 4, the fluorescence quenching of the hetero-PBI aggregates was analyzed by using the Stern–Volmer equation,56 leading to a Stern–Volmer constant of 1.25 × 105 M−1. Taking into account the concentration of PBI 1 (1.25 × 10−4 M) and 1/Ksv, we evaluate that one quencher PBI 4 interacts with at least 16 PBI units in the hetero-PBI aggregates. This suggests that the PBI 4 moiety efficiently quenches the exciton which is migrating along the rod-like columnar aggregate structure in spite of the energetic similarities between the two PBI dyes.
Additionally, we have investigated the excitation power dependences on the TA measurements for the hetero-PBI aggregates (the ratio of PBIs 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 was changed gradually from 1000:1 to 5:1). Fig. 4 reveals the exciton–exciton annihilation dynamics of PBI 1–PBI 4 hetero-aggregates for which we assume a statistical incorporation of the minor component PBI 4 within the PBI 1 stack. This assumption appears reasonable because both dyes form stacks of identical columnar diameter by non-directional van der Waals interactions.29,34 Interestingly, the exciton–exciton annihilation processes are restricted as the concentration of quenchers increases. When the ratio of PBIs 1:4 was above 10:1, we clearly observed pump-power dependent fast decay dynamics. As the concentration of PBI 4 was increased, the excitation power dependences, the fast decay dynamics responsible for exciton–exciton annihilation process, completely vanished. Based on these results, we can suggest that the exciton diffusion length does not exceed about ten PBI units.
:4 was changed gradually from 1000:1 to 5:1). Fig. 4 reveals the exciton–exciton annihilation dynamics of PBI 1–PBI 4 hetero-aggregates for which we assume a statistical incorporation of the minor component PBI 4 within the PBI 1 stack. This assumption appears reasonable because both dyes form stacks of identical columnar diameter by non-directional van der Waals interactions.29,34 Interestingly, the exciton–exciton annihilation processes are restricted as the concentration of quenchers increases. When the ratio of PBIs 1:4 was above 10:1, we clearly observed pump-power dependent fast decay dynamics. As the concentration of PBI 4 was increased, the excitation power dependences, the fast decay dynamics responsible for exciton–exciton annihilation process, completely vanished. Based on these results, we can suggest that the exciton diffusion length does not exceed about ten PBI units.
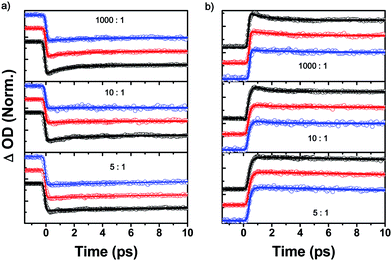 | ||
| Fig. 4 S1–S1 annihilation process in PBI 1–4 hetero-aggregates observed at (a) 490 nm and at (b) 730 nm. The relative ratio between PBIs 1 and 4 were controlled (1000:1, 10:1 and 5:1). The excitation powers are 9.0, 2.8 and 1.00 mW for black, red and blue circles, respectively. | ||
Discussion
There are several excellent studies on the excited-state dynamics of dimeric and trimeric PBI dye-aggregates which include vibrational relaxation process and singlet- (or triplet) excited-state behavior.22–24 For the highly aggregated PBI systems investigated in this work, totally different spectroscopic features as compared with their monomeric forms, are revealed. This suggests that the transition dipoles located on the long molecular axis of PBI monomers interact with each other in their dimeric or columnar structures, resulting in spectral shift and broadening of the absorption band. In the case of PBI 1, the highly aggregated form (10−3 M) reveals complicated absorption spectra arising simultaneously from J- and H-type couplings. The fluorescence spectra of these aggregates (10−3 M) reveal the characteristic features of an excimer state; broader spectra with much longer lifetime (>30 ns) than those of the monomer. Neither temporal nor spectral evolution corresponding to the decay dynamics appears in the transient absorption measurements. Since new transient species, excimers, are not optically accessible from the ground state,10 there was no reason for the temporal and spectral shift in GSB signals. Interestingly, however, the fluorescence temporal profile of the aggregate exhibits a rise profile with a time constant of ∼300 ps. It should be emphasized that there is no corresponding transient feature responsible for the monomeric fluorescence emission. Moreover, the highly concentrated PBI 2 exhibiting dimer feature does not show such rise dynamics. Despite the helically stacked columnar aggregates of PBI 1 and the dimer stacks of PBI 2 exhibiting similar excimeric features in the stationary fluorescence spectra, there are certain processes related to the rise profile only in the fluorescence of extended columnar aggregate structures. Compared to the ground-state stacks, the π–π distances between the neighboring PBI units should be reduced in the relaxed structures of the excited species. Further, the resonance contributions of exciton and charge transfer states affect the structural displacement.29,42–44 Thus, we conclude that the fluorescence rise profiles of large aggregates can be affected by structural rearrangement processes as well as excitonic interactions.However, the exciton dynamics from its initial generation to the eventually formed excimer species is still somewhat obscured and several questions arise for the relaxation processes of nascent excitons. The TAA measurements reveal the exciton delocalization within the aggregate system. There is ultrafast depolarization process faster than our experimental time resolution (∼40 fs) that mitigates the initial anisotropy values; this process is more operative in the helically stacked systems than for dimers; while this process diminishes the initial anisotropy value of PBI 1 aggregates to 0.03, the value of dimerized PBI 2 is estimated to be 0.08. It might be reasonable to consider that the decreases of initial anisotropy values originate from the formation of excitons and their interactions. Moreover, based on the steady-state absorption features and calculation results,29,42–44 we can predict that the excimers are composed of more than two PBI units for the PBI 1 π-stacks. In our observation, the transient anisotropy decay profiles also exhibit significant differences between the two PBI self-assembly systems. The anisotropy decay time of dimerized PBI 2 (∼20 ps) is shorter than that of PBI 1 aggregate system (40–50 ps). These data indicate that the anisotropy decay process in PBI 1 aggregates is attributable to excitonic interactions such as formation of an excited multimer state among PBI units. In other words, highly aggregated structures of PBI 1 exhibit a longer formation time to build up its excimer state than the dimerized PBI 2 as well as more extended exciton size. We conjecture that the excimer formation process, including excitonic interactions accompanied by structural rearrangement/ordering processes, should be operative, which is a crucial factor to determine the decrease of anisotropy values.
Transient absorption measurements also reveals the exciton dynamics in various aggregate structures. Owing to the broad transient signals, it might be difficult to resolve the complex exciton dynamics based on their temporal and spectral changes. At least, however, we can discern two distinct decay processes; an ultrafast (∼0.8 ps) excitation power dependent signal and 30 ns time components. Under high excitation power, a more pronounced ultrafast decay profile was observed, which provides evidence for exciton–exciton annihilation. This process takes place when two excitons approach each other and their annihilation allows an access to higher electronic states that have fast electronic relaxation channels. In the columnar aggregate structures, it is relevant that the over-populated excitons can easily interact with each other. The long-lived transient signal corresponds to the lifetime of the excimer state, because identical decay was observed in fluorescence temporal profiles. Moreover, we observed weak rise dynamics at the spectral region of excimeric ESA signals at 830 nm although it was difficult to clarify their time profiles (Fig. S12, ESI†). It may be interpreted by that the weak intensity of the rise dynamics could not overcome the broad and relatively strong ESA signals. However, the rise dynamics independently appear upon photoexcitation at various excitation wavelengths (490, 510 and 550 nm), but does not appear in the monomeric form of PBIs dissolved in chloroform. We expect that the rise profiles correspond to the exciton diffusion process within the excimer formation process as observed in the anisotropy decay measurements.
The interactions among delocalized excitons become manifest as demonstrated by excitation power dependent TA decay signals. Although the exciton diffusion process is restricted by excimer formation processes, the exciton–exciton annihilation process indicates that the delocalized excitons are instantaneously generated and they migrate freely in whole aggregate structures before the formation of a trap state. Thus, we have attempted to substantiate and at least suggest a certain boundary in the exciton diffusion length. Considering the results of quencher embedment experiments, we were able to evaluate a certain boundary for the coherence length of excitons. Based on the Stern–Volmer analysis, we have estimated that one quencher PBI 4 efficiently quenches the fluorescence from nearby 16 PBI molecules. The exciton–exciton annihilation processes originated from the interaction between more than two excitons reveal that the bound exciton can spread out less than 10 monomer units.
The self-assembly systems are defined as regular assemblies composed of repeating molecular entities.57 The difference between a polymer and a self-assembly system is the linkage among their repeating structural units.57 Whereas the former utilizes solid covalent chemical bonds, the latter utilizes directional and reversible secondary interactions such as π–π interactions, hydrogen bonding, metal coordination, hydrophobic forces, van der Waals forces, and/or electrostatic forces. Owing to these differences, self-assembly systems are more sensitive to their internal and external conditions, e.g. sterics of the monomeric building block, concentration, solvent polarity and temperature dependences. Here, in this paper, on the basis of previous researches for the exciton dynamics in covalent polymers,58–60 we suggest the exciton dynamics of PBI aggregates as a series of successive processes such as photoexcitation (delocalization), self-localization, exciton formation, exciton migration, and change of aggregate conformation.
In the case of covalent polymers, the ultrafast (<100 fs) decay dynamics is related to the photogeneration of mobile electrons and holes that experience self-localization, driven by local structural lattice distortions coupled to vibrational relaxations.41 We can assume that the photogenerated excitons in PBI aggregates experience similar relaxation pathways (Scheme 1), but it is difficult to recognize initial self-localization process and the formation of bound singlet exciton states. In our observations, there are no specific temporal and spectral changes during the initial ultrafast time range, which may be considered by two factors, structural effects and experimental limitations. While the covalent polymers reveal randomly distorted local geometries such as kink structures to allow for intermolecular π–π stacking interactions in the excited state, self-assembled aggregates already attain the π-stacked structures with the monomer units rotationally displaced toward each other. There is no need for structural distortions among the monomer units in the columnar aggregates when the bound singlet exciton state is generated. In terms of linkages among unit molecules, the aggregate systems are more homogeneous than conventional covalent polymers. Thus, the self-localization process accompanied by the formation of bound singlet exciton states in the PBI aggregates occur on an ultrafast time scale that exceeds our time resolution. Following the self-localization process, excitons in covalent polymers undergo diffusion or migration among the localized exciton segments along with the delocalization processes.41 In addition to these processes, slow conformational relaxation on the timescale of hundreds of picoseconds occurs in the covalent polymers.41 In the case of helically stacked columnar aggregate systems, however, photogenerated excitons evolve quickly into the excimer state and as a consequence, there is not enough time for diffusion processes within the whole aggregate structures. Furthermore, huge dynamic conformational rearrangement in covalent polymers does not appear in the helically stacked aggregates system due to their homogeneous linkages.
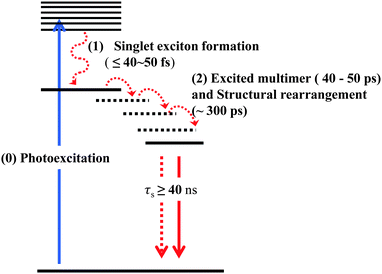 | ||
| Scheme 1 Exciton dynamics in helically stacked aggregates of PBI dyes. | ||
Conclusions
Until now, there have been numerous investigations that are mainly focused on the photophysical properties of slipped stacking structures of J-aggregate PBI systems. However, most self-assembled structures composed of PBI dyes exhibit columnar stacks with complex photophysical properties such as in helically stacked PBI 1 aggregates. In this regard, it was relevant to explore the exciton diffusion and its migration length, and the role of photoexcited species in the exciton relaxation dynamics of helically stacked columnar PBI aggregate systems. In contrast to their monomer forms, the self-assembled PBI systems reveal characteristic excitonic behaviors such as exciton formation and diffusion dynamics as well as excimer formation processes. Referring to the photophysical properties of dimerized PBI 2 and monomeric PBI 3, we can figure out the exciton delocalization and excited multimer formation processes in the helically stacked PBI 1 aggregates. The initial anisotropy values of PBI 1 aggregates indicate the exciton formation process and their interactions. Owing to the attained rod-like aggregates structures and strong interactions among PBI units, the exciton formation and delocalization occur immediately upon photoexcitation. The formation time for excimers in the helical aggregates of PBI 1 is longer than that of excited dimerization process for PBI 2. This feature corresponds to the observation of fluorescence emission of helically stacked PBI 1 in MCH (10−3 M) which shows a rise dynamics (∼300 ps) for the excimeric fluorescence emission without a corresponding decay profile at the spectral region of monomeric emission. In other words, these features indicate that the excimer in the helically stacked PBI 1 is composed of more than two PBI unit molecules. Furthermore, by using quencher embedment and pump-power dependent experiments, we were able to estimate a certain length of exciton diffusion that could be extended up to about 10 monomer units. Namely, the exciton formed in the helically stacked PBI 1 aggregates experiences the diffusion process within the length of 10 monomer units and evolves into the excimer state.Acknowledgements
This work was financially supported by the Mid-career Researcher (2010-0029668) and World Class University (R32-2010-000-10217) Programs of National Research Foundation (NRF) grant funded by the Ministry of Education, Science and Technology (MEST) of Korea and AFSOR/AOARD Grant (FA2386-10-1-4080) (D. K.). The work at Würzburg was financially supported by the DFG for the research unit “Light induced dynamics in molecular aggregates”.Notes and references
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS.
- J. Wang, A. Kulago, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2010, 132, 4191–4196 CrossRef CAS.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef.
- D. A. Doval, J. Areephong, E.-K. Bang, L. Bertone, P. Charbonnaz, A. Fin, N.-T. Lin, M. Lista, S. Matile, J. Montenegro, E. Orentas, N. Sakai, D.-H. Tran and A. V. Jentzsch, Langmuir, 2011, 27, 9696–9705 CrossRef CAS.
- P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 20009–20029 CrossRef CAS.
- V. Faramarzi, F. Niess, E. Moulin, M. Maaloum, J.-F. Dayen, J.-B. Beaufrand, S. Zanettini, B. Doudin and N. Giuseppone, Nat. Chem., 2012, 4, 485–490 CrossRef CAS.
- M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910–1921 CrossRef CAS.
- L. D. Bakalis and J. Knoester, J. Phys. Chem. B, 1999, 103, 6620–6628 CrossRef CAS.
- V. Dehm, Z. Chen, U. Baumeister, P. Prins, L. D. A. Siebbeles and F. Würthner, Org. Lett., 2007, 9, 1085–1088 CrossRef CAS.
- R. F. Fink, J. Seibt, V. Engel, M. Renz, M. Kaupp, S. Lochbrunner, H.-M. Zhao, J. Pfister, F. Würthner and B. Engels, J. Am. Chem. Soc., 2008, 130, 12858–12859 CrossRef CAS.
- T. E. Kaiser, I. G. Scheblykin, D. Thomsson and F. Würthner, J. Phys. Chem. B, 2009, 113, 15836–15842 CrossRef CAS.
- A. N. Lebedenko, R. S. Grynyov, G. Y. Guralchuk, A. V. Sorokin, S. L. Yefimova and Y. V. Malyukin, J. Phys. Chem. C, 2009, 113, 12883–12887 CAS.
- I. G. Scheblykin, O. Y. Sliusarenko, L. S. Lepnev, A. G. Vitukhnovsky and M. Van der Auweraer, J. Phys. Chem. B, 2001, 105, 4636–4646 CrossRef CAS.
- T. Kobayashi, Supramol. Sci., 1998, 5, 343–347 CrossRef CAS.
- Y. Hamanaka, H. Kurasawa, A. Nakamura, Y. Uchiyama, K. Marumoto and S. Kuroda, J. Lumin., 2001, 94–95, 451–455 CrossRef.
- M. van Burgel, D. A. Wiersma and K. Duppen, J. Chem. Phys., 1995, 102, 20–33 CrossRef CAS.
- J. Moll, S. Daehne, J. R. Durrant and D. A. Wiersma, J. Chem. Phys., 1995, 102, 6362–6370 CrossRef CAS.
- L. D. Bakalis, M. Coca and J. Knoester, J. Chem. Phys., 1999, 110, 2208–2218 CrossRef CAS.
- T. Ogawa, E. Tokunaga and T. Kobayashi, Chem. Phys. Lett., 2005, 410, 18–23 CrossRef CAS.
- V. Percec, S. D. Hudson, M. Peterca, P. Leowanawat, E. Aqad, R. Graf, H. W. Spiess, X. Zeng, G. Ungar and P. A. Heiney, J. Am. Chem. Soc., 2011, 133, 18479–18494 CrossRef CAS.
- Z. Chen, A. Lohr, C. R. Saha-Moller and F. Wurthner, Chem. Soc. Rev., 2009, 38, 564–584 RSC.
- D. Veldman, S. M. A. Chopin, S. C. J. Meskers, M. M. Groeneveld, R. M. Williams and R. A. J. Janssen, J. Phys. Chem. A, 2008, 112, 5846–5857 CrossRef CAS.
- C. Hippus, I. H. M. van Stokkum, E. Zangrando, R. M. Williams, M. Wykes, D. Beljonne and F. Würthner, J. Phys. Chem. C, 2008, 112, 14626–14638 Search PubMed.
- J. M. Giaimo, J. V. Lockard, L. E. Sinks, A. M. Scott, T. M. Wilson and M. R. Wasielewski, J. Phys. Chem. A, 2008, 112, 2322–2330 CrossRef CAS.
- M. J. Tauber, R. F. Kelley, J. M. Giaimo, B. Rybtchinski and M. R. Wasielewski, J. Am. Chem. Soc., 2006, 128, 1782–1783 CrossRef CAS.
- A. Sautter, B. K. Kaletas, D. G. Schmid, R. Dobrawa, M. Zimine, G. Jung, I. H. M. van Stokkum, L. De Cola, R. M. Williams and F. Würthner, J. Am. Chem. Soc., 2005, 127, 6719–6729 CrossRef CAS.
- X.-Q. Li, X. Zhang, S. Ghosh and F. Würthner, Chem.–Eur. J., 2008, 14, 8074–8078 CrossRef CAS.
- H. Marciniak, X.-Q. Li, F. Würthner and S. Lochbrunner, J. Phys. Chem. A, 2011, 115, 648–654 CrossRef CAS.
- Z. Chen, V. Stepanenko, V. Dehm, P. Prins, L. D. A. Siebbeles, J. Seibt, P. Marquetand, V. Engel and F. Würthner, Chem.–Eur. J., 2007, 13, 436–449 CrossRef CAS.
- F. Wurthner, Z. Chen, V. Dehm and V. Stepanenko, Chem. Commun., 2006, 1188–1190 RSC.
- F. C. Spano, S. C. J. Meskers, E. Hennebicq and D. Beljonne, J. Am. Chem. Soc., 2007, 129, 7044–7054 CrossRef CAS.
- W. Liu, V. Settels, P. H. P. Harbach, A. Dreuw, R. F. Fink and B. Engels, J. Comput. Chem., 2011, 32, 1971–1981 CrossRef CAS.
- Z. Chen, U. Baumeister, C. Tschierske and F. Würthner, Chem.–Eur. J., 2007, 13, 450–465 CrossRef CAS.
- F. Würthner, C. Thalacker, S. Diele and C. Tschierske, Chem.–Eur. J., 2001, 7, 2245–2253 CrossRef.
- F. C. Spano, Acc. Chem. Res., 2010, 43, 429–439 CrossRef CAS.
- H.-M. Zhao, J. Pfister, V. Settels, M. Renz, M. Kaupp, V. C. Dehm, F. Würthner, R. F. Fink and B. Engels, J. Am. Chem. Soc., 2009, 131, 15660–15668 CrossRef CAS.
- Z. Chen, B. Fimmel and F. Wurthner, Org. Biomol. Chem., 2012, 10, 5845–5855 CAS.
- E. Fron, G. Schweitzer, P. Osswald, F. Wurthner, P. Marsal, D. Beljonne, K. Mullen, F. C. De Schryver and M. Van der Auweraer, Photochem. Photobiol. Sci., 2008, 7, 1509–1521 CAS.
- C. Hippius, I. H. M. v. Stokkum, E. Zangrando, R. M. Williams and F. Würthner, J. Phys. Chem. C, 2007, 111, 13988–13996 CAS.
- R. M. Williams, Turk. J. Chem., 2009, 33, 727–737 CAS.
- N. Banerji, S. Cowan, M. Leclerc, E. Vauthey and A. J. Heeger, J. Am. Chem. Soc., 2010, 132, 17459–17470 CrossRef CAS.
- J. Seibt, V. Dehm, F. Wurthner and V. Engel, J. Chem. Phys., 2007, 126, 164308 CrossRef.
- J. Seibt, P. Marquetand, V. Engel, Z. Chen, V. Dehm and F. Würthner, Chem. Phys., 2006, 328, 354–362 CrossRef CAS.
- J. Seibt, T. Winkler, K. Renziehausen, V. Dehm, F. Würthner, H. D. Meyer and V. Engel, J. Phys. Chem. A, 2009, 113, 13475–13482 CrossRef CAS.
- V. Sundström, T. Gillbro, R. A. Gadonas and A. Piskarskas, J. Chem. Phys., 1988, 89, 2754–2762 CrossRef.
- F. J. M. Hoeben, L. M. Herz, C. Daniel, P. Jonkheijm, A. P. H. J. Schenning, C. Silva, S. C. J. Meskers, D. Beljonne, R. T. Phillips, R. H. Friend and E. W. Meijer, Angew. Chem., Int. Ed., 2004, 43, 1976–1979 CrossRef CAS.
- D. Beljonne, E. Hennebicq, C. Daniel, L. M. Herz, C. Silva, G. D. Scholes, F. J. M. Hoeben, P. Jonkheijm, A. P. H. J. Schenning, S. C. J. Meskers, R. T. Phillips, R. H. Friend and E. W. Meijer, J. Phys. Chem. B, 2005, 109, 10594–10604 CrossRef CAS.
- C. Daniel, F. Makereel, L. M. Herz, F. J. M. Hoeben, P. Jonkheijm, A. P. H. J. Schenning, E. W. Meijer and C. Silva, J. Chem. Phys., 2008, 129, 104701–104707 CrossRef.
- S. Kirstein and S. Daehne, Int. J. Photoenergy, 2006, 2006, 1–21 Search PubMed.
- T. Kobayashi, T. Taneichi and S. Takasaka, J. Chem. Phys., 2007, 126, 194705 CrossRef.
- D. L. Huber, Phys. Rev. B, 1979, 20, 2307–2314 CrossRef CAS.
- R. M. Jones, L. Lu, R. Helgeson, T. S. Bergstedt, D. W. McBranch and D. G. Whitten, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14769–14772 CrossRef CAS.
- L. Lu, R. Helgeson, R. M. Jones, D. McBranch and D. Whitten, J. Am. Chem. Soc., 2002, 124, 483–488 CrossRef CAS.
- R. S. Grynyov, A. V. Sorokin, G. Y. Guralchuk, S. L. Yefimova, I. A. Borovoy and Y. V. Malyukin, J. Phys. Chem. C, 2008, 112, 20458–20462 CAS.
- A. V. Sorokin, I. I. Filimonova, R. S. Grynyov, G. Y. Guralchuk, S. L. Yefimova and Y. V. Malyukin, J. Phys. Chem. C, 2010, 114, 1299–1305 CAS.
- J. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, Berlin, 2006 Search PubMed.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS.
- H. Bässler and B. Schweitzer, Acc. Chem. Res., 1999, 32, 173–182 CrossRef.
- E. Busby, E. C. Carroll, E. M. Chinn, L. Chang, A. J. Moulé and D. S. Larsen, J. Phys. Chem. Lett., 2011, 2, 2764–2769 CrossRef CAS.
- L. M. Herz, C. Daniel, C. Silva, F. J. M. Hoeben, A. P. H. J. Schenning, E. W. Meijer, R. H. Friend and R. T. Phillips, Phys. Rev. B: Condens. Matter, 2003, 68, 045203 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2sc21178e |
| This journal is © The Royal Society of Chemistry 2013 |